
Abstract
Clusterin (CLU) is one of the most significant genetic risk factors for late onset Alzheimer’s disease (AD). However, the mechanisms by which CLU contributes to AD development and pathogenesis remain unclear. Studies have demonstrated that the trafficking and localisation of glycosylated CLU proteins is altered by CLU-AD mutations and amyloid-β (Aβ), which may contribute to AD pathogenesis. However, the roles of non-glycosylated and glycosylated CLU proteins in mediating Aβ toxicity have not been studied in human neurons. iPSCs with altered CLU trafficking were generated following the removal of CLU exon 2 by CRISPR/Cas9 gene editing. Neurons were generated from control (CTR) and exon 2 −/− edited iPSCs and were incubated with aggregated Aβ peptides. Aβ induced changes in cell death and neurite length were quantified to determine if altered CLU protein trafficking influenced neuronal sensitivity to Aβ. Finally, RNA-Seq analysis was performed to identify key transcriptomic differences between CLU exon 2 −/− and CTR neurons. The removal of CLU exon 2, and the endoplasmic reticulum (ER)-signal peptide located within, abolished the presence of glycosylated CLU and increased the abundance of intracellular, non-glycosylated CLU. While non-glycosylated CLU levels were unaltered by Aβ25–35 treatment, the trafficking of glycosylated CLU was altered in control but not exon 2 −/− neurons. The latter also displayed partial protection against Aβ-induced cell death and neurite retraction. Transcriptome analysis identified downregulation of multiple extracellular matrix (ECM) related genes in exon 2 −/− neurons, potentially contributing to their reduced sensitivity to Aβ toxicity. This study identifies a crucial role of glycosylated CLU in facilitating Aβ toxicity in human neurons. The loss of these proteins reduced both, cell death and neurite damage, two key consequences of Aβ toxicity identified in the AD brain. Strikingly, transcriptomic differences between exon 2 −/− and control neurons were small, but a significant and consistent downregulation of ECM genes and pathways was identified in exon 2 −/− neurons. This may contribute to the reduced sensitivity of these neurons to Aβ, providing new mechanistic insights into Aβ pathologies and therapeutic targets for AD.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and is the most common cause of dementia1. AD is characterised by the presence of two pathological hallmarks: extracellular Aβ aggregates known as Aβ plaques and intracellular neurofibrillary tau tangles (NFTs) formed of hyperphosphorylated tau proteins. A prevailing hypothesis of AD is the Amyloid Cascade Hypothesis that postulates Aβ plays a central role in initiating AD development2,3,4. Aggregation of Aβ may trigger downstream events, including synaptic dysfunction, neurite damage, formation of NFTs and cognitive decline that ultimately result in AD. Major support for this hypothesis arises from the genetics of familial AD (fAD)2 where several mutations in genes encoding proteins involved in Aβ production, including amyloid precursor protein (APP), presenilin 1 and 2 (PSEN 1/2)5,6,7,8,9,10,11,12 have been identified. However, fAD represents only a small number of AD cases (< 1%), and it is not clear if fAD offers mechanistic insight into sporadic AD (sAD). Nevertheless, such studies provide important insight into a crucial role of Aβ in AD development. Numerous risk genes have been identified that alter an individual’s susceptibility to sAD, of which CLU is the third most significant genetic risk factor after APOE and BIN113,14,15. However, little is known of the mechanisms by which single nucleotide polymorphisms (SNPs) and mutations in CLU alter AD risk13,14,16. Although rare CLU mutations have been identified to alter CLU protein localisation, these are non-coding and located in exons 5 and 616,17,18. To date no AD-related CLU variant has been described in exon 219.
In addition to CLU acting as a genetic risk factor for sAD, much interest has been placed on CLU’s relationship with Aβ. Secreted CLU binds to Aβ altering its solubility and aggregation13,14,15,16 and contributes to the clearance of Aβ by astrocytes and through the blood–brain barrier21. In contrast, data from animal studies have identified a role for CLU in facilitating the deposition of Aβ and the formation of plaques20,22,23, suggesting CLU may facilitate AD pathology in vivo. It therefore remains unclear if CLU proteins facilitate or protect against Aβ toxicity in AD.
Clusterin is a highly glycosylated, cleaved and secreted protein (sCLU) that is considered to be cytoprotective21,22,23. The biogenesis of sCLU is well described24,25,26,27,28. Initially, CLU mRNA is translated into a clusterin preproprotein that is trafficked to the ER and the Golgi apparatus, where it is cleaved, phosphorylated and glycosylated29. Located within exon 2 is the endoplasmic reticulum (ER) signal peptide that directs the trafficking of immature CLU protein to the ER and subsequently to the Golgi apparatus where it is cleaved, phosphorylated and glycosylated to generate mature and cleaved, secreted CLU proteins28. Additionally, inside the ER, the ER signal peptide is removed to generate an N-terminal truncated protein. Mature clusterin (75/80 kDa) is secreted as a heterodimeric protein consisting of an α chain and a β chain that are linked by 5 disulphide bonds24,27,28,30,31,32. Although typically considered secreted, glycosylated CLU proteins have also been identified within cells, typically in the cytoplasm, although the function, origin and biogenesis of intracellular, glycosylated CLU proteins remains debated33.
Stress has been demonstrated to induce localisation and trafficking of glycosylated CLU into the cytoplasm. This may be mediated through the binding of sCLU to cell surface receptors resulting in its uptake back into cells34,35, or via premature release of CLU from the secretory pathway prior to secretion36, both of which result in reduced secretion and increased intracellular retention of CLU proteins35,36,37,38,39,40. Altered CLU trafficking in vitro has been demonstrated in response to a variety of stressors, including ER stress36,38,40, proteasomal inhibition41, nerve growth factor39. With relevance specifically to AD, the treatment of cells with Aβ35,37 and CLU-AD mutations17,18 have both been demonstrated to alter CLU trafficking suggesting CLU trafficking contributes to AD pathogenesis. Clusterin trafficking is altered by Aβ25–35 treatment of rodent neurons35 and by Aβ1–42 treatment of astrocytes in vitro37, both of which lead to increased intracellular abundance and reduced CLU secretion. The observed alteration of CLU trafficking in rodent neurons suggested CLU trafficking may mediate Aβ induced toxicity in these cells, which was further supported by the demonstration that CLU knockdown protects rodent neurons against Aβ induced cell death35. Furthermore, several CLU mutations enriched in AD cohorts16, were demonstrated to alter the trafficking and subcellular localisation of clusterin demonstrating a potential mechanism by which CLU variants may increase the risk of developing AD17,18. Thus, several types of stressors in different cell types have been shown to alter CLU trafficking, however, it remains unclear if this alteration in trafficking facilitates cellular stress or a protective response in human neurons.
Although often referred to as a cytoprotective protein, CLU has also been shown to play a pro-apoptotic role in the human breast cancer cell line MCF-742,43. Alternative splicing of CLU generates a minor CLU variant lacking exon 2 (also referred to as CLU variant 1 Δ exon 2)41,42,43. Translation of this variant generates a non-glycosylated, uncleaved CLU protein that resides intracellularly, typically in the cytoplasm and nuclei of cells41,42,43. Although this shorter CLU isoform has low abundance in cells41,43 or is not expressed at all44, expression is upregulated in cancer cell lines by stress and following induction of the cell death cascade, suggesting a role for these proteins in mediating cellular response to stress41,43. However, their role in human neurons and AD has not yet been explored.
In this study, we used CRISPR/Cas9 genome engineering and human iPSC technologies to examine the relationship between CLU proteins and Aβ induced toxicity in human neurons in vitro. Given the importance of the ER-signal peptide in the generation of glycosylated, secreted CLU proteins in cells, we targeted this domain of CLU to prevent sCLU biogenesis. We generated human neurons lacking glycosylated CLU proteins, which displayed altered sensitivity to Aβ. We also used RNA-Seq analysis to identify gene pathways that may underly this altered sensitivity and contribute to our understanding of AD pathogenesis and identification of novel AD therapeutics.
Materials and methods
CRISPR/Cas9 gene editing to generate exon 2 −/− iPSC lines
For all work CTR M3 36S (CTR) iPSCs were used. The CTR iPSCs were previously generated from the Price lab at King’s College London from human keratinocytes obtained from a healthy, neurotypical adult male, as previously described45,46,47. CTR iPSCs were used to generate exon 2 −/− iPSCs (A4 and D1 clones), a dual single guide RNA (sgRNA) approach was chosen to recruit Cas9-2NLS (Synthego) to cut within intron 1 and 2 to remove CLU exon 2 (Supplementary Table 1)48. Transfection of CTR iPSCs was performed using Nucleofection and an optimised protocol (Synthego). Briefly, ribonucleotide protein complexes (RNPs) were assembled by mixing sgRNA and Cas9 in a 7.5:1 ratio: 3 µl (300 pmol) sgRNA was added to 2 µl (40 pmol) Cas9 in a total volume of 5 µl per well. CTR iPSCs were detached from wells using Accutase, and 500,000 cells were transferred to a falcon tube for transfection. Cells were centrifuged at 115g for 3 min and resuspended in 20 µl Lonza P3 Nucleofector™ solution (Lonza). 20 µl of cells were mixed with 5 µl of pre-prepared RNP complex solution and the total volume (25 µl) was transferred to a Nucleocuvette™ strip and placed into a 4D-X Core unit. Electroporation was performed using protocol ‘CA137’. Following transfection, cells were returned to a well of a 6-well plate into pre-warmed E8 containing 10 µM Rock inhibitor. Cells were incubated overnight, and media was replaced daily until cells reached 70–80% confluence and quality control assays were performed.
