
Abstract
We describe an approach based on non-equilibrium molecular dynamics (NEMD) simulations to calculate the ionic mobility of solid ion conductors such as solid electrolytes from first-principles. The calculations are carried out in finite slabs of the material, where an electric field is applied and the dynamic response of the mobile ions is measured. We compare our results with those obtained from diffusion calculations, under the non-interacting ion approximation, and with experiment. This method is shown to provide good quantitative estimates for the ionic mobilities of two silver conductors, \(\alpha\)-AgI and \(\alpha\)-RbAg\(_4\)I\(_5\). In addition to being convenient and numerically robust, this method accounts for ion-ion correlations at a much lower computational cost than exact approaches.
Similar content being viewed by others


Introduction
The successful expansion of renewable energies and the corresponding liberation from the dependence on fossil fuels require the development of both mobile and stationary storage energy solutions. Li-ion batteries, which store electrical energy directly and are light and compact, have been the solution of choice for consumer electronics, hybrid cars, wearables and medical devices1,2. However, improvements are needed with regard to safety, cost, storage capacity and lifetime.
Batteries are typically composed of a cathode, an anode and an electrolyte. Commercial Li-ion batteries have a separator to prevent the contact between the cathode and the anode, and a liquid electrolyte solution3. The presence of the liquid electrolyte and the possibility of unexpected chemical reactions in the battery leads to safety concerns with regards to swelling and fire hazard. All-solid-state batteries, in contrast, employ a solid electrolyte instead of a liquid organic electrolyte. For this reason, they are more stable and need less safety components, leaving room for increasing the packing density2,4,5,6,7.
The main constraint is the lower ionic conductivity (\(\sigma\)) of solid electrolytes, compared with liquid electrolytes. The ionic conductivity is a characteristic of the material, and can be directly related to the ionic mobility (\(\mu\)), \(\sigma =nq\mu\), where n is the density of ions that carry charge and q is the ion charge. A theoretical description of ion drag in solids relating a higher \(\mu\) to fundamental properties such as lower density and stiffness has recently been proposed8. Experimental development have also lead to the discovery of new solid electrolyte materials with ionic mobilities rivaling those of liquids, including garnets, perovskites and NASICON-based materials7.
In practice, there are diverse ionic conduction mechanisms, some not yet completely understood. One of the most studied archetype superionic conductors is \(\alpha\)-AgI. This is a ‘molten’ sublattice type superionic conductor, where the charge conducting ions, making up a disordered sublattice, are able to diffuse in a liquid-like fashion, while the remaining atoms display crystalline order9. This \(\alpha\) phase is stable at ambient pressure only between 147 and 555 \(^\circ\)C10. The extraordinarily high ionic conductivity of \(\alpha\)-AgI and its relatively weak temperature dependence are comparable with those of liquid electrolytes making it a paradigmatic example of a solid electrolyte11.
Computational methods have been used both to understand the ion conduction mechanisms and to predict which materials are good ionic conductors. Notably, molecular dynamics simulations under electric field have been a source of insight on the ion dynamics12,13,14, on interface morphology and behaviour15,16, and have been used to estimate the ionic conductivity17. Nevertheless, the ionic conductivity is instead often estimated based on equilibrium molecular dynamics calculations, using the Nernst-Einstein relationship18, which was originally established for gases, and fails when the ionic motion is correlated19,20,21. Calculations of the mobility based on an exact relationship between the conductivity and particle-particle velocity correlation functions, obtained from linear response theory20,20,22,23, are notoriously difficult to evaluate, requiring large time sampling21 and careful correction for the drift of the origin of coordinates in calculations with periodic boundary conditions20. Since taking advantage of correlations may be a way to surpass the current conductivity limits, it is desirable to be able to preform calculations of the conductivity that include correlations and are at the same time simple and numerically robust.
