
Abstract
We investigated the doping and temperature evolutions of the optical response of Sr3(Ir1−xMnx)2O7 single crystals with 0 ≤ x ≤ 0.36 by utilizing infrared spectroscopy. Substitution of 3d transition metal Mn ions into Sr3Ir2O7 is expected to induce an insulator-to-metal transition via the decrease in the magnitude of the spin–orbit coupling and the hole doping. In sharp contrast, our data reveal the resilience of the spin–orbit coupling and the incoherent character of the charge transport. Upon Mn substitution, an incoherent in-gap excitation at about 0.25 eV appeared with the decrease in the strength of the optical transitions between the effective total angular momentum Jeff bands of the Ir ions. The resonance energies of the optical transitions between the Jeff bands which are directly proportional to the magnitude of the spin–orbit coupling hardly varied. In addition to these evolutions of the low-energy response, Mn substitution led to the emergence of a distinct high-energy optical excitation at about 1.2 eV which is larger than the resonance energies of the optical transitions between the Jeff bands. This observation indicates that the Mn 3d states are located away from the Ir 5d states in energy and that the large difference in the on-site energies of the transition metal ions is responsible for the incoherent charge transport and the robustness of the spin–orbit coupling. The effect of Mn substitution was also registered in the temperature dependence of the electronic response. The anomaly in the optical response of the parent compound observed at the antiferromagnetic transition temperature is notably suppressed in the Mn-doped compounds despite the persistence of the long-range antiferromagnetic ordering. The suppression of the spin-charge coupling could be related to charge disproportionation of the Ir ions.
Similar content being viewed by others

Introduction
Layered perovskite iridates of the Srn+1IrnO3n+1 (n = 1, 2) have attracted a great deal of attention as candidate systems from which unconventional superconductivity may emerge1,2,3,4. Srn+1IrnO3n+1 is an effective total angular momentum Jeff = 1/2 Mott insulator realized by the cooperation of the moderate electronic correlations and strong spin–orbit coupling (SOC)5,6,7. The Jeff = 1/2 Mott state of the iridates shares close similarities in its electromagnetic properties with the cuprates, which motivated intensive efforts to search for novel phases of the iridates by means of charge carrier doping. The representative cuprate phenomenology including the pseudogap8,9, the d-wave gap9,10, charge density wave11 was also observed in doped iridates.
Since the Mott state of the iridates is stabilized by the strong SOC, it is expected that the reduction of the SOC may lead to an insulator-metal transition and associated novel phases. Theoretical studies suggested that the singlet d-wave and triplet p-wave pairing state could appear with the control of SOC2,4. The magnitude of the relativistic SOC is known to be proportional to the Z2 (Z: atomic number)12, thus it can be controlled via substitution of Ir ions with other transition metal ions. A recent angle-resolved photoemission spectroscopy (ARPES) study on Sr2(Ir1−xRhx)O4 and Sr2(Ir1−xRux)O4 suggested that the Rh/Ru doping resulted in the reduction of the SOC via hybridization between doped Rh/Ru ions and the host Ir ions, and this SOC reduction played a decisive role for their insulator-metal transition13. It was also suggested that the efficiency in the SOC dilution depended on the closeness of the on-site energies of the states of the Ir and Rh/Ru ions. On the other hand, another ARPES measurement indicated that the SOC did not control the insulator-metal transition of Sr2(Ir1−xRhx)O4 and Sr2(Ir1−xRux)O4. Instead, this study showed that the rigid band shift by hole doping and the appearance of the sets of bands with mostly Ru character generated by the hybridization between Ru and Ir ions drove the insulator-metal transition in Sr2(Ir1−xRhx)O4 and Sr2(Ir1−xRux)O4, respectively14. The distinct origins of the insulator-metal transitions in the two systems were attributed to the difference in the on-site energies of the Rh and Ru ions.
Doping of 3d transition metal ions may be more effective in studying the effects of the change in the SOC to the electronic response of the iridates and the roles of the on-site energy of the host and impurity ions for the SOC dilution. Several studies investigated the transport and magnetic properties of Fe- or Co-doped Sr2IrO4, which showed that Fe or Co doping induced insulator-metal transitions15,16. The insulator-metal transition was attributed to the formation of the impurity states close to the Fermi level. The effects of the Fe/Co doping on the electronic structure and the SOC, which can be gained by spectroscopy studies, were not discussed in these reports15,16. To the best of our knowledge, there is no optical spectroscopy reported on the 3d transition-metal-doped iridates.