PCR amplification was used to confirm the homozygous knockout (KO) of CLU exon 2 in A4 and D1 iPSCs. For each clonal iPSC line, genomic DNA was extracted using the QuickExtract™ DNA Extraction Solution (Lucigen, QE09050) and was amplified using the AmpliTaq Gold™ 360 Master Mix according to manufacturer’s instructions (ThermoFisher, 4398881) with 200 nM forward and reverse primers (Supplementary Table 2). PCR amplifications were performed using the following parameters: 95 °C for 10 min, 95 °C for 30 s, 55 °C for 30 s, 72 °C for 45 s and a final extension at 72 °C for 7 min, a total of 39 cycles were used. PCR products were then used in agarose electrophoretic analysis by running on a 1% agarose gel and imaged using a BioRad Gel Doc (Supplementary Fig. 1). Following confirmation that A4 and D1 iPSCs were homozygous exon 2 KO clonal lines, quality control experiments were performed to check karyotype and pluripotency of the iPSC lines. Synthego provided karyotype data for A4 and D1 iPSCs, which were confirmed to be normal for both lines (Supplementary Fig. 2). For pluripotency analysis, A4 and D1 iPSCs were plated on 96-well plates, fixed, and stained with nuclear marker DAPI, and immunostaining for pluripotency markers OCT4 and SSEA4 to confirm the retention of pluripotency of A4 and D1 iPSCs following removal of CLU exon 2 (Supplementary Fig. 3).
Neuronal differentiation
Human iPSCs were maintained in Essential 8 media (Life Technologies) on Geltrex coated plates (Nunc) until 100% confluent, at which point cells were differentiated according to a protocol adapted from Shi and colleagues49,50. iPSCs were cultured in 50% Neurobasal (Life Technologies) and 50% DMEM-F12 media (Life Technologies) supplemented with 1% N2 (Life Technologies), 2% B27 (Life Technologies), 1% Glutamax (Life Technologies), 10 µM SB-431542 (ApexBIO) and 1 µM LDN-193189 (Sigma) for 7 days with daily 100% media changes. Cells were dissociated with Accutase (Life Technologies) and replated on days 7, 12, 15 and 18 in 50% Neurobasal and 50% DMEM-F12 media supplemented with 1% N2 (Life Technologies), 2% B27 (Life Technologies) and 1% Glutamax (Life Technologies). On day 21, neural progenitor cells were replated on poly-ornithine and laminin coated plates at a density of 10,000 cells/well (96-well plate) and 200,000 cells/well (12-well plate) in B27 media (100% Neurobasal media supplemented with 2% B27 and 1% Glutamax). Media was changed after 24 h to remove cell debris. A full media change was performed on day 25 when 0.1 µM cytosine arabinoside (Sigma) was added to inhibit the growth of glial cells. A further full media change was performed 3 days later, and a half media change was performed another 3 days later. For all experiments, control and exon 2 −/− iPSCs were differentiated three independent times and neuronal identity was assessed by immunostaining (details of antibodies used can be found in Supplementary Table 3). The expression of neuronal marker MAP2 and cortical lineage marker CTIP2 was confirmed in all lines, and the numbers of MAP2 positive neurons and of CTIP2-positive MAP2 neurons were quantified to determine the percentage of neuronal cells in control and exon 2 −/− neuron cultures that were CTIP2 positive (Supplementary Fig. 4).
Aβ25–35 oligomer preparation
Aβ25–35 Trifluoroacetate salt (BACHEM AG, Bubendorf, Switzerland, #4030205) was supplied at 5 mg with a purity of 97% (molecular weight 1060.28). Nuclease-free water was added to make up a solution of 2 mg/ml and stored at − 20 °C. Prior to use, aliquots were defrosted on ice at room temperature (RT). For measuring Aβ toxicity, three concentrations of Aβ25–35 (500 nM, 2 µM, 20 µM) was added to cells for 24 h prior to experimentation. For Western blotting and RNA-Seq, cells were treated for 24 h with 20 µM Aβ25–35 only.
Western blotting
Prior to cell lysis, media samples were collected on ice and concentrated 45 times using Amicon Ultra-2 ml 10K Centrifugal Filter Units (Sigma, Aldrich, UFC201024) according to the manufacturer’s instructions. Protein content in the concentrated media samples was then measured following the addition of 150 µl Pierce 660 nm™ reagent (ThermoFisher, 22662). Protein content was measured relative to samples of Bovine Serum Albumin of known concentrations on a CLARIOstar Plus Plate reader (BMG LABTECH). For total cell lysates, 4 × 200 µl of laemmli buffer (BioRad) supplemented with HALT™ inhibitor (ThermoFisher, 78429) was added directly to cells. Lysates were collected, vortexed and boiled at 95 °C for 5 min at which point the supernatant was collected. For each sample, 4 µg of concentrated media was prepared in DPBS (Life Technologies, 14190094) and equal volumes of cell lysate and Laemmli buffer were separated by SDS-PAGE electrophoresis on 10% gels and transferred onto nitrocellulose membranes. The membranes were blocked with 5% BSA (made in 1 × TBST, tris-buffered saline and 0.1% Tween-20) for 1 h in agitation at RT. After blocking, membranes were incubated at 4 °C overnight in agitation with goat anti-clusterin primary antibody in 5% BSA (Santa Cruz, SC-6420, diluted 1:500). Membranes were then washed three times with 1 × TBST, incubated with secondary antibody for 2 h at RT in agitation (goat anti-donkey, IRDye® 800 Li-COR Biosciences 926-32214, diluted 1:10,000 in 5% BSA). Membranes were again washed three times and imaged using an infrared scanner (LI-COR Biosystems). To confirm equal protein loading across samples, membranes were finally incubated with a rabbit anti-β-actin primary antibody (Cell Signalling Technology, D6A8, diluted in 1:1000 in 5% BSA) for 1 h at RT in agitation. Membranes were washed and incubated for 1 h at RT in agitation with a secondary antibody prior to imaging (donkey anti-rabbit, IRDye® 680, Li-COR Biosciences 926-68073, diluted 1:10,000 in 5% BSA).
Cell death assay
Cell death was quantified using the CytoTox-Glo Cytotoxicity Assay (Promega, G9291) according to manufacturer's instructions. For all treatment conditions, cell death was compared relative to neurons that were untreated for 24 h. Following Aβ25–35 treatment (500 nM, 2 µM, 20 µM) 50 μl AAF-Glo was added to each well for 15 min. Luminescence as a result of apoptosis induced loss of membrane integrity was read on a PHERAstar FSX Microplate reader (BMG LABTECH) giving a measure of cell death induced by Aβ25–35 treatment. Then, 50 μl lysis reagent containing digitonin and AAF-Glo was added to wells for 15 min. The addition of these reagents was used to induce death in remaining live cells in each well, quantified as the total dead count. Luminescence was again recorded and was used as a measure of the total number of cells in each well. Dead/Total Dead Ratios were then calculated and compared to the ratios of untreated neuron cultures; an increase in this ratio following treatment with Aβ25–35 would reflect an increase in the number of dead cells in the culture indicating Aβ25–35 treatment had caused significant increases in apoptosis and cell death.
Neurite length assay
To quantify neurite damage following Aβ25–35 treatment (500 nM, 2 µM, 20 µM), neurons were fixed with 4% paraformaldehyde-DPBS at RT for 15 min. Cells were washed with DPBS three times prior to immunostaining. Cells were simultaneously permeabilised and blocked in DPBS containing 0.25% Triton-X and 10% donkey serum (Life Technologies) for 1 h at RT. Chicken anti-MAP2 (Abcam, ab5392, 1:1000) primary antibodies were diluted in DPBS containing 0.25% Triton-X and 5% donkey serum. Cells were washed three times with TBST and incubated for 1 h at RT in secondary antibody goat anti-chicken 647 diluted in blocking solution (Life Technologies, A-21449, 1:500). Cells were then washed once with Hoechst 33342 for 30 s (Thermo Fisher, 0.01 μg/ml diluted in DPBS). Cells were then washed a further three times with DPBS prior to imaging on an Opera Phenix High Content Screening System (Perkin Elmer) under 20 × magnification. Immunofluorescent images were analysed using the Harmony High-Content Analysis Software (Perkin Elmer) using a neurite length assay developed in the analysis interface. First, intact nuclei were identified following positive staining by Hoechst. Neurites protruding from cell bodies stained positive for MAP2 in the 647 channel were then traced and quantified.