In this article, we revisit the use of non-equilibrium molecular dynamics simulations in superionic conductors12,13,14,17 and show that the ionic mobility, the main figure of merit for solid electrolytes, can be directly calculated from first-principles, modelling the drift under an electric field, rather than the diffusion in the absence of the electric field.
Results
We have performed a direct calculation for two silver superionic conductors, \(\alpha\)-AgI and \(\alpha\)-RbAg\(_4\)I\(_5\).
Both the \(\alpha\)-AgI and the \(\alpha\)-RbAg\(_4\)I\(_5\) structures have relatively less mobile I or Rb lattices, forming a matrix throughout which the Ag\(^+\) ions distribute statistically among a multiplicity of available sites24. \(\alpha\)-AgI belongs to the Im\(\bar{3}\)m space group, with two I atoms per unit cell forming a body-centered cubic lattice, and the two Ag atoms distributed over the 36 available sites, 24h and 12d, with probabilities of 0.07 and 0.027, respectively (Fig. 1a)24. The \(\alpha\)-RbAg\(_4\)I\(_5\) crystal belongs to the P4\(_1\)32 space group, which has four formula units per primitive cell (Fig. 1b), with 16 Ag\(^+\) ions occupying the available Ag sites, which have been proposed to be 5611,25 or more26,27,28. We have adopted the structure obtained by Spencer et al.25 as a starting point.
Our calculations employ slabs of material of length \(\sim\) 30–50 Å along the direction of the electric field (z), and periodic along the perpendicular directions (Fig. 1). We will start by examining in detail the behaviour of the 30 Å slab, which is sufficient to obtain quantitative predictions. More details can be found under the Methods section.
Our direct calculation of the mobility under the application of a constant electric field resembles the experimental Transient Ionic Current (TIC) technique, which employs DC (direct current) voltage across an electrolyte connected to two blocking electrodes29. Thus, in respect to boundary conditions, we maintain the atoms at the surfaces of the slab fixed, which is equivalent to having perfectly blocking electrodes, preventing mobile ions from escaping to the vacuum spacing. Calculations where all atoms are free give identical results, for small voltages, provided that we correct for the arbitrary translation of the centre of mass due to the periodic boundary conditions.
Some DFT codes apply by default a slab dipole correction, which creates vanishing internal electric field conditions. Such correction was designed to model ferroelectrics in short-circuit conditions30. However, in the case of the electrolyte, the internal electric field is not vanishing (except for a fully discharged battery). Thus, a slab dipole correction should not be applied for this particular purpose. The external electric field in the TIC experiments is simply determined by the external DC source, and the internal electric field by the polarisation response of the material.

Structure of Ag\(^+\) superionic conductors: (a) \(\alpha\)-AgI, and (b) \(\alpha\)-RbAg\(_4\)I\(_5\), showing all available positions for Ag. (c) Example of 2 × 2 × 6 slab used for calculations.
Electric field response
We are interested in the linear response to the electric field, where conductivity is defined as \(\vec {j}=\sigma \vec {E}=nq\vec {v}\), with \(\vec {j}\) is the current density, \(\vec {E}\) is the electric field, and \(\vec {v}\) is the velocity of the ions, and n is the density of ions carrying charge, which we assume for simplicity to be of only one type.
The external electric field \(E_0\) is turned on at \(t=0\) in a slab that previously was in thermal equilibrium. Thus, for \(t\sim 0\), the depolarising field can be neglected. To keep to a definition that is consistent to what is measured experimentally, we take E to be the external electric field \(E_0\). The largest electric field that can be applied in the simulation can be estimated using the expression \(eE_{\mathrm{max}}=E_g(L_z)/L_z\), where \(E_g(L_z)\) and \(L_z\) are the bandgap and the length of the slab, respectively, and e is the electron charge. The bandgap decreases with the slab length due to effect of electron confinement. Under application of an external electric field, the coordinates of the Ag\(^+\) ions change linearly with time during the first 10 ps (Fig. 2a). Exceptions are (i) for very small times (<1 ps), where transport exhibits characteristics of the ballistic regime31, and (ii) for high fields close to \(E_{\mathrm{max}}\), where electronic transitions may already take place due to the presence of defect states. The calculated velocities, obtained by linear regression, are in turn proportional the electric field (Fig. 2b).