In this paper, we studied the doping and temperature evolutions of the electronic response of Mn-doped Sr3Ir2O7 single crystals, Sr3(Ir1−xMnx)2O7 with x = 0, 0.09, 0.18, 0.36 by means of infrared spectroscopy. The substitution of Mn ions, Mn3+ (3d4) or Mn4+ (3d5), is expected to dope holes to the system and to dilute the SOC, which can induce an insulator–metal transition. Upon Mn doping, the optical excitations between the Jeff bands were suppressed and a low-energy in-gap excitation at the energies \(\hslash \omega \le\) 0.25 eV appeared. The low-energy in-gap excitation reflects hole doping and is universally observed in doped iridates17,18,19,20. While the in-gap excitation was enhanced with Mn doping, it did not evolve into the coherent Drude-like response in Sr3(Ir1−xMnx)2O7, which is in sharp contrast to other doped iridates. In addition, the resonance energies of the optical excitations between the Jeff bands hardly changes with Mn doping, indicating the stiffness of the SOC. At \(\hslash \omega \ge\) 1 eV, a distinct optical excitation which has not been observed in 4d transition-metal doped iridates, such as Sr2(Ir,Rh)O4 and Srn+1(Ir,Ru)nO3n+1, emerged with Mn doping. The resonance energy of this optical excitation is higher than those of the transitions between Jeff bands. The observation of the high-energy optical transition suggests that the on-site energy of Mn ions is quite different from that of Ir ions. The large difference in the on-site energies can lead to a strong disorder and a resilience of the SOC and thus can be associated with the absence of the insulator–metal transition in Sr3(Ir1−xMnx)2O7. Mn doping also alters the temperature evolution of the electronic response. The strong anomaly in the optical response of the parent compound at the antiferromagnetic transition temperature TN disappeared upon Mn doping, which may be associated with charge disproportionation of the Ir ions.
Results and discussion
Infrared-active phonon modes
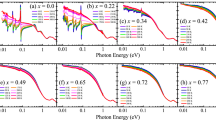
The inset of Fig. 1a shows the optical conductivity data at 10 K in the energy region below 100 meV where the infrared-active phonon modes are observed. For the parent compound, the six phonon modes expected for the layered perovskite structure are registered: three external modes below 20 meV, two bending modes at 32 and 46 meV, and a stretching mode at 78 meV21. Upon 9% Mn substitution, the stretching mode (78 meV) and one of the bending modes (46 meV) of the parent compound become weaker and broader, and new modes appear at about 41 and 71 meV. The former and latter energies are close to the resonance energies of the bending mode and the stretching mode of the parent compound, respectively. This suggests that the modes at 41 and 71 meV can be attributed to the bending and the stretching modes associated with the Mn–O bonds. Further Mn substitution induces gradual suppression and enhancement of the modes related to the Ir–O and Mn–O bonds, respectively, which are demonstrated most clearly in the stretching modes. Further Mn substitution leads to gradual suppression and enhancement of the phonon modes associated with the Ir–O and Mn–O bonds, respectively. This observation indicates a continuous evolution of the lattice properties with Mn substitution, which is consistent with the results of the previous neutron diffraction measurements22.

Temperature dependent optical conductivity spectra σ1(ω) of Sr3(Ir1-xMnx)2O7 for (a) x = 0, (b) x = 0.09, (c) x = 0.18, (d) x = 0.36 from T = 10 to 300 K. The inset of (a) displays the σ1(ω) below 100 meV. The baseline of each spectrum is shifted for clarity. Fitting results of the Drude-Lorentz oscillator model (dashed lines: Drude oscillators, solid lines: Lorentz oscillators) for (e) x = 0, (f) x = 0.09, (g) x = 0.18, (h) x = 0.36 at T = 10 K.
An exception to the gradual evolutions of the phonon modes is observed in the x = 0.09. A mode at 35 meV is observed only in the x = 0.09 compound. The resonant elastic X-ray scattering (REXS) data showed the pronounced splitting of the nuclear Bragg reflections of the x = 0.09 in the antiferromagnetic phase22. The neutron diffraction data also revealed an enhancement of the monoclinic distortion in the compounds with low-level of Mn doping (x = 0.11)22. At higher Mn concentrations, the enhancement of the monoclinic distortion disappeared, which was attributed to disorder due to increased Mn substitution or the disappearance of the antiferromagnetic order22. It was suggested that for low-level of Mn substitution where the long-range antiferromagnetic order remains intact a strong magnetostructural coupling may play an important role for the enhancement of the monoclinic distortion22. The close similarities between the doping evolutions of the infrared-active phonon modes and the structural anomalies in the REXS and the neutron diffraction data suggest that the magnetostructural coupling may be responsible for the 35 meV mode in the x = 0.09 compound.