RNA-Seq and analysis
For RNA-Seq experiments, neurons were treated with 20 µM Aβ25–35 for 24 h. Total RNA was collected using the Qiagen RNeasy Mini PLUS Kit (Qiagen) according to the manufacturer’s instructions from untreated and Aβ25–35 treated control and exon 2 −/− neurons from three independent differentiations. 100 ng of RNA were used for library preparation and analysis (Wellcome Trust Centre for Human Genetics, University of Oxford). Only samples with an RNA integrity number greater than 7 were used for subsequent sequencing. TruSeq RNA Library Preparation Kit (Illumina) was used, and mRNA was enriched by polyA selection. The mRNA fraction was selected from total RNA and converted to cDNA. cDNA was then end83 repaired, A-tailed and adaptor ligated. Samples were sequenced at 150 paired ends on a NovaSeq6000 for PolyA-RNA-Seq. We conducted quality control (QC) of the RNA-seq data using FastQC (version 0.11.5)51, for annotation of low-quality adapters and reads followed by trimming in Cutadapt (version 1.13)52, and then aligned by BWA-MEM (version 0.7.15)53 to the human reference genome GRCh37.EBVB95-8wt.ERCC. The average sequencing quality of all reads across samples was above 36 (Sanger scording, > Q30). Reads that overlap multiple features were resolved and summarised to the gene-level using the ‘summarizeOverlaps’ function of the (GenomicAlignments R package)54, with the follow settings: (i) mode = Union, (ii) singleEnd = F, (iii) ignore.strand = T, (iv) fragments = T and features were summarised to the gene-level. This yielded an Ensembl gene ID by sample counting matrix of the class S4 (R-object). The latter counting matrix was modelled using the DESeq2 pipeline which performs (i) size factors estimation, (ii) dispersion estimation and (iii) Gamma-Poisson Generalized Linear Model (GLM) fitting55. We report differentially expressed (DE) genes based on log2 fold-change (abs(FC) > 1) and p-values (p < 0.05) adjusted with Benjamini–Hochberg false discovery rate (FDR). Over-representation analysis (ORA) of ontology and pathway terms performed with hypergeometric tests (clusterProfiler, PMID:22455463)56 using gene sets encompassing Gene Ontology (GO) subsets such as biological process (BP), molecular function (MF), and cellular component (CC). Also, incorporating biological pathways gene sets from the MSigDB Collections (version c2.cp.kegg.v7.5.1, BROAD Institute)57,58 and topological signalling pathway enrichment implemented in SPIA59. Dimension reduction techniques such as Principal Component analysis (PCA) and the Uniform Manifold Approximation and Projection (UMAP) were applied to the normalised counts across all samples. The normalised counts were mean-centred and standardized, then dimension reduction with PCA, and afterwards using as input the first 20 principal components (PC) for the computation of UMAP60.
Statistical analysis
Statistical significance was set as p < 0.05 for all assays and experiments used in this study. Statistical tests were performed using GraphPad Prism (v. 7.02). A normal distribution in cell populations was assumed and two-tailed parametric tests were used. For experiments examining changes in CLU protein abundance in control neurons, analysis was performed using an unpaired t test (Fig. 1). For comparisons of CLU protein abundance between untreated and Aβ treated control and exon 2 −/− neurons analysis was performed by One-way ANOVA with Tukey’s Multiple Comparisons post hoc test (Fig. 1). Additionally, One-way ANOVA with Tukey’s Multiple Comparisons post hoc test was used to analyse differences in CTIP2 expression in control, A4 and D1 neurons (Supplementary Fig. 4d,e). For comparisons of changes in cell death and neurite length between untreated and Aβ25–35 treated control, A4 and D1 neurons, analysis was performed using a Two-way ANOVA with Tukey’s multiple comparisons post hoc test (Figs. 2 and 3). In analysis where post hoc analysis was performed adjusted p values are reported. Data are shown as mean ± standard error of the mean (SEM) unless otherwise indicated.

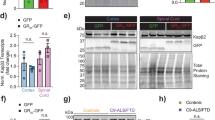
Aβ25–35 treatment alters trafficking of glycosylated CLU in human neurons. CTR and exon 2 −/− neurons (A4 and D1) were treated for 24 h with 20 μM Aβ25–35. Concentrated media and total cell lysate samples were collected, ran on a western blot and immunostained for CLU. (a) Western blot analysis of clusterin expression in cell lysate and culture media of untreated and Aβ25–35 treated CTR and A4 neurons (b) and untreated and Aβ25–35 treated CTR and D1 neurons. In both (a) and (b) β-actin were used as a loading control for protein loading of cell lysate samples. The absence of β-actin expression was confirmed in all culture media samples. (c,d) CLU immunoreactivity in cell lysates was normalised to β-actin immunoreactivity for each sample individually. This was then normalised to the immunoreactivity of the untreated CTR samples. (c) Intracellular, glycosylated CLU immunoreactivity was significantly increased (p = 0.0221*) while (d) secreted, glycosylated CLU was significantly decreased in CTR neurons (p = 0.0432*) following treatment with Aβ25–35. Exon 2 −/− neurons were demonstrated to not express glycosylated CLU proteins, either intracellularly or secreted into the media. (e,f) Loss of CLU exon 2 increased immunoreactivity of non-glycosylated CLU proteins in exon 2 −/− neurons at basal conditions (A4: p = 0.0077** and D1: p = 0.0348*) but Aβ25–35 did not alter the immunoreactivity of non-glycosylated, intracellular CLU species in either CTR or exon 2 −/− neurons (CTR: p = 0.09785 and A4: p = 0.7991, CTR: 0.9255 and D1: p = 0.9154). Immunoreactivity is reported as mean ± standard error of the mean (SEM). Mean values were calculated from neuronal samples collected from three independent differentiations of neurons, of which each contained triplicate samples (n = 3). Two-Way ANOVAs were performed for all comparisons (*p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001). Western blot images used in this figure have been cropped for clarity, all original blots are presented in Supplementary Fig. 5.

Exon 2 −/− neurons exhibit lower cell death rates when treated with Aβ25–35. CTR and exon 2 −/− neurons were treated with a one of three concentrations of Aβ25–35 (500 nM, 2 μM or 20 μM). After 24 h of treatment, cell death induced by Aβ25–35 was quantified. AAF-Glo reagent was added to cultures to measure protease activity released from apoptotic cells, giving a measurement of death induced by Aβ25–35 treatment. Death was then induced in the remaining, live cells by addition of digitonin and AAF-Glo. Death was quantified again giving a measurement of total number of death cells in each culture, representing the total number of cells. For each condition, the Dead/Total Dead Ratio was calculated and compared. Before Aβ25–35 treatment, the ratios of exon 2 −/− and CTR neurons were comparable (p > 0.9999 for all comparisons). 20 μM Aβ25–35 significantly increased the Dead/Total Dead Ratios of both CTR and exon 2 −/− neurons (CTR; p < 0.0001****, A4; p = 0.0200* and D1; p = 0.0254*. The percentage increase in number ratios for each cell line: CTR: 658.67% increase, A4: 208.24% increase and D1: 209.61% increase). 500 nM and 2 μM Aβ25–35 treatments did not induce significant cell death in any culture. Dead/Total Dead Ratios of 20 μM Aβ25–35 treated CTR cultures were significantly higher than the Ratios demonstrated in exon 2 −/− cultures (20 μM Aβ25–35 CTR vs A4: p < 0.0001**** and 20 μM Aβ25–35 CTR vs D1: p < 0.0001****) but were similar between the exon 2 −/− cultures (A4 vs D1: p = 0.9952). This reflects less cell death quantified in exon 2 −/− cultures than in CTR neurons. Dead/Total Dead Ratios are reported as mean ± SEM and Two-Way ANOVAs were performed for all concentrations (*p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001). Mean values were calculated from samples collected from three independent differentiations of neurons (n = 3), of which each contained 6 replicant samples.

Aβ25–35 does not induce neurite retraction in exon 2 −/− neurons. CTR and exon 2 −/− (A4 and D1) neurons were treated for 24 h with one of three concentrations of Aβ25–35. Following treatment, neurons were fixed, immunostained for DAPI (blue) and MAP2 (red) and imaged. Representative images of (a) CTR, (b) A4 and (c) D1 neurons. (d–f) Changes in length of neuronal processes were then quantified using an automated analysis software pipeline that identified MAP2 positive areas. Before treatment, mean segment length (CTR vs A4: p = 0.0310** and CTR vs D1: p = 0.0379*) and maximum segment length were significantly reduced in exon 2 −/− neurons than CTR neurons (CTR vs A4: p = 0.0161* and CTR vs D1: p = 0.0487*). 20 μM Aβ25–35 induced significant reductions in neurite length parameters in CTR neurons but not in exon 2 −/− neurons (CTR: mean segment length; p < 0.0001****, maximum segment length; p = 0.0009***, mean segment length to cell; p = 0.0010***). Exon 2 −/− neurons did not display evidence of Aβ25–35 induced neurite damage at any concentration of Aβ25–35. Values are reported as mean ± SEM and Two-Way ANOVAs were performed for all time points (*p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001). Mean values were calculated from samples collected from three independent differentiations of neurons (n = 3), of which each contained 6 replicant samples.
Results
Glycosylated CLU protein trafficking is altered by Aβ25–35 in human neurons
While Aβ1–42 is considered to the pathological Aβ species its use in vitro is impaired due to its instability when used in an aggregated fibrillary state61. Numerous studies have demonstrated similarities in activity in vitro between Aβ25–35 and Aβ1–4262,63,64, making Aβ25–35 a suitable alternative for use in this work. A variety of concentrations of Aβ25–35 have been demonstrated to induce cell death previously35,65,66. Given our group has demonstrated significant alterations in CLU trafficking in rodent neurons treated with Aβ25–3535 our first aim was to determine if Aβ25–35 treatment alters trafficking of glycosylated CLU in human neurons. Control (CTR) and exon 2 −/− neurons were treated with 20 μM Aβ25–35 for 24 h, after which media and cell lysates were collected, ran on a western blot and immunostained for CLU. The abundance of CLU proteins was first quantified in all samples separately and normalised to the abundance of β-actin for all samples. Finally, this was then normalised to CTR samples to compare abundance of CLU proteins in exon 2 −/− neurons to that of CTR neurons. Thus, we have compared CLU levels between treated and untreated neurons, as well as between exon 2 −/− and CTR neurons to examine the effect of both, Aβ25–35 and the loss of CLU exon 2.