Calculation of the ionic conductivity of AgI at 450 K. (a) linear dependence of z on t, (b) calculation of the mobility including all atoms in the slab; (c) dependence of the mobility on the initial position of the atoms in the slab, and (d) calculation of the mobility, excluding the atoms at the end of the slab. (e) Illustration of a cooperative jump observed for drift under an electric field of 0.075 V/Å. The Ag\(^+\) ions involved in mechanism are highlighted in yellow, orange and red (each one being previously knocked-on by the previous, in this order).
Some approximations that are implicit to this simulation design are adopting the nominal charge of \(+e\) for Ag\(^+\), which may be underestimated32, and neglecting charge transport by I\(^-\) ions, which may also lead to an underestimation of the conductivity. Both approximations have been commonly employed by previous studies of \(\alpha\)-AgI.
Finite length correction
Due to the finite length of the model slab, atoms near the positive end (\(z=L\)) will have a short distance to run than atoms nearer to the negative end (\(z=0\)). Hence, if \(L-z_0^i<\mu E\Delta t\), where \(z_0^i\) is the initial coordinate of atom i \(\Delta t\) is the simulation time, the mobility will be underestimated. Additionally, the interaction with clamped atoms affects the mobility of Ag\(^+\) ions. As can be seen in Fig. 2c, the calculated mobility is approximately constant in the first three-fifths of the slab, but it decays rapidly afterwards. Thus, a more accurate value for the mobility can be obtained if the atoms with \(z_0^i>18\) Å are disregarded. The corresponding results are shown in Fig. 2d. The calculated mobility, 6.34 \(\times 10^{-4}\) cm\(^2\)/Vs is very close to the experimental value (Table 1).
To check for the convergence with respect to the slab length, we have performed additional calculations in slabs consisting of 2 × 2 × 10 unit cells. In this case, we have obtained \(\mu\) = 5.18 ± 1.19 \(\times 10^{-4}\) cm\(^2\)/V.s for \(z_0^i<20\) Å and \(\mu\) = 8.50 ± 0.89 \(\times 10^{-4}\)cm\(^2\)/V.s for \(z_0^i<30\) Å. We thus see that the calculated mobility increases with the slab length, because the interaction of the Ag ions with the fixed atoms at the \(z=0\) and \(z=L\) ends decreases, in average. The final value, \(\mu\) = 8.50 ± 0.89 \(\times 10^{-4}\)cm\(^2\)/V.s, converged within \(\sim\)2 \(\times 10^{-4}\)cm\(^2\)/V.s, is consistent with experiment.
Temperature dependence
The ionic conductivity of \(\alpha\)-AgI has a weak temperature dependence33, due to the small activation energy for Ag\(^+\) hopping between sites (see Supplementary Information S2). Hall effect measurements also find a weak temperature dependence of the mobility34. We have performed calculations for samples equilibrated to 450, 550 and 650 K. After thermal equilibration using a Nosé thermostat35, the dynamical evolution of the system under electric field is calculated using a Verlet algorithm36. There is a slight increase of the mobility with temperature (Fig. 3), however not conclusive in comparison with the error bars (Table 1). Above 500 K, z(t) becomes non-linear for the highest electric field (0.1 V/Å), likely due to the increased defect density and resulting narrowing of the bandgap. The mobility values presented in Table 1 for \(T>\)450 K are obtained using \(E<0.075\) V/Å.