Doping evolution of the electronic response
Figure 1 shows the real part of the optical conductivity spectra σ1(ω) of Sr3(Ir1−xMnx)2O7. In the σ1(ω) of the parent compound, two prominent peaks, labeled as α and β are observed. The peak α corresponds to the optical transition from the Jeff = 1/2 lower Hubbard band (LHB) to the Jeff = 1/2 upper Hubbard band (UHB). The peak β corresponds to the optical transitions from the Jeff = 3/2 bands to the Jeff = 1/2 UHB5,7,23. We note that the Jeff = 3/2-to-Jeff = 1/2 UHB transition is composed of two peaks, i.e., β and γ, as shown in Fig. 1e. Upon Mn doping, the peaks α and β are suppressed and an in-gap excitation labeled as A appears below about 0.25 eV. In addition to the change in the low-energy response, Mn doping induces an enhancement of σ1(ω) in the energy region between 1 and 2 eV, where in the parent compound the peak γ, the optical transition from the Jeff = 3/2 bands to the Jeff = 1/2 upper Hubbard band, is located. Since the peak γ should be suppressed in the Mn-doped samples, similar to the peaks α and β, the majority of the spectral weight in the energy region between 1 and 2 eV for the Mn-doped compound could not be attributed to the optical transition between the Jeff bands but should be assigned as a transition associated with the Mn states. To reflect its distinct nature, we labeled the peak at about 1.2 eV in the Mn-doped samples as C.
Optical conductivity data at 10 K in Fig. 2a illustrate the evolution of the electronic structure with Mn doping more clearly. One of the most prominent changes is the drastic suppression of the peak α. Part of the spectral weight of the peak α is shifted across an isosbestic point at about 0.25 eV to the lower-energy in-gap excitation, peak A, which is one of the universal characteristic features of the filling-controlled insulator–metal transition in correlated electron systems24,25. For a system exhibiting the filling-controlled insulator–metal transition, charge carrier doping leads to a suppression of the optical transitions across the gap and spectral weight shift to fill the gap with an incoherent in-gap excitations. Upon further doping, a weak coherent Drude-like response appears before a fully coherent Drude-like peak develops with the merger between the coherent and the incoherent responses. These universal behaviors were registered in the optical response of Rh- or Ru-doped Srn+1IrnO3n+1 (n = 1, 2). Both the Rh and Ru substitution result in hole doping and induces an insulator–metal transition. In the optical conductivity data of Sr2(Ir,Rh)O4 and Sr3(Ir,Ru)2O7, a clear Drude-like peak appeared upon 5% Rh and 34% Ru doping, respectively17,19. In sharp contrast, for Sr3(Ir1-xMnx)2O7, the incoherent in-gap excitation does not evolve into the coherent Drude-like peak but remains incoherent up to the highest Mn concentration of x = 0.36, which suggests that the doped holes remain localized.

(a) Optical conductivity spectra of Sr3(Ir1−xMnx)2O7 at 10 K. The inset shows the imaginary parts of the dielectric constant. The spectra are shifted vertically for clarity. (b) Doping dependence of the resonance energies of the peaks α, β, A, B, C (γ) at 10 K. (c) Doping dependence of the spectral weights of the peaks α, β, A, B, C (γ). The inset shows the doping dependence of the SW of the peak C. Schematic band diagram of Sr3(Ir1−xMnx)2O7 with the cases where (d) the Mn4+ and (e) Mn3+ ions dominate. The bands drawn with dashed lines in (d) represent the \({e}_{g,1}^{\uparrow }\) and \({e}_{g,2}^{\uparrow }\) states of the minority Mn3+ ions in the x = 0.09 and 0.18 compounds. The LHB (UHB) in (d) and (e) denotes the Jeff = 1/2 lower (upper) Hubbard bands.
Anderson localization can be responsible for the incoherent nature of the low-energy optical response26,27. Anderson localization is mainly due to disorders in the system. In a correlated Mott system, the cooperation between the randomness and the electronic correlations can induce a soft gap in the electronic density of states27,28,29,30,31, thus hindering coherent charge transport. Accordingly, the dc resistivity follows variable-range hopping (VRH)32. The resistivity data of Sr3(Ir1−xMnx)2O7 indeed display the VRH behavior in a wide range of temperature22. We note that the VRH behavior of the dc resistivity was also registered in insulating/bad metallic Rh- and Tb-doped Sr2IrO4 compounds33,34.