Exon 2 −/− neurons did not express glycosylated CLU proteins, confirming previous reports that CLU exon 2 is required for the generation of mature, cleaved and glycosylated CLU proteins (Fig. 1a,b, full-length blots for CLU, both intracellular and secreted, are provided in Supplementary Fig. 5. Raw image files β-actin are also provided, these have not been altered/cropped, full-length blots for β-actin were not taken originally and thus, cannot be provided in all cases). To examine if Aβ25–35 altered glycosylated CLU trafficking, we compared levels of sCLU and intracellular (iCLU) clusterin protein expression between untreated and 20 μM Aβ25–35 treated CTR neurons and showed that intracellular CLU was significantly increased (Fig. 1a–c, p = 0.0221*) while secretion of CLU was significantly decreased (Fig. 1a,b,d, p = 0.0432*, original blots are presented in Supplementary Fig. 5). This response was absent in exon 2 −/− neurons. This is the first demonstration that Aβ25–35 exposure alters CLU trafficking in human neurons and supports previous studies that demonstrated CLU trafficking is altered in a variety of cell types/lines including rodent neurons, U251, SH-SY5Y, N2a and LNCaP cell lines following the induction of several stressors including Aβ25–35 treatment and ER stress (induced by MG132 proteasome inhibitor)35,36,38,40.
The translation of CLU variant 1 Δ exon 2 mRNA generates intracellular CLU proteins that are neither cleaved nor glycosylated, due to the absence of the ER-signal peptide41,43,67. Abundance of these CLU proteins is increased following stress43, and is thought to facilitate cell death42,43. Therefore, we next aimed to examine whether Aβ25–35 treatment induces an upregulation of these proteins in human neurons. First, abundance of non-glycosylated CLU proteins was compared between CTR and exon 2 −/− neurons prior to treatment with Aβ25–35. Unsurprisingly, abundance of non-glycosylated CLU proteins was significantly higher in exon 2 −/− neurons than in CTR neurons following the removal of CLU exon 2 (Fig. 1a,e,f). Next, the effect of Aβ25–35 treatment on non-glycosylated CLU abundance was examined. 20 μM Aβ25–35 did not alter the abundance of non-glycosylated CLU in either CTR or exon 2 −/− neurons (Fig. 1a,e,f). This contrasts with previous research that demonstrated that stress in cancer cell lines induces an upregulation of non-glycosylated CLU proteins. Our data show that stress induced by Aβ25–35 treatment in human neurons has a specific effect on the trafficking of glycosylated CLU proteins, but not on the overall abundance of the non-glycosylated CLU proteins.
Exon 2 −/− neurons display less Aβ25–35 induced cell death than CTR neurons
Having demonstrated that Aβ25–35 induces alterations in CLU trafficking in CTR neurons but does not alter abundance of non-glycosylated CLU, we next aimed to determine if this resulted in altered sensitivity of exon 2 −/− neuron to Aβ25–35. To do this, we first treated CTR and exon 2 −/− neurons with increasing concentrations of Aβ25–35 for 24 h (500 nM, 2 μM or 20 μM, respectively). After treatment, cell death was quantified by luminescence using the CytoTox-Glo Cytotoxicity Assay (Fig. 2).
Previous studies have identified a potential role for non-glycosylated CLU in facilitating cellular response to stress and apoptosis42,43. Additionally, overexpression of non-glycosylated CLU has been demonstrated to sensitise cells to stress42. Therefore, we aimed to determine if the loss of CLU exon 2 and the increased abundance of non-glycosylated CLU proteins at basal conditions in exon 2 −/− neurons altered their sensitivity to Aβ25–35 induced cell death. Dead/Total Dead Ratios were compared across all treatment conditions and between genotypes to determine if exon 2 −/− neurons were more sensitive to Aβ25–35 induced cell death (Fig. 2). In untreated conditions, Dead/Total Dead Ratios of exon 2 −/− neurons did not significantly differ to that of the CTR neurons, indicating that the proportion of dead neurons at basal conditions was not influenced by the genotype. No significant increase in ratios was observed at either 500 nM or 2 μM Aβ25–35 treatment for either CTR or exon 2 −/− neurons, indicating that these concentrations were not sufficient to induce a significant increase in cell death. However, a significant increase in ratios, reflecting a significant increase in the number of dead cells, was observed using 20 μM Aβ25–35 treated neuron cultures. This was observed in both, CTR and exon 2 −/− neuron cultures. Interestingly higher amounts of cell death were quantified in CTR cultures treated with 20 μM Aβ25–35 compared to exon 2 −/− cultures, since the Dead/Total Dead ratios of the latter where significantly lower. This finding indicates significantly fewer exon 2 −/− neurons underwent cell death because of Aβ25–35 treatment. These data demonstrate that the loss of CLU exon 2 and the increased abundance of non-glycosylated CLU proteins in human neurons reduces sensitivity to Aβ25–35.
Exon 2 −/− neurons are protected against Aβ25–35 induced neurite retraction
The observed reduction in Aβ25–35 induced cell death in exon 2 −/− neurons suggests that the lack of glycosylated CLU proteins renders neurons less sensitive to Aβ25–35 induced toxicity. To further explore this, we examined Aβ25–35 induced neurite retraction by measuring changes in neurite length in CTR and exon 2 −/− neurons.
As before, CTR and exon 2 −/− neurons were treated with one of three concentrations of Aβ25–35. After 24 h, neurons were fixed, immunostained and imaged. Neurons that were positively stained for nuclear marker DAPI and neuronal marker MAP2 were imaged (Fig. 3a–c) and an automated pipeline was used to identify MAP2 positive neurite processes and quantify their length. Neurite length parameters were quantified and compared between genotypes and Aβ25–35 treatment conditions (Fig. 3d–f).
Previously, we demonstrated that cell death prior to treatment was comparable between CTR and exon 2 −/− neurons, indicating that loss of CLU exon 2 did not impact neuronal death in the absence of an Aβ challenge. Accordingly, we aimed to determine if this was similar in the case of neurite length. Surprisingly, two neurite length parameters measured were significantly reduced in exon 2 −/− neurons prior to treatment with Aβ25–35, suggesting that loss of CLU exon 2 may impact neurite length even at basal conditions.
Next, we aimed to determine what concentration, if any, is sufficient to induce neurite damage in human neurons resulting in significant neurite retraction. For all parameters, 20 μM Aβ25–35 consistently caused a significant reduction in neurite length in CTR neurons, no significant changes in neurite length were identified in CTR neurons treated with 500 nM or 2 μM Aβ25–35, except in the case of mean segment length, which was significantly reduced in CTR neurons treated with 2 μM Aβ25–35. Finally, we explored whether the observed changes in neurite length in CTR neurons could also be detected in exon 2 −/− neurons. Interestingly, exon 2 −/− neurons did not demonstrate significant neurite retraction at any concentration of Aβ25–35, although did demonstrate reduced neurite length in basal conditions. Taken together, our data show that exon 2 −/− neurons are protected against both, Aβ25–35 induced neurite retraction and Aβ25–35 induced neuronal death.
CLU exon 2 removal downregulates extracellular matrix (ECM) pathways
Having identified differences in the responses of exon 2 −/− and CTR neurons to Aβ25–35 treatment, we aimed to use RNA-Seq analysis to identify changes in gene expression that may contribute to the altered sensitivity of exon 2 −/− neurons to Aβ25–35 induced toxicity. Differences in gene expression were compared between CTR and exon 2 −/− neurons and between untreated and Aβ25–35 treated neurons giving a total of 7 comparisons (Table 1 and Supplementary Table 4). UMAP analysis revealed distinct cell cultured based on treatment condition for CTR, A4 and D1 neurons (untreated vs Aβ25–35), additionally, separation of cell clusters based on neuronal genotype (CTR vs A4/D1) were observed with A4 and D1 counts clustering more closely to each other than with CTR counts (Supplementary Fig. 6). Differentially expressed genes in each comparison were grouped according to GO pathway enrichment to identify significantly altered pathways (Fig. 4a). Ten pathways were differentially activated between exon 2 −/− and CTR neurons in basal conditions prior to treatment with Aβ25–35 (res 4 and 5). Genes implicated in extracellular matrix (ECM) organisation, extracellular structure organisation, homophilic cell adhesion via plasma membrane adhesion molecules, cell–cell adhesion via plasma membrane adhesion molecules and several chondrocyte morphogenesis pathways were all were downregulated in exon 2 −/− neurons. Several ECM implicated genes were identified in a heatmap displaying the top 20 most highly variable genes (Fig. 4b), including COL1A1, COL1A2, COL3A1 and PCDHGA3. These were demonstrated to be more highly expressed in CTR neurons than in A4 and D1 neurons, implicating a depression of ECM-related gene expression. Functional enrichment analysis was performed (Supplementary Table 5). Functional directionality analysis of KEGG pathway enrichment (Fig. 4c) identified focal adhesion and extracellular matrix receptor interaction pathways and pointed to a general downregulation of ECM in exon 2 −/− neurons. Importantly, this was unaltered by Aβ25–35 in exon 2 −/− neurons. Topology analysis performed on KEGG pathway enrichment data supported these findings (Fig. 4d) and we could identify a suppression of ECM receptor interaction in exon 2 −/− neurons independent of treatment condition. Additionally, this analysis identified suppression of PI3K-Akt signalling in exon 2 −/− neurons, also independent of the treatment condition. These data suggest that loss of exon 2 and the subsequent alterations in CLU localisation in human neurons may result in reduced expression of genes encoding proteins that perform functions in the ECM and in the PI3K-Akt pathway. These findings indicate that the loss of CLU exon 2 and the subsequent changes in CLU localisation is sufficient to reduce sensitivity of human neurons in vitro to Aβ25–35 induced toxicity and downregulate genes performing ECM functions. It is unclear as to whether this reduced sensitivity is attributed to the loss of glycosylated CLU proteins or the observed increase in non-glycosylated intracellular CLU proteins in exon 2 −/− neurons.