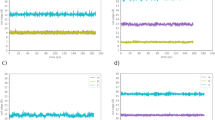
Ionic mobility at different temperatures. For \(\alpha\)-AgI: (a) dependence of the drift velocity on the electric field and (b) temperature changes during the simulation due to Joule heating, for a starting temperature of 450 K and an electric field of 0.05 V/Å. The green line is a linear fit. For RbAg\(_4\)I\(_5\) at different temperatures: (a) at 400 K and (b) at 298 K. At 298 K, the ionic mobility of \(\sim\) 3 × 10\(^{-4}\) cm\(^2\)/V s is close to the lowest value that we can estimate using this method, with a reasonable number of samples.
In the case of RbAg\(_4\)I\(_5\) at 400 K, non-linear behaviour starts to be observed for an electric field of 0.055 V/Å, due to the proximity of the valence and conduction band edges for this value of the field (Fig 3c). For smaller electric fields, we obtain a mobility in excellent agreement with experiment (Table 1). At 300 K, conduction is slower. For the ensemble of 10 starting points that we use, the error bars of \(\langle v(E) \rangle\) are comparable to the velocity increment \(\frac{dv}{dE}\Delta E\) (Fig 3d). Still, the calculated mobility is surprisingly close to experiment (Table 1). This example illustrates the limitations of this method for conditions of low mobility, which require the use of larger ensemble and higher integration times. Nevertheless, an advantage of this method is that it allows a quick and unambiguous identification of low mobility materials and/or conditions that are of no technological interest.
The lattice constant changes with temperature were neglected in our calculations here39. Additionally, one of the reasons why it is difficult to determine the temperature dependencee of the mobility is that the temperature itself is changing during the application of the electric field, due to the inelastic collisions of the mobile ions with the matrix and with other mobile ions (Joule effect).
Joule effect
The heat generation rate by Joule effect is given by \(P=VI\), where V is the voltage and I the current, or, in its volumetric form,
The rate of temperature change is then given by
where N is the number of moles of material and \(C_v\) is the heat capacity at constant volume. We observe, as expected, an increase in temperature that is approximately linear in time, as shown in Fig. 3b (see Supplementary Information S3). From there we obtain \(C_v=169\) J/mol K, which is higher than the experimental value 80 J/mol K39.
Tracer diffusion
We now compare the mobility obtained from NEMD to that obtained from equilibrium molecular dynamics simulations, using the Einstein relation
where \(\vec {r(t)}\) are the positions of the silver ions, and \(\langle ... \rangle\) is the ensemble average. This approach assumes that all atoms are independent, and the diffusion coefficient D thus defined corresponds experimentally to what is measured by tracer experiments, with very dilute tracers.
The diffusion coefficient we have obtained for \(\alpha\)-AgI over 50 ps of diffusion is in good agreement with experiment, showing the same temperature dependence, though systematically underestimated (Fig. 4). For RbAg\(_4\)I\(_5\), the diffusion constant, estimated for a time of 30–60 ps of diffusion, is also in good agreement with experiment.
Assuming that the atomic displacements are independent and random, the diffusion coefficient can be related to the conductivity via the Nernst-Einstein relation
Conversely, the mobility can be obtained from the diffusion coefficient:
This mobility is not equivalent to the mobility \(\mu\) measured in conduction experiments; still, it is often used as an approximation for \(\mu\). The ratio from the diffusion and conduction mobilities is the Haven ratio, \(H=\mu _D/\mu\), which is determined experimentally by comparing the mobility obtained from tracer diffusion or conductivity experiments. Our calculated values are in reasonable agreement, although smaller than the experimental ones (Table 2).
Since conductivity and tracer diffusivity were calculated by different methods, the calculated Haven ratio may suffer from the lack of error cancellation. Still, it is consistently \(<1\), which indicates that the motion of Ag\(^+\) ions is correlated. The cooperative ‘caterpillar’ mechanism, whereby Ag\(^+\) ions knock-on each other successively, either in a straight line or in zig-zag fashion40, has been previously used to justify the ionic conductivity measurements deviating from the non-interacting Einstein model. Such knock-on events can easily be observed in our drift trajectories, like the one shown in Fig. 2e.