In order to obtain more quantitative information on the evolution of the electronic structure with Mn doping, we analyzed the σ1(ω) using Drude-Lorentz oscillator model:
ωp and γD are the plasma frequency and the scattering rate of the Drude oscillator, respectively. Sk, γk, and ω0,k are the strength, the width, and the resonant frequency of the Lorentz oscillator, respectively. The result of the Drude-Lorentz oscillator model fit for the 10 K data of Sr3(Ir1−xMnx)2O7 is displayed in Fig. 1e–h. One Drude (D) oscillator, three (α, β, γ) and five (α, β, A, B, C) Lorentz oscillators are needed to reproduce the σ1(ω) data of the parent and Mn-doped compounds, respectively. The peak A represents the in-gap excitation. The peak B is required to account for the little change in σ1(ω) at about 0.6–0.7 eV despite the suppression of the peak β. The parameters extracted from the fitting are summarized in Fig. 2b and c.
The Lorentz oscillator model analysis provides important information on the evolution of the electronic structure of Sr3(Ir1−xMnx)2O7. We note that the resonance energies of the peak α and β hardly change with Mn doping, as shown in Fig. 2b. It should be mentioned that, whereas the resonance energy of the peak β is not clearly resolved in σ1(ω) due to its overlap with the neighboring optical transitions, it can be identified in the imaginary part of the dielectric function ε2(ω) [inset of Fig. 2a]. Since the separation of the peaks α and β is proportional to the magnitude of the SOC, λSO, this observation indicates that the SOC is resilient against Mn doping. The robustness of the SOC in the Mn-doped compounds samples is further supported by the large branching ratio from a recent x-ray absorption spectroscopy study22.
A recent ARPES study on Sr2(Ir,Ru)O4 and Sr2(Ir,Rh)O4 suggested that the reduction of the SOC is strongly dependent on the impurity potential13. It was shown that the large difference between the on-site energies of the host and impurity states can prevent their hybridization and thus the dilution of the SOC. Our observation of the robustness of the SOC as well as the incoherent charge transport of Sr3(Ir1−xMnx)2O7 suggests that the Mn states may be located away from the Ir states in energy.
The strong enhancement of σ1(ω) in the energy region between 1 and 1.5 eV, which leads to the development of the peak C, with Mn doping indicates that the Mn states are indeed located away from the Ir Jeff = 1/2 states in energy: the resonance energy of the peak C is larger than those of the optical transitions between the Jeff =1/2 bands. In Sr3(Ir,Ru)2O7 and Sr2(Ir,Rh)O4 where the Ru or Rh states were closer in energy to the Ir Jeff = 1/2 bands13,14 than the Mn states, Ru or Rh substitution do not induce the enhancement of σ1(ω) in the energy region between 1 and 1.5 eV17,18,19,35, as shown in Fig. 3a and b.

Optical conductivity spectra of (a) Sr3Ir2O7, Sr3(Ir0.78Ru0.22)2O7, Sr3(Ir0.64Mn0.36)2O7 and (b) Sr2IrO4, Sr2Ir0.96Rh0.04O4. The data of Sr3(Ir0.78Ru0.22)2O7 and Sr2IrO4/Sr2Ir0.96Rh0.04O4 are from refs. 19 and 17, respectively.
The assignment of the peak C should depend on the valence state of Mn ions36,37,38. In perovskite manganites, Mn ions can have 4 + (3d3) or 3 + (3d4) valence state. For Mn4+ ions, the doubly-degenerate eg spin-up (\({e}_{g}^{\uparrow }\)) bands are closest to the Fermi level: it is located above the Fermi level by about 1.71 eV in Sr3Mn2O739. For Mn3+ ions, the degeneracy of the \({e}_{g}^{\uparrow }\) bands is lifted by Jahn–Teller effects and the split \({e}_{g,1}^{\uparrow }\) and \({e}_{g,2}^{\uparrow }\) bands are located above and below the Fermi level by at least about 0.6 eV40.