Extracellular matrix (ECM) genes and pathways are downregulated in exon 2 −/− neurons. RNA-Seq analysis of exon 2 −/− and CTR neurons reveal suppression of ECM related genes and pathways in exon 2 −/− neurons. (a) GO (biological process) enrichment analysis reveal downregulation of collagen and ECM genes in exon 2 −/− neurons (res 4 and 5). (b) Heatmap visualisation of top 20 most variable genes clustered with complete hierarchical clustering using z-scored counts. Column annotations represent Aβ25–35 treatment condition and neuron genotype (CTR, A4 or D1). (c) Functional directionality analysis identified focal adhesion and ECM receptor interaction were suppressed in exon 2 −/− neurons, which was not altered by Aβ25–35 treatment. (d) Topology identified a significant suppression of ECM-receptor interaction and PI3K-Akt signalling pathways were suppressed in exon 2 −/− neurons, again unaltered by Aβ25–35. Figure legend outlines comparisons made in this analysis: res1; Aβ25–35 treated vs untreated CTR neurons, res2; Aβ25–35 treated vs untreated A4 neurons, res3; Aβ25–35 treated vs untreated D1 neurons, res4; untreated A4 vs untreated CTR neurons, res5; untreated D1 vs untreated CTR neurons, res6; Aβ25–35 treated A4 vs CTR neurons and res7; Aβ25–35 treated D1 vs CTR neurons. Differential gene expression satisfied thresholds for both fold change (> 1.5) and adjusted p value (< 0.05) were included in this analysis. Numbers of each res represent the number of differentially expressed genes enriched in each pathway for each comparison. Gene set enrichment satisfied adjusted p value (< 0.05). NES (enrichment score normalised to mean enrichment of random samples of the same size). tA the observed total perturbation accumulation in the pathway, pPERT probability to observe a total accumulation more extreme than tA only by chance. pPERT* < 0.05, ** < 0.01 and *** < 0.001.
Identification of gene expression changes induced by Aβ25–35 treatment
Having identified transcriptomic changes arising in neurons following removal of CLU exon 2, we next used our RNA-Seq dataset to identify changes in gene expression arising in neurons following treatment with Aβ25–35 that may be regulating neurite damage and cell death induced by Aβ25–35. Using topology-based analysis of KEGG pathway changes, several pathways were found altered in both, CTR and exon 2 −/− neurons, including apoptosis (activated), MAPK signalling (activated), p53 signalling (activated), cholinergic synapse (activated), glutamatergic synapse (inhibited) and axon guidance (inhibited) (Table 2 and Supplementary Fig. 7). Given these pathways were regulated by Aβ25–35 in CTR and exon 2 −/− neurons, it is likely that these contribute to cell death observed in both lines. For example, it is reasonable to suggest the observed activation in p53 signalling, apoptosis and MAPK signalling are related to Aβ25–35 induced cell death in both, exon 2 −/− and CTR neurons. However, it is not clear why cell death is induced by exon 2 −/− neurons but only to a lesser extent than in CTR neurons. Interestingly, although neurite retraction was absent in Aβ25–35 treated exon 2 −/− neurons, like in CTR neurons, axon guidance and glutamatergic synapse pathways were inhibited while cholinergic synapses were activated. These observations would be predicted in CTR neurons displaying neurite damage but since neurite retraction is absent in exon 2 −/− neuron it remains unclear why these pathways appear altered. We hypothesise that this could be due to differences in the extent of activation and inhibition of the pathways highlighted by our analysis in CTR and exon 2 −/− neurons, which may be reflected in the reduced sensitivity of exon 2 −/− neurons to cell death and neurite retraction; this is something to be explored in further studies. Therefore, examination of levels of activation and inhibition of pathways including axon guidance and cholinergic and glutamatergic synapses will help identify if they play a crucial role in mediating Aβ25–35 induced neurite retraction.
Finally, no pathway was identified as significantly altered in both A4 and D1 neurons that were not altered in CTR neurons following Aβ25–35 treatment. This suggests that exon 2 −/− neurons’ altered sensitivity to Aβ25–35 induced toxicity may arise due to an absence of pathway activation/inhibition observed in CTR neurons. As supporting evidence, three further pathways were altered in CTR neurons and not in exon 2 −/− neurons. Cell cycle and complement and coagulation cascades were all significantly activated in CTR neurons following Aβ25–35 treatment, while long term depression (LTD) was suppressed (Supplementary Fig. 7). This could be an indication that one or more of these pathways plays a crucial role in mediating Aβ25–35 induced neurite retraction, given that activation of these pathways was not observed in exon 2 −/− neurons in response to Aβ25–35 treatment.
Discussion
CLU has long been implicated in AD, however, its exact role in AD pathogenesis and disease development remains unclear. Our study demonstrates a central role for CLU proteins in facilitating Aβ25–35 induced cell death and neurite damage in human neurons in vitro.
Previous studies have examined the cytoprotective role of secreted CLU and the pro-apoptotic role of non-glycosylated CLU proteins43,68,69,70. However, these studies have focussed on the role of CLU proteins in cancer pathways in immortal cancer cell lines and few studies have directly manipulated the expression of non-glycosylated CLU proteins in human neurons to explore their importance in neurodegeneration. In contrast to previous work, we demonstrate that stress induced by Aβ25–35 does not induce increased expression of non-glycosylated CLU in human neurons. Therefore, the previously identified pro-apoptotic role of non-glycosylated CLU proteins may not be universal and may be dependent on factors such as the type of cell and source of stress. Yang et al., demonstrated that overexpression of non-glycosylated CLU proteins in cancer cells sensitizes cells to stress and induces more death in basal conditions42. This is not observed in human neurons where we have demonstrated that exon 2 −/− neurons displayed similar levels of cell death in basal conditions, indicating that increased expression of non-glycosylated CLU did not result in increased neuronal death, although changes to basal neurite length were demonstrated in these same cells. Further examination of the role of non-glycosylated CLU species may reveal their role in altering basal neurite length in exon 2 −/− neurons and if this is contributing to the reduced sensitivity of these neurons to Aβ25–35 or if there is a masking of the effect induced by Aβ25–35 on neurite length due to the reduction in basal conditions. It would therefore be valuable to assess the effect of CLU exon 2 removal on neuron development in vitro; the neurons used in this research are developmentally immature and thus, culturing for longer periods of time would be beneficial to compare the expression of neurons maturity markers and synapse formation in CTR and exon 2 −/− neurons. This would provide clarity on the development of neurons lacking glycosylated CLU and its effect on neuronal sensitivity to Aβ25–35. To the best of our knowledge, the authors are unaware of any study identifying CLU or Aβ25–35 as a regulator of neuron maturity.
We cannot rule out a combinatory effect on Aβ25–35 sensitivity reflected by both the loss of glycosylated CLU and increase in non-glycosylated CLU proteins in the same neurons. Very little is known of the role of non-glycosylated CLU proteins in cells, especially in human neurons, it is suggested that non-glycosylated CLU proteins are induced by stress and apoptosis41,42,43, however, these studies failed to categorically show that these CLU proteins are needed before cell death can occur, or their increased expression arises following the induction and initiation of apoptosis. Indeed, there may be a role for the non-glycosylated CLU proteins in neurite development that is influencing their sensitivity to Aβ25–35 induced toxicity. Although others have suggested a role of non-glycosylated CLU proteins generated from CLU mRNA lacking exon 2, this has only been demonstrated in immortal cancer cell lines using overexpression studies41,42,43, we are the first to generate iPSCs and iPSC-neurons lacking exon 2 and increased non-glycosylated CLU proteins in the absence of non-glycosylated CLU proteins. It is reasonable to suggest that the role of these proteins will likely differ between cell types, especially in cancer derived lines and neurons and is likely influenced by the mechanism of the stressor.
Exon 2 −/− iPSC-neurons displayed reduced sensitivity to Aβ25–35 induced cell death and neurite damage, suggesting that it is the glycosylated version of CLU that facilitate Aβ25–35 toxicity. We have therefore demonstrated that non-glycosylated CLU proteins likely do not play a critical role in mediating neurodegeneration in human neurons induced by Aβ25–35.