Discussion
The ionic mobilities calculated for \(\alpha\)-AgI and \(\alpha\)-RbAg\(_4\)I\(_5\) by the NEMD method agree well with experimental values reported by both conductivity and Hall effect measurements. However, there are some considerations that we have to be aware of when comparing the simulations with experiments. Firstly, our simulations model the drift and can be directly compared to conduction experimental setup. In contrast, there is still no theoretical basis to assume that the Hall mobility is equal to the drift mobility in a solid, especially since ionic Hall effect has only been observed for superionic conductors where cooperative effects seem to play an important role46. Thus, we note the agreement with Hall effect measurements even though a formal justification for that is missing.
Additionally, since DC measurements are scarce, we compare our calculated mobilities with those obtained from AC (alternate current) measurements, assuming that the ionic conductivity is independent on the AC frequency near the frequency range used in the Hall and conductivity measurements, as it is believed to be the case below 10\(^6\) Hz47, justifying a direct comparison with our DC simulation.
We found a report of a DC measurement for \(\alpha\)-AgI48, where the conductivity at 450 K was found to be a few times lower than those reported by AC measurements. Addtionally, we note that Ref.48 uses a different definition of \(\mu\) per mobile carrier—the carriers in the ‘ion cloud’ first arriving to the end of the pellet. Since we averaged the velocity over all silver ions, for a direct comparison we have to renormalise the mobility in that reference to assume the participation of all silver ions, obtaining \(\mu =1.3\times 10^{-4}\) cm\(^2\)/V.s, considerably smaller than other reports. A clarification on whether this difference arises from the frequency dependence of the conductivity or other differences in the method (eg. the presence of memory effects) is needed.
Further, the ionic conductivity in general depends on crystallinity49. In the case of \(\alpha\)-RbAg\(_4\)I\(_5\), the conductivity of μm-sized polycrystalline samples has been measured to be 3.3 % higher than that of single-crystals50. For \(\alpha\)-AgI however, above the temperature of transition to \(\alpha\)-AgI, the conductivity converges to an unique value independently of the sample shape and size of the crystals51,52. This invariance, and the fact that the conductivity of \(\alpha\)-AgI has been consistently measured since the earliest experiments, makes \(\alpha\)-AgI an excellent benchmark system to test our simulation method.
Conclusions
The NEMD method employed here is able to quantify the ionic mobility in solids and therefore can be used to screen and evaluate potential electrolyte materials, using first-principles or classical molecular dynamics simulations. We now discuss some important considerations when chosing a method to compute the ionic mobility. Firstly, the advantages of the NEMD method we propose here are as follows:
-
Ion-ion correlations are fully taken into account, in contrast with calculations using the Nernst-Einstein relationship.
-
The NEMD method is easy to converge with a small ensemble and moderate simulation times (provided that the mobility is high enough), and it is numerically very robust with regards to the choice of integration timestep, the presence of anomalous starting points for the trajectory, etc. This makes it easier to use than linear response theory or Green-Kubo methods.
-
The NEMD simulations offer more insight into the physics of the response to the electric field, compared to statistical methods based on equilibrium molecular dynamics. For example, deviations from the linear response to the electric field, or interaction with the surface can be readily observed. Cooperative jumps can be observed and how their direction is determined by the electric field is apparent.
However, these are some of the limitations of the current method:
-
It is difficult to obtain the temperature dependence of the mobility, due to the heating of the slab as a result of the Joule effect (similar to experiments). Although the present calculations have not taken into account the changes of the lattice parameter with temperature, these can readily be introduced using either ab-initio or experimental parameters. Still, the calculated activation energy is close to experiment for \(\alpha\)-AgI, but comparatively smaller for \(\alpha\)-RbAg\(_4\)I\(_5\). It can be reasonably assumed that the temperature dependence would be more pronounced for electrolyte materials with higher activation energy, such as non Ag-based electrolytes2.