Magnetization measurements of Sr3(Ir1−xMnx)2O7 suggested a shift in the Mn valence from 4+ to 3+ at x \(\approx\) 0.2522. However, a comparison between the low-energy optical response of Sr3(Ir1−xMnx)2O7 and Sr3(Ir,Ru)2O7/Sr2(Ir,Rh)O4 suggests that Mn3+ ions should exist in x = 0.09 and 0.18 samples. As shown in Fig. 3a, the conductivity data of Sr3(Ir,Ru)2O7 barely changes even upon 22% Ru doping19. The peak α remains robust against Ru doping and in-gap excitations do not emerge. The sizeable changes in σ1(ω) occur only beyond the critical Ru concentration of about 0.35 at which the transport data indicates an insulator-to-metal transition41. This behavior suggests that the charge transfer between Ir4+ and Ru4+ ions is prohibited, thus protecting 4+ valence of Ir ions41,42,43. In contrast, it is known that Rh substitution induces hole doping on the Ir sites via electron transfer from Ir ions to Rh at low Rh concentrations, introducing Ir5+ and Rh3+ pairs44,45,46,47. The corresponding σ1(ω) of Sr2(Ir,Rh)O4 exhibits the suppression of the peak α and the emergence of an in-gap excitation17. The close similarities between the optical responses of Sr3(Ir1−xMnx)2O7 and Sr2(Ir,Rh)O4 suggest that Mn substitution changes the filling of the Jeff = 1/2 LHB via hole doping and induces the formation of Mn3+ and Ir5+ ions. Therefore, we suggest that Mn3+ ions coexist with majority Mn4+ ions at low Mn concentrations and the portion of Mn3+ ions increases with increasing Mn doping. Based on the inference of the valence of Mn ions from the magnetization measurements22 and our optical data, the peak C can be assigned mainly as a transition from Jeff = 3/2 state to Mn \({e}_{g}^{\uparrow }\) state for x = 0.09 and 0.18 and a transition from Jeff = 3/2 to Mn \({e}_{g,2}^{\uparrow }\) state for x = 0.36 compound. Then, the peak B at about 0.68 eV can be assigned as a transition from Jeff = 1/2 LHB to Mn \({e}_{g}^{\uparrow }\) or \({e}_{g,2}^{\uparrow }\) state. The corresponding schematic diagrams of the electronic density of states are displayed in Fig. 2d and e.
The variation of the SW of the peaks B and C with Mn doping supports the assignments. As shown in Fig. 2c, the peak C is enhanced with Mn doping up to x = 0.18 but is suppressed with further Mn doping. The peak B exhibits a rapid enhancement as x changes from 0.18 to 0.36. When the valence of Mn ions changes from 4+ to 3+, the Mn \({e}_{g}^{\uparrow }\) state can split into \({e}_{g,1}^{\uparrow }\) and \({e}_{g,2}^{\uparrow }\) due to the Jahn–Teller effect, resulting in the decrease in the density of states of the \({e}_{g}^{\uparrow }\) state. Therefore, the SW of the peak C, a transition from the Jeff = 3/2 state to Mn \({e}_{g}^{\uparrow }\) state should be suppressed. In order to account for the enhancement of the peak B, the split \({e}_{g,1}^{\uparrow }\) state should be located between the Jeff = 3/2 and the Jeff = 1/2 LHB, so that an optical transition from the Mn \({e}_{g,1}^{\uparrow }\) state to the Jeff = 1/2 UHB contributes to the spectral weight at the energies where the peak B is located. Further studies are desired to investigate the effects of the Jahn–Teller splitting of Mn \({e}_{g}^{\uparrow }\) states.
Temperature evolution of the optical response
Having identified the effects of Mn doping to the ground-state electronic structure, we discuss the temperature evolution of the low-energy optical response. In the parent compound, the optical conductivity data show clear anomalies at the antiferromagnetic transition temperature TN, implying a strong spin-charge coupling18. As the temperature increases across TN, the peak α exhibits an abrupt redshift [Fig. 4a]. In addition, the in-gap spectral weight obtained by the integration of σ1(ω) up to the isosbestic point of ωc = 0.35 eV, \(SW\left({\omega }_{\mathrm{c}}\right)={\int }_{0}^{{\omega }_{\mathrm{c}}}{\sigma }_{1}\left(\omega \right)d\omega\), is significantly enhanced at TN [Fig. 4e]. A combined optical spectroscopy and ARPES study demonstrated that these changes in σ1(ω) were attributed to a magnetically driven band shift toward the Fermi level with the suppression of the antiferromagnetic order18.

Temperature dependence of the peak α of Sr3(Ir1−xMnx)2O7 with (a) x = 0, (b) x = 0.09, (c) x = 0.18, (d) x = 0.36. Temperature dependence of the SW(ωc) for (e) x = 0 (ωc = 0.34 eV), (f) x = 0.09 (ωc = 0.34 eV), (g) x = 0.18 (ωc = 0.36 eV) and (h) x = 0.36 (ωc = 0.33 eV). The dashed lines in (e)–(h) denote the Néel temperature for each sample.
The temperature evolutions of the low-energy optical responses of the Mn-doped compounds suggest that Mn-doping suppresses the spin-charge coupling. Figure 4b–d show that the resonance energy of the peak α, which corresponds to the distance between the Jeff = 1/2 LHB and UHB, is independent of temperature. This observation indicates that the shift of the Jeff = 1/2 Hubbard bands in energy due to the antiferromagnetic order does not occur in the Mn-doped compounds. In addition, the in-gap spectral weights SW(ωc) of Sr3(Ir1−xMnx)2O7 calculated with ωc = 0.34 (x = 0.09), 0.36 (x = 0.18), 0.33 eV (x = 0.36) show gradual changes with the variation in temperature without any anomaly [Fig. 4e–h]. In addition to the absence of the anomaly at TN, the magnitude of the change in the SW decreases with Mn doping. It is worth pointing out that the long-range antiferromagnetic order of the Jeff = 1/2 pseudospin survives up to x = 0.2522.