Although typically considered a secreted protein, glycosylated CLU proteins have been shown to localise within cells35,36,40, however, the mechanisms leading to intracellular retention of sCLU have not been described. Numerous studies have demonstrated that trafficking of glycosylated CLU proteins is altered by stress in a variety of cell types/lines36,37,38,39,40,41, resulting in reduced secretion and increased intracellular retention of CLU. Additionally, CLU-AD mutations and Aβ25–35 treatment of rodent neurons have been shown to alter CLU trafficking, providing a potential mechanistic role of CLU in mediating neurodegeneration17,35.
To date, few studies have attempted to dissect whether stress induced alterations in CLU trafficking act to facilitate cell stress or act to protect cells from further damage. Gregory et al. demonstrated altered CLU trafficking by ER stress in N2a cells resulting in increased binding of intracellular CLU and TDP-43, which reduced both the aggregation and toxicity of TDP-43, thus suggesting altered CLU trafficking in this context provides a protection to cells40. In comparison, previous work by our group found evidence that altered CLU trafficking facilitates Aβ25–35 induced cell death in rodent neurons and that Aβ25–35 increased intracellular CLU retention and CLU knockdown provided protection from Aβ25–35 induced neurodegeneration35. It is therefore likely that stress-induced CLU trafficking alterations and subsequent increases in intracellular CLU as well as reduced secretion can be both protective and detrimental to cells dependent on the type of stress and cell. Understanding the importance of glycosylated CLU proteins is particularly important since several studies have identified plasma CLU as a promising marker for AD71; higher plasma CLU levels are associated with increased hippocampal atrophy and increased rate of clinical progression72,73.
Our data clearly show that loss of glycosylated CLU protein in human neurons in vitro provides partial protection against Aβ25–35 induced toxicity, which we postulate may arise from ECM remodelling (Fig. 5). Although cell death was still induced by Aβ25–35 in exon 2 −/− neurons, it was significantly less than control neurons and neurite damage was absent in exon 2 −/− neurons. What remains unclear is whether the observed protection arises because of the loss of intracellular glycosylated CLU proteins or the loss of secreted glycosylated CLU proteins, or both. Although previous studies have suggested a pro-apoptotic role of non-glycosylated CLU proteins in cells42,43, little is really known of their physiological and pathological functions in cells. We demonstrate an increase in the abundance of non-glycosylated CLU proteins in exon 2 −/− neurons, which is not altered by Aβ25–35 treatment. It would be crucial to determine if protection observed in exon 2 −/− is conferred by the increased abundance of these CLU proteins.

Summary diagram. Graphic illustration highlighting key results observed in this study. To alter the type and localisation of CLU proteins generated by iPSCs CRISPR/Cas9 gene editing was used; iPSCs lacking CLU exon 2 do not produce mature, glycosylated and cleaved CLU proteins and therefore do not secrete CLU proteins. Instead, exon 2 −/− iPSCs generate single polypeptide CLU protein that does not undergo glycosylated and remains intracellular. Following differentiation of CTR and exon 2 −/− iPSCs into neurons, neurons were treated with Aβ25–35 and phenotypes compared to assess if altered CLU protein production alters the sensitivity of neurons to Aβ25–35 induced toxicity. Aβ25–35 treatment induced significant cell death and neurite damage to control neurons, this was accompanied by an alteration in CLU trafficking resulting in increased intracellular retention and reduced secretion of glycosylated CLU proteins. CLU trafficking was not altered in exon 2 −/− neurons since they do not express glycosylated CLU proteins. In contrast to previous reports, no evidence was found that non-glycosylated CLU protein abundance is increased following induction of stress or cell death. Exon 2 −/− neurons displayed significant cell death following Aβ25–35 treatment but no significant neurite damage was observed. We suggest that the partial protection displayed by exon 2 −/− neurons may be attributed to the altered expression and activity in extracellular matrix genes and pathways as highlighted by RNA-seq analysis. Diagram created with BioRender.com.
It is therefore crucial to determine the roles played by intracellular and secreted CLU proteins in Aβ25–35 induced toxicity to determine the exact roles played by CLU proteins in Aβ25–35 induced cell death and neurite retraction and the underlying mechanisms. Several studies have shown that CLU proteins bind directly to Aβ peptides20,37 to regulate Aβ uptake and clearance by astrocytes37. This role is attributed to secreted glycosylated CLU proteins. We hypothesise that in control neurons in response to Aβ25–35 treatment, secreted CLU is taken back into cells, potentially as a method of protecting cells. However, this role may facilitate toxicity if concentrations of Aβ25–35 peptides are high in the cellular media. sCLU proteins may bind to Aβ25–35 peptides and facilitate the uptake of CLU-Aβ25–35 complexes into neurons, thereby promoting entry of Aβ25–35 peptides into cells. Once inside the cell, Aβ25–35 peptides can then induce cytotoxicity74,75,76,77. Thus, in this context CLU is facilitating Aβ25–35 toxicity. Further interrogation of this relationship is required. Several receptors for CLU have been identified, including the megalin receptor21,34, but this interaction is not well described and has not been analysed in iPSC-neurons. In exon 2 −/− neurons, the absence of secreted CLU may result in reduced Aβ25–35 uptake into neurons, resulting in a lower concentration of Aβ25–35 peptides accumulating in the neurons and thus, less cell damage. Secreted clusterin has been shown to bind Aβ peptides and may mediate the internalisation and clearance of Aβ following binding to cell surface receptors34,37,78,79. Clusterin has been demonstrated to facilitate the uptake of Aβ by astrocytes37 but others have demonstrated clusterin may hamper Aβ uptake80,81. This interaction so far has not been explored in human neurons, and the model generated by our group would facilitate this; CTR and exon 2 −/− neurons could be treated with Aβ by addition to the media and imaging could then be used to measure Aβ uptake in both sets of neurons, to determine if glycosylated CLU proteins are facilitating Aβ uptake into neurons. Since we described only partial protection in exon 2 −/− neurons, other mechanisms must also facilitate the uptake of Aβ25–35 peptides and Aβ25–35 toxicity in human neurons, in addition to CLU proteins.
Interestingly, our RNAseq data has identified only three pathways that were altered by Aβ25–35 treatment in CTR and not in exon 2 −/− neurons: cell cycle, complement and coagulation cascades and LTD. The absence of response in these pathways in exon 2 −/− neurons suggest a role for CLU in these pathways, exploration of the role of these pathways in mediating Aβ25–35 toxicity in vitro may aid in understanding CLU role in facilitating Aβ25–35 neurotoxicity but also the role of these individual pathways in contributing to both cell death and neurite damage induced by Aβ25–35. It would be interesting to determine if the targeting of these pathways in CTR neurons to prevent their activation (in the case of cell cycle and complement/coagulation cascades) and prevent their suppression (LTD) would manifest as a reduction in Aβ25–35 induced phenotypes like those demonstrated in our exon 2 −/− neurons. The complement system is heavily implicated in AD and although it may not initiate disease pathology it is considered to be a driver of neurodegeneration82. Additionally, clusterin is a key inhibitor of the complement system34,83. Further exploration of the effect CLU exon 2 knockout has on the complement system in exon 2 −/− neurons will be invaluable in evaluating the source of their partial protection to Aβ25–35. The complement system however, is complex and it may be that certain complement components are driving pathology while others are protective. Nevertheless, our data implicate a key role of complement activation in mediating Aβ25–35 toxicity in vitro. Additionally, exon 2 −/− knockout resulted in a suppression of the PI3K-Akt pathway, a known regulator of GSK-3β; activation of Akt inhibits GSK-3β and thus, downstream reduces tau hyperphosphorylation84. The suppression of PI3K-Akt in exon 2 −/− neurons may thus, have effects in vitro beyond Aβ induced cell death and neurite damage; it would be interesting to determine if the suppression of PI3K-Akt has any effect on tau phosphorylation and synapse integrity following Aβ treatment in exon 2 −/− neurons.
Changes to the ECM were demonstrated in exon 2 −/− neurons and we postulate that ECM remodelling contributes to the partial protection observed in exon 2 −/− neurons to Aβ25–35 providing evidence for targeting the ECM as a therapeutic option for AD and neurodegeneration. The ECM is composed of a range of diverse proteins that serve a multitude of functions85. Those include regulation of cell death and survival86, albeit this is not clearly described in neurons, synaptic function87,88 and neurite outgrowth89,90, all three of which are significantly altered in AD and by Aβ25–35. ECM remodelling is associated with several diseases including AD91,92,93,94. Numerous ECM proteins have been described as upregulated or downregulated in AD brains94. For example, Collagen V and fibronectin are upregulated in the cerebral cortex of AD patients in early stages of disease progression and may arise from a reduction in proteolytic activity and may contribute to early changes in the AD brain95. Several ECM proteins play a crucial role in regulating synaptic transmission and thus, changes to these proteins may result in a disruption in synaptic function and result in abnormal synaptic activity, and impaired learning and memory96,97. ECM proteins have also been shown to alter Aβ toxicity. Heparan and chondroitin sulfate proteoglycans bind to Aβ peptides reducing Aβ25–35 induced toxicity in hippocampal cultures and PC12 cells. Indeed, reducing the expression of chondroitin sulfate proteoglycans enhances Aβ1–42 toxicity in neurons98. Other ECM proteins have been shown to promote Aβ aggregation and plaque formation99,100. Most recently, a large scale deep multi-layer analysis study of AD brains identified the ECM to strongly correlate with AD traits101, providing strength to our observations that the ECM may play a key role in mediating Aβ25–35 induced toxicity in human neurons. More work needs to be done to fully understand the role of the ECM in AD since both, protective and facilitatory roles have been identified. In our study it is apparent that the ECM directly contributes to toxicity induced by Aβ25–35 in vitro in human iPSC-neurons since ECM remodelling is associated with neuroprotective properties to iPSC-neurons. Given the varied functions of ECM proteins in neurons, it is essential to identify the key proteins that are central to the partial protection observed in exon 2 −/− neurons to determine if they can be utilised as a therapeutic target to provide neuroprotection.