-
For poor ionic conductors, it is difficult to estimate the mobility due to the long simulation times involved (see RbAg\(_4\)I5 at 300 K as an example). Fortunately, the solid electrolytes of technological interest have at present mobilities at least two orders of magniture higher53. Still, poor ionic conductors can immediately be screened using this present method. For the actual evaluation of the mobility across different orders of magnitude it may be better to use a method based on the evaluation of the energy surface8.
-
For a more accurate calculation of the Heaven ratio, Green-Kubo or Einstein methods may be more appropriate, as the difference between the non-interacting and interacting ion diffusivities can be calculated directly.
Increasing the probability of cooperative jump mechanisms may be one of the ways to engineer solid electrolytes with improved conductivity. The NEMD method is a powerful ally in the design of such materials, and bears testimony to the predictive power of first-principles calculations.
Methods
Molecular dynamics simulations were carried out using the SIESTA code54. The forces were calculated using the local density approximation (LDA) of density functional theory55, and a Harris functional was used for the first step of the self-consistency cycle. The core electrons are represented by pseudopotentials of the Troullier-Martins scheme56. The basis sets for the Kohn-Sham states are linear combinations of numerical atomic orbitals, of the polarized double-zeta type57,58. The \(\Gamma\)-point is used for Brillouin zone sampling. The integration time step used is 1 fs.
The approach used to calculate the ionic conductivity can be summarised as follows (Fig. 5):
-
1.
The slab was constructed and randomly populated with Ag atoms according to the respective site occupations;
-
2.
The maximum electric field was estimated from the bandgap and slab length, \(eE_{\max } = E_{g}(L_z)/L_z\);
-
3.
The atoms at both surfaces of the slab were mechanically clamped to prevent drifting atoms from breaking free into the vacuum regions;
-
4.
The system was equilibrated to the target temperature, in the absence of an electric field, by using a Nosé thermostat35 (NVT). Different samples/starting points were generated for different equilibration times between 1 ps and 11 ps. The thermostat was turned off at the end of the equilibration (\(t=0\))
-
5.
At \(t=0\), the electric field was imposed and the system dynamic equations were integrated using a Verlet algorithm. Then, we selected the time interval for which the ion mobility is constant (linear regime of v(E))
-
6.
The ion mobility was analyzed as a function of the initial coordinate \(z_0\) and average \(\mu\) over the region that is not affected by the proximity to \(z_0=L_z\)
The total integration time for the production runs was at least 10 ps. The integration time should be long enough to observe ion migration, but short enough so that \(E\sim E_{\rm ext}\), where \(E_{\rm ext}\) is the external applied electric field (see Supplementary Information S1).

Schematic illustration of the calculation steps (clockwise): construction of the slab; estimation of the maximum electric field from the bandgap; clamping the extremities; equilibration to target temperature; Verlet integration under electric field, and selection of the time interval where \(\mu\) is constant; average \(\mu\) for the ions that did not start too close to \(z_0=L_z\).
The diffusion simulations(equilibrium molecular dynamics) were carried out using a Nosé thermostat35, with an equilibration time of at least 5 ps and a simulation time of at least 50 ps. The equilibration time varies between different temperatures and systems and was determined from the mean square displacement, using the Einstein relation.
Data availibility
The datasets generated during and/or analysed during the current study are available from the corresponding author [AC] upon request.
References
Li, M., Lu, J., Chen, Z. & Amine, K. 30 years of lithium-ion batteries. Adv. Mater. 30, 1800561 (2018).
Murugan, R. & Weppner, W. Solid Electrolytes for Advanced Applications (Springer, 2019).
Deng, D. Li-ion batteries: Basics, progress, and challenges. Energy Sci. Eng. 3, 385–418 (2015).