The temperature dependence of the optical response of Sr3(Ir1−xMnx)2O7 should be contrasted to that of Sr3(Ir,Ru)2O7. In the latter system, the anomaly in σ1(ω) at TN, related to the redshift of the peak α and the increase in the SW(ωc), remained robust in the antiferromagnetic and insulating compounds18,19. The clear distinction between the Mn- and Ru-doped compounds may be attributed to the different valence states of the dopant ions. Since Ru ion (4+) is isovalent with Ir ions in Sr3(Ir,Ru)2O7, the holes tend to be localized at the Ru sites41. Therefore, the Jeff = 1/2 pseudospin can remain intact despite a substantial Ru doping, which may be associated with the survival of the long-range antiferromagnetic ordering of the Jeff = 1/2 moments up to about 70% Ru doping. Conversely, as inferred from our optical data, Mn doping induces formation of Ir5+ ions. Thus, the Ir ions near the doped Mn ions can lose their Jeff = 1/2 pseudospin. This parallels the observation of the rather quick suppression of the antiferromagnetic ordering of the Ir moments in Sr3(Ir1−xMnx)2O722. We conjecture that this charge disproportionation and the resulting loss of the pseudospin may be responsible for the rapid suppression of the spin-charge coupling in Sr3(Ir1−xMnx)2O7.
Conclusion
We studied the doping and temperature dependence of the optical response of Sr3(Ir1−xMnx)2O7 with 0 ≤ x ≤ 0.36. We observed that the Mn substitution resulted in the suppression of the Mott gap excitation and the appearance of an in-gap excitation, which is one of the characteristic features of charge carrier doping. The in-gap excitation did not evolve into the Drude-like peak, but remained incoherent up to the highest Mn concentration, which is possibly due to the Anderson localization. While the optical excitations between the Ir Jeff bands were suppressed, their resonance energies did not change, indicating the robustness of the SOC. Mn doping induced an enhancement of the optical conductivity for energies above 1 eV, which shows that the on-site energy of the Mn states is quite different from that of the Ir Jeff = 1/2 bands. The doping evolution of the optical response suggests that the Anderson localization and the robustness of the SOC are likely to be attributed to the large difference between the on-site energies of the Mn and Ir states. The temperature dependence of the electronic response was significantly affected by Mn doping. The anomalies of the optical response at the antiferromagnetic ordering temperature, the energy shift of the optical transition between the Jeff = 1/2 Hubbard bands and the abrupt change in the SW of the in-gap excitation observed in the parent compound, disappeared in the Mn-doped compounds. We ascribe this phenomenon of the spin-charge decoupling to the charge disproportionation due to Mn substitution, resulting in the loss of Jeff = 1/2 pseudospin.
Methods
High-quality single crystals of Sr3(Ir1−xMnx)2O7 with x = 0, 0.09, 0.18, 0.36 were grown using a halide flux growth technique. Details of the single crystal growth were described in Ref. 22. We measured near-normal incidence reflectivity spectra R(ω) in the energy region between 5 meV and 1 eV using Fourier transform infrared spectrometer (Vertex 70v, Bruker). We employed in-situ gold overcoating technique to obtain accurate reflectivity data48. Complex optical constants in the energy region between 0.74 and 5 eV were obtained using spectroscopic ellipsometer (M-2000, J. A. Woollam Co.). Optical conductivity was calculated from the R(ω) data through Kramers-Kronig transformation.
Data availability
The data used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Wang, F. & Senthil, T. Twisted hubbard model for Sr2IrO4: Magnetism and possible high temperature superconductivity. Phys. Rev. Lett. 106, 136402 (2011).
Meng, Z. Y., Kim, Y. B. & Kee, H. Y. Odd-parity triplet superconducting phase in multiorbital materials with a strong spin-orbit coupling: Application to doped Sr2IrO4. Phys. Rev. Lett. 113, 177003 (2014).
Sumita, S., Nomoto, T. & Yanase, Y. Multipole superconductivity in nonsymmorphic Sr2IrO4. Phys. Rev. Lett. 119, 027001 (2017).
Watanabe, H., Shirakawa, T. & Yunoki, S. Monte Carlo study of an unconventional superconducting phase in iridium oxide Jeff = 1/2 Mott insulators induced by carrier doping. Phys. Rev. Lett. 110, 027002 (2013).