Conclusions
Our data show that glycosylated CLU proteins facilitate neurotoxicity induced by Aβ25–35 in human neurons. In fact, we demonstrate a role for CLU in mediating both Aβ25–35 induced neurite retraction and cell death; the absence of glycosylated CLU and increased non-glycosylated CLU proteins provided partial protection against Aβ25–35 to neurons in vitro. Previous research has identified the role of non-glycosylated CLU proteins in promoting cell death and increasing cell sensitivity to stress. However, these studies had been carried out in cancer cell lines and not neurons. Our work clearly shows the importance of cellular context and clearly demonstrates that, in neurons, non-glycosylated CLU does not promote neuronal degeneration but that increased expression results in a protection against neurite loss and neuronal death. We have identified several genes and pathways that potentially underlie this neuroprotection including those that may play a critical role in regulating cell death and neurite damage induced by Aβ25–35. Our data will facilitate the identification of key components underlying Aβ25–35 toxicity and identify potential therapeutic targets to prevent neurodegeneration in AD.
Data availability
The RNA-Seq datasets generated and analysed during this current study are available in the NCBI GEO repository (GSE207466), which can be accessed here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE207466). For data, please contact corresponding author Professor Noel Buckley.
References
National Institute on Aging. Alzheimer’s Disease Fact Sheet. http://www.nia.nih.gov/health/alzheimers-disease-fact-sheet (2019).
Hardy, J. A. & Higgins, G. A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 256, 184–185 (1992).
Musiek, E. S. & Holtzman, D. M. Three dimensions of the amyloid hypothesis: Time, space, and “wingmen”. Nat. Neurosci. 18, 800–806 (2015).
Selkoe, D. J. & Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608 (2016).
Goate, A. et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706 (1991).
Murrell, J., Farlow, M., Ghetti, B. & Benson, M. A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science 254, 97–99 (1991).
Mullan, M. et al. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of β-amyloid. Nat. Genet. 1, 345–347 (1992).
Sherrington, R. et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760 (1995).
Scheuner, D. et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 2, 864–870 (1996).
Bertram, L. The genetic epidemiology of neurodegenerative disease. J. Clin. Investig. 115, 1449–1457 (2005).
Cruts, M., Theuns, J. & Van Broeckhoven, C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 33, 1340–1344 (2012).
Tanzi, R. E. & Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 120, 545–555 (2005).
Lambert, J.-C. et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1099 (2009).
Harold, D. et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 41, 1088–1093 (2009).
Lambert, J.-C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Bettens, K. et al. Both common variations and rare non-synonymous substitutions and small insertion/deletions in CLU are associated with increased Alzheimer risk. Mol. Neurodegener. 7, 3 (2012).
Bettens, K. et al. Reduced secreted clusterin as a mechanism for Alzheimer-associated CLU mutations. Mol. Neurodegener. 10, 30 (2015).
Zhou, Y. et al. Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule-associated protein tau in Alzheimer’s disease. PLoS One 9, e103187 (2014).
AlzGene. http://www.alzgene.org/geneoverview.asp?geneid=323.
Wojtas, A. M. et al. Loss of clusterin shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways. Proc. Natl. Acad. Sci. 114, E6962–E6971 (2017).
Zlokovic, V. Cerebrovascular transport of Alzheimer’s amyloidp and apolipoproteins J and E: Possible anti-amyloidogenic role of the blood-brain barrier. Life Sci. 59, 15 (1996).
DeMattos, R. et al. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. 99, 10843–10848 (2002).
DeMattos, R. et al. ApoE and clusterin cooperatively suppress Abeta levels and deposition: Evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron 41, 193–202 (2004).
Kapron, J. T. et al. Identification and characterization of glycosylation sites in human serum clusterin. Protein Sci. Publ. Protein Soc. 6, 2120–2133 (1997).
Lakins, J. et al. Clusterin biogenesis is altered during apoptosis in the regressing rat ventral prostate. J. Biol. Chem. 273, 27887–27895 (1998).
Sabatte, J. et al. Semen clusterin is a novel DC-SIGN ligand. J. Immunol. 187, 5299–5309 (2011).
Urban, J., Parczyk, K., Leutz, A., Kayne, M. & Kondor-Koch, C. Constitutive apical secretion of an 80-kD sulfated glycoprotein complex in the polarized epithelial Madin–Darby canine kidney cell line. J. Cell Biol. 105, 2735–2743 (1987).
Wong, P. et al. Genomic organization and expression of the rat TRPM-2 (clusterin) gene, a gene implicated in apoptosis. J. Biol. Chem. 268, 5021–5031 (1993).
Rohne, P., Prochnow, H. & Koch-Brandt, C. The CLU-files: Disentanglement of a mystery. Biomol. Concepts 7, 1–15 (2016).
Choi-Miura, N.-H. & Oda, T. Relationship between multifunctional protein “clusterin” and Alzheimer disease. Neurobiol. Aging 17, 717–722 (1996).
Jenne, D. E. & Tschopp, J. Molecular structure and functional characterization of a human complement cytolysis inhibitor found in blood and seminal plasma: Identity to sulfated glycoprotein 2, a constituent of rat testis fluid. Proc. Natl. Acad. Sci. U.S.A. 86, 7123–7127 (1989).
Kirszbaum, L., Bozas, S. E. & Walker, I. D. SP-40,40, a protein involved in the control of the complement pathway, possesses a unique array of disulphide bridges. FEBS Lett. 297, 70–76 (1992).
Foster, E. M., Dangla-Valls, A., Lovestone, S., Ribe, E. M. & Buckley, N. J. Clusterin in Alzheimer’s disease: Mechanisms, genetics, and lessons from other pathologies. Front. Neurosci. 13, 164 (2019).
Ghiso, J., Matsubara, E., Koudinov, A. & Frangione, B. The cerebrospinal-fluid soluble form of Alzheimer’s amyloid beta is complexed to SP-40,40 (apolipoprotein J), an inhibitor of the complement membrane-attack complex. Biochem. J. 1, 27–30 (1993).
Killick, R. et al. Clusterin regulates β-amyloid toxicity via Dickkopf-1-driven induction of the Wnt–PCP–JNK pathway. Mol. Psychiatry 19, 88–98 (2014).
Nizard, P. et al. Stress-induced retrotranslocation of clusterin/ApoJ into the cytosol. Traffic 8, 554–565 (2007).
Nuutinen, T. et al. Amyloid-β 1–42 induced endocytosis and clusterin/apoJ protein accumulation in cultured human astrocytes. Neurochem. Int. 50, 540–547 (2007).
Li, N., Zoubeidi, A., Beraldi, E. & Gleave, M. E. GRP78 regulates clusterin stability, retrotranslocation and mitochondrial localization under ER stress in prostate cancer. Oncogene 32, 1933–1942 (2013).
O’Sullivan, J., Whyte, L., Drake, J. & Tenniswood, M. Alterations in the post-translational modification and intracellular trafficking of clusterin in MCF-7 cells during apoptosis. Cell Death Differ. 10, 914–927 (2003).
Gregory, J. M. et al. Clusterin protects neurons against intracellular proteotoxicity. Acta Neuropathol. Commun. 5, 81 (2017).
Prochnow, H. et al. Non-secreted clusterin isoforms are translated in rare amounts from distinct human mRNA variants and do not affect Bax-mediated apoptosis or the NF-κB signaling pathway. PLoS One 8, e75303 (2013).
Yang, C.-R. et al. Nuclear clusterin/XIP8, an X-ray-induced Ku70-binding protein that signals cell death. Proc. Natl. Acad. Sci. 97, 5907–5912 (2000).
Leskov, K. S., Klokov, D. Y., Li, J., Kinsella, T. J. & Boothman, D. A. Synthesis and functional analyses of nuclear clusterin, a cell death protein. J. Biol. Chem. 278, 11590–11600 (2003).
Andersen, C. L. et al. Clusterin expression in normal mucosa and colorectal cancer. Mol. Cell. Proteom. MCP 6, 1039–1048 (2007).
Cocks, G. et al. The utility of patient specific induced pluripotent stem cells for the modelling of Autistic Spectrum Disorders. Psychopharmacology 231, 1079–1088 (2014).
Nishimura, K. et al. Development of defective and persistent Sendai virus vector. J. Biol. Chem. 286, 4760–4771 (2011).
Adhya, D. et al. Atypical neurogenesis in induced pluripotent stem cells from autistic individuals. Biol. Psychiatry 89, 486–496 (2020).
Blayney, J. et al. Unexpectedly high levels of inverted re-insertions using paired sgRNAs for genomic deletions. Methods Protoc. 3, 53 (2020).