Kim, B. J. et al. Novel Jeff = 1/2 Mott state induced by relativistic spin-orbit coupling in Sr2IrO4. Phys. Rev. Lett. 101, 076402 (2008).
Kim, B. J. et al. Phase-sensitive observation of a spin-orbital mott state in in Sr2IrO4. Science 323, 1329–1332 (2009).
Moon, S. J. et al. Dimensionality-controlled insulator-metal transition and correlated metallic state in 5d transition metal oxides Srn+1Ir nO3n+1 (n = 1, 2, and ∞). Phys. Rev. Lett. 101, 226402 (2008).
Kim, Y. K. et al. Superconductivity. Fermi arcs in a doped pseudospin-1/2 Heisenberg antiferromagnet. Science 345, 187–190 (2014).
Yan, Y. J. et al. Electron-doped Sr2IrO4: An analogue of hole-doped cuprate superconductors demonstrated by scanning tunneling microscopy. Phys. Rev. X 5, 041018 (2015).
Kim, Y. K., Sung, N. H., Denlinger, J. D. & Kim, B. J. Observation of a d-wave gap in electron-doped Sr2IrO4. Nat. Phys. 12, 37–41 (2015).
Chu, H. et al. A charge density wave-like instability in a doped spin-orbit-assisted weak Mott insulator. Nat. Mater. 16, 200–203 (2017).
Shanavas, K. V., Popović, Z. S. & Satpathy, S. Theoretical model for Rashba spin-orbit interaction in d electrons. Phys. Rev. B 90, 165108 (2014).
Zwartsenberg, B. et al. Spin-orbit-controlled metal–insulator transition in Sr2IrO4. Nat. Phys. 16, 290–294 (2020).
Brouet, V. et al. Origin of the different electronic structure of Rh- and Ru-doped Sr2IrO4. Phys. Rev. B 104, L121104 (2021).
Hu, B. et al. Correlation between antiferromagnetic and Mott states in spin orbit coupled Sr2IrO4: A study of Sr2Ir−xMxO4 (M = Fe or Co). Phys. Rev. B 103, 115122 (2021).
Samanta, K. et al. Quantum criticality in a layered iridate. Commun. Phys. 4, 89 (2021).
Xu, B. et al. Optical signature of a crossover from Mott- to Slater-type gap in Sr2Ir1-xRhxO4. Phys. Rev. Lett. 124, 027402 (2020).
Song, S. et al. Magnetically driven band shift and metal-insulator transition in spin-orbit-coupled Sr3(Ir1−xRux)2O7. Phys. Rev. B 98, 035110 (2018).
Ahn, G., Schmehr, J. L., Porter, Z., Wilson, S. D. & Moon, S. J. Doping and temperature evolutions of optical response of Sr3(Ir1-xRux)2O7. Sci. Rep. 10, 22340 (2020).
Lee, S. et al. Evolution of the electronic structure of Ru-doped single-crystal iridates Sr2Ir1−xRuxO4. Phys. Rev. B 104, 165106 (2021).
Seo, J. H. et al. Infrared probe of pseudogap in electron-doped Sr2IrO4. Sci. Rep. 7, 10494 (2017).
Schmehr, J. L. et al. Preferential quenching of 5d antiferromagnetic order in Sr3(Ir1−xMnx)2O7. J. Phys. Condens. Matter 31, 244003 (2019).
Moon, S. J. et al. Temperature dependence of the elctronic structure of the Jeff = 1/2 Mott insulator Sr2IrO4 studied by optical spectroscopy. Phys. Rev. B 80, 195110 (2009).
Basov, D. N., Averitt, R. D., van der Marel, D., Dressel, M. & Haule, K. Electrodynamics of correlated electron materials. Rev. Mod. Phys. 83, 471–541 (2011).
Imada, M., Fujimori, A. & Tokura, Y. Metal-insulator transitions. Rev. Mod. Phys. 70, 1039–1263 (1998).
Anderson, P. W. Absence of Diffusion in Certain Random Lattices. Phys. Rev. 109, 1492–1505 (1958).
Shinaoka, H. & Imada, M. Theory of electron transport near anderson-mott transitions. J. Phys. Soc. Jpn. 79, 113703 (2010).
Chiesa, S., Chakraborty, P. B., Pickett, W. E. & Scalettar, R. T. Disorder-induced stabilization of the pseudogap in strongly correlated systems. Phys. Rev. Lett. 101, 086401 (2008).
Fazileh, F., Gooding, R. J., Atkinson, W. A. & Johnston, D. C. Role of strong electronic correlations in the metal-to-insulator transition in disordered LiAlyTi2-yO4. Phys. Rev. Lett. 96, 046410 (2006).