Shi, Y., Kirwan, P. & Livesey, F. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 7, 1836–1846 (2012).
Shi, Y., Kirwan, P., Smith, J., Robinson, H. & Livesey, F. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 15, 477–486 (2012).
Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data (2010).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at http://arxiv.org/abs/1303.3997 (2013).
Lawrence, M. et al. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287 (2012).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 102, 15545–15550 (2005).
Tarca, A. L. et al. A novel signaling pathway impact analysis. Bioinformatics 25, 75–82 (2009).
McInnes, L., Healy, J. & Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. Preprint at http://arxiv.org/abs/1802.03426 (2020).
Blasko, I., Wagner, M., Whitaker, N., Grubeck-Loebenstein, B. & Jansen-Du, P. The amyloid β peptide (25–35) induces apoptosis independent of p53. FEBS Lett. 470, 221–225 (2000).
Copani, A. et al. Mitotic signaling by β-amyloid causes neuronal death. FASEB J. 13, 2225–2234 (1999).
Li, Y. P., Bushnell, A. F., Lee, C. M., Perlmutter, L. S. & Wong, S. K. Beta-amyloid induces apoptosis in human-derived neurotypic SH-SY5Y cells. Brain Res. 738, 196–204 (1996).
Michaelis, M. L. et al. Protection against beta-amyloid toxicity in primary neurons by paclitaxel (Taxol). J. Neurochem. 70, 1623–1627 (1998).
Morishima, Y. et al. β-Amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J. Neurosci. 21, 7551–7560 (2001).
Yankner, B. A., Duffy, L. K. & Kirschner, D. A. Neurotrophic and neurotoxic effects of amyloid, beta protein: Reversal by tachykinin neuropeptides. Science 250, 279–282 (1990).
Reddy, K. B., Jin, G., Karode, M. C., Harmony, J. A. K. & Howe, P. H. Transforming growth factor β (TGFβ)-induced nuclear localization of apolipoprotein J/clusterin in epithelial cells. Biochemistry 35, 6157–6163 (1996).
Zhang, H. et al. Clusterin inhibits apoptosis by interacting with activated Bax. Nat. Cell Biol. 7, 909–915 (2005).
Trougakos, I. P. et al. Intracellular clusterin inhibits mitochondrial apoptosis by suppressing p53-activating stress signals and stabilizing the cytosolic Ku70-Bax protein complex. Clin. Cancer Res. 15, 48–59 (2009).
Trougakos, I. P., Lourda, M., Agiostratidou, G., Kletsas, D. & Gonos, E. S. Differential effects of clusterin/apolipoprotein J on cellular growth and survival. Free Radic. Biol. Med. 38, 436–449 (2005).
Liang, H.-C. et al. Glycosylation of human plasma clusterin yields a novel candidate biomarker of Alzheimer’s disease. J. Proteome Res. 14, 5063–5076 (2015).
Thambisetty, M. et al. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch. Gen. Psychiatry 67, 739–748 (2010).
Thambisetty, M. et al. Plasma biomarkers of brain atrophy in Alzheimer’s disease. PLoS One 6, e28527 (2011).
Sato, K. et al. Correlation among secondary structure, amyloid precursor protein accumulation, and neurotoxicity of amyloid β(25–35) peptide as analyzed by single alanine substitution. J. Biochem. (Tokyo) 118, 1108–1111 (1995).
Clementi, M. E. et al. Aβ(31–35) and Aβ(25–35) fragments of amyloid beta-protein induce cellular death through apoptotic signals: Role of the redox state of methionine-35. FEBS Lett. 579, 2913–2918 (2005).
Takashima, A. et al. Amyloid β peptide induces cytoplasmic accumulation of amyloid protein precursor via tau protein kinase I/glycogen synthase kinase-3β in rat hippocampal neurons. Neurosci. Lett. 198, 83–86 (1995).
Millucci, L., Ghezzi, L., Bernardini, G. & Santucci, A. Conformations and biological activities of amyloid beta peptide 25–35. Curr. Protein Pept. Sci. 11, 54–67 (2010).
Bartl, M. M., Luckenbach, T., Bergner, O., Ullrich, O. & Koch-Brandt, C. Multiple receptors mediate apoJ-dependent clearance of cellular debris into nonprofessional phagocytes. Exp. Cell Res. 271, 130–141 (2001).
Bell, R. D. et al. Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. J. Cereb. Blood Flow Metab. 27, 909–918 (2007).
Mulder, S. D., Nielsen, H. M., Blankenstein, M. A., Eikelenboom, P. & Veerhuis, R. Apolipoproteins E and J interfere with amyloid-beta uptake by primary human astrocytes and microglia in vitro: ApoE and ApoJ hamper amyloid-beta uptake. Glia 62, 493–503 (2014).
Nielsen, H. M. et al. Astrocytic Aβ1–42 uptake is determined by Aβ-aggregation state and the presence of amyloid-associated proteins: Uptake of Aβ1–42 oligomers and fibrils. Glia 58, 1235–1246 (2010).
Morgan, B. P. Complement in the pathogenesis of Alzheimer’s disease. Semin. Immunopathol. 40, 113–124 (2018).
Tschopp, J., Chonn, A., Hertig, S. & French, L. E. Clusterin, the human apolipoprotein and complement inhibitor, binds to complement C7, C8 beta, and the b domain of C9. J. Immunol. (Baltim. Md.) 1950(151), 2159–2165 (1993).
Baki, L. et al. PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: Effects of FAD mutations. EMBO J. 23, 2586–2596 (2004).
Barros, C. S., Franco, S. J. & Muller, U. Extracellular matrix: Functions in the nervous system. Cold Spring Harb. Perspect. Biol. 3, a005108–a005108 (2011).
Makino, H., Sugiyama, H. & Kashihara, N. Apoptosis and extracellular matrix–cell interactions in kidney disease. Kidney Int. 58, S67–S75 (2000).
Ferrer-Ferrer, M. & Dityatev, A. Shaping synapses by the neural extracellular matrix. Front. Neuroanat. 12, 40 (2018).
Dityatev, A. & Schachner, M. The extracellular matrix and synapses. Cell Tissue Res. 326, 647–654 (2006).
Letourneau, P. C., Condic, M. L. & Snow, D. M. Extracellular matrix and neurite outgrowth. Curr. Opin. Genet. Dev. 2, 625–634 (1992).
Srinivasan, P., Zervantonakis, I. K. & Kothapalli, C. R. Synergistic effects of 3D ECM and chemogradients on neurite outgrowth and guidance: A simple modeling and microfluidic framework. PLoS One 9, e99640 (2014).
Bonneh-Barkay, D. & Wiley, C. A. Brain extracellular matrix in neurodegeneration. Brain Pathol. 19, 573–585 (2009).
Morawski, M. et al. Involvement of perineuronal and perisynaptic extracellular matrix in Alzheimer’s disease neuropathology: Perineuronal and perisynaptic matrix in AD. Brain Pathol. 22, 547–561 (2012).
Soria, F. N. et al. Synucleinopathy alters nanoscale organization and diffusion in the brain extracellular space through hyaluronan remodeling. Nat. Commun. 11, 3440 (2020).
Sun, Y. et al. Role of the extracellular matrix in Alzheimer’s disease. Front. Aging Neurosci. 13, 554 (2021).
Lepelletier, F.-X., Mann, D. M. A., Robinson, A. C., Pinteaux, E. & Boutin, H. Early changes in extracellular matrix in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 43, 167–182 (2017).
Lindahl, B., Eriksson, L., Spillmann, D., Caterson, B. & Lindahl, U. Selective loss of cerebral keratan sulfate in Alzheimer’s disease. J. Biol. Chem. 271, 16991–16994 (1996).
Buckley, K. & Kelly, R. B. Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J. Cell Biol. 100, 1284–1294 (1985).
Miyata, S., Nishimura, Y. & Nakashima, T. Perineuronal nets protect against amyloid β-protein neurotoxicity in cultured cortical neurons. Brain Res. 1150, 200–206 (2007).
Gupta-Bansal, R., Frederickson, R. C. & Brunden, K. R. Proteoglycan-mediated inhibition of A beta proteolysis. A potential cause of senile plaque accumulation. J. Biol. Chem. 270, 18666–18671 (1995).
Bruinsma, I. B. et al. Sulfation of heparan sulfate associated with amyloid-beta plaques in patients with Alzheimer’s disease. Acta Neuropathol. (Berl.) 119, 211–220 (2010).
Johnson, E. C. B. et al. Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat. Neurosci. 25, 213–225 (2022).
Acknowledgements
We thank members of Synthego Corporation generating the CLU exon 2 knockout iPSC lines and their support in this research. This work was supported by AstraZeneca as part of a CASE studentship and the Valat-Jones Foundation (Nigel and Françoise Jones).
Author information
Authors and Affiliations
Contributions
S.L., N.J.B. and E.M.R. conceived the project. M.P. provided external funding and support for the project. P.H. and A.D.V. provided technical assistance during the development and optimisation of the project. E.M.F. carried out experiments, performed the data analysis and wrote the manuscript. RNA-Seq analysis was performed by M.F. E.M.F. and N.J.B. edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Foster, E.M., Fernandes, M., Dangla-Valls, A. et al. Glycosylated clusterin species facilitate Aβ toxicity in human neurons. Sci Rep 12, 18639 (2022). https://doi.org/10.1038/s41598-022-23167-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-23167-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