Byczuk, K., Hofstetter, W. & Vollhardt, D. Mott-Hubbard transition versus Anderson localization in correlated electron systems with disorder. Phys. Rev. Lett. 94, 056404 (2005).
Shinaoka, H. & Imada, M. Single-particle excitations under coexisting electron correlation and disorder: A numerical study of the Anderson-Hubbard model. J. Phys. Soc. Jpn. 78, 094708 (2009).
Efros, A. L. & Shklovskii, B. I. Coulomb gap and low temperature conductivity of disordered systems. J. Phys. C: Solid State Phys. 8, L49–L51 (1975).
Qi, T. F. et al. Spin-orbit tuned metal-insulator transitions in single-crystal Sr2Ir1−xRhxO4 (0≤x≤1). Phys. Rev. B 86, 125105 (2012).
Wang, J. C. et al. Decoupling of the antiferromagnetic and insulating states in Tb-doped Sr2IrO4. Phys. Rev. B 92, 214411 (2015).
Lee, J. S., Krockenberger, Y., Takahashi, K. S., Kawasaki, M. & Tokura, Y. Insulator-metal transition driven by change of doping and spin-orbit interaction in Sr2IrO4. Phys. Rev. B 85, 035101 (2012).
Jung, J. H. et al. Determination of electronic band structures of CaMnO3 and LaMnO3 using optical-conductivity analyses. Phys. Rev. B 55, 15489–15493 (1997).
Lee, Y. et al. Correlation of electronic structure and ordered charge and orbital patterns for single-layered manganites in a wide hole-doping range (0≤x≤1). Phys. Rev. B 75, 144407 (2007).
Ishikawa, T., Tobe, K., Kimura, T., Katsufuji, T. & Tokura, Y. Optical study on the doping and temperature dependence of the anisotropic electronic structure in bilayered manganites La2–2xSr1+2xMn2O7 (0.3<~x<~0.5). Phys. Rev. B 62, 12354–12362 (2000).
Meskine, H., Popović, Z. S. & Satpathy, S. Electronic structure and exchange interaction in the layered perovskite Sr3Mn2O7. Phys. Rev. B 65, 094402 (2002).
Park, K. T. Electronic structure calculations for layered LaSrMnO4 and Ca2RuO4. J. Condens. Matter Phys. 13, 9231 (2001).
Dhital, C. et al. Carrier localization and electronic phase separation in a doped spin-orbit-driven Mott phase in Sr3(Ir1–xRux)2O7. Nat. Commun. 5, 1–7 (2014).
Schmehr, J. L. et al. Overdamped antiferromagnetic strange metal state in Sr3IrRuO7. Phys. Rev. Lett. 122, 157201 (2019).
Cava, R. J. et al. Localized-to-itinerant electron transition in Sr2Ir1-xRuxO4. Phys. Rev. B 49, 11890–11894 (1994).
Cao, Y. et al. Hallmarks of the Mott-metal crossover in the hole-doped pseudospin-1/2 Mott insulator Sr2IrO4. Nat. Commun. 7, 11367 (2016).
Chikara, S. et al. Charge partitioning and anomalous hole doping in Rh-doped Sr2IrO4. Phys. Rev. Lett. 95, 060407 (2017).
Clancy, J. P. et al. Dilute magnetism and spin-orbital percolation effects in Sr2Ir1−xRhxO4. Phys. Rev. B 89, 054409 (2014).
Louat, A. et al. Formation of an incoherent metallic state in Rh-doped Sr2IrO4. Phys. Rev. B 97, 161109 (2018).
Homes, C. C., Reedyk, M., Cradles, D. A. & Timusk, T. Technique for measuring the reflectance of irregular, submillimeter-sized samples. Appl. Opt. 32, 2976–2983 (1993).
Acknowledgements
This research was supported by the National Research Foundation grant of Korea (NRF) funded by the Korean government (MSIT) (2022R1F1A1072865 and RS-2022-00143178) and BrainLink program funded by the Ministry of Science and ICT through the National Research Foundation of Korea (2022H1D3A3A01077468). S.D.W. and J.S. acknowledge support from National Science Foundation award DMR-1905801.
Author information
Authors and Affiliations
Contributions
D. W. K. and S. J. M. conceived the project. D. W. K. carried out the optical measurements. D. W. K., G. A., and S. J. M. analyzed the optical data. J. S and S. D. W grew and characterized the single crystals. All authors participated in discussing the results and writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, D., Ahn, G., Schmehr, J. et al. Effects of the on-site energy on the electronic response of Sr3(Ir1−xMnx)2O7. Sci Rep 12, 18957 (2022). https://doi.org/10.1038/s41598-022-23593-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-23593-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