Varzi, A., Raccichini, R., Passerini, S. & Scrosati, B. Challenges and prospects of the role of solid electrolytes in the revitalization of lithium metal batteries. J. Mater. Chem. A 4, 17251–17259 (2016).
Fergus, J. W. Ceramic and polymeric solid electrolytes for lithium-ion batteries. J. Power Sources 195, 4554–4569 (2010).
Zheng, F., Kotobuki, M., Song, S., Lai, M. O. & Lu, L. Review on solid electrolytes for all-solid-state lithium-ion batteries. J. Power Sources 389, 198–213 (2018).
Wu, Z. et al. Utmost limits of various solid electrolytes in all-solid-state lithium batteries: A critical review. Renew. Sustain. Energy Rev. 109, 367–385 (2019).
Rodin, A., Noori, K., Carvalho, A. & Castro Neto, A. H. Microscopic theory of ionic motion in solids. Phys. Rev. B 105, 224310 (2022).
Geisel, T. The nature of the molten sublattice in superionic conductors. Solid State Ionics 3, 103–107 (1981).
Mellander, B. E. Electrical conductivity and activation volume of the solid electrolyte phase \(\alpha\)-AgI and the high-pressure phase fcc AgI. Phys. Rev. B 26, 5886–5896 (1982).
Hull, S., Keen, D., Sivia, D. & Berastegui, P. Crystal structures and ionic conductivities of ternary derivatives of the silver and copper monohalides: I. Superionic phases of stoichiometry \({{\rm MAg}}_4{{\rm I}}_5\): \({{\rm RbAg}}_4{{\rm I}}_5\), \({{\rm KAg}}_4{{\rm I}}_5\), and \({{\rm KCu}}_4{{\rm I}}_5\). J. Solid State Chem. 165, 363–371 (2002).
Nakamura, N. Nuclear quadrupole interaction and ionic dynamics in sodium superionic conductors. Zeitschrift für Naturforschung A 49, 337–344 (1994).
Futera, Z., Tse, J. S. & English, N. J. Possibility of realizing superionic ice vii in external electric fields of planetary bodies. Sci. Adv. 6, eaaz2915 (2020).
Rozhkov, A., Ignatov, S. & Suleimanov, E. Effect of an external electric field on the diffusion of oxygen ions in zirconium dioxide doped with yttrium oxide. Molecular dynamics study. Solid State Ionics 371, 115758 (2021).
Galvez-Aranda, D. E. & Seminario, J. M. Solid electrolyte interphase formation between the Li\(_{0.29}\)La\(_{0.57}\)TiO\(_3\) solid-state electrolyte and a Li-metal anode: An ab initio molecular dynamics study. RSC Adv. 10, 9000–9015 (2020).
Galvez-Aranda, D. E. & Seminario, J. M. Ab initio study of the interface of the solid-state electrolyte Li\(_9\)N\(_2\)Cl\(_3\) with a Li-metal electrode. J. Electrochem. Soc. 166, A2048 (2019).
Das, T., Merinov, B. V., Yang, M. Y. & Goddard, W. A. III. Structural, dynamic, and diffusion properties of a Li\(_6\)(PS\(_4\)) SCl superionic conductor from molecular dynamics simulations; prediction of a dramatically improved conductor. J. Mater. Chem. A 10, 16319–16327 (2022).
Zhu, Z., Deng, Z., Chu, I.-H., Radhakrishnan, B. & Ping Ong, S. Ab initio molecular dynamics studies of fast ion conductors. In Computational Materials System Design, 147–168 (Springer, 2018).
Pang, M.-C., Marinescu, M., Wang, H. & Offer, G. Mechanical behaviour of inorganic solid-state batteries: Can we model the ionic mobility in the electrolyte with Nernst-Einstein’s relation?. Phys. Chem. Chem. Phys. 23, 27159–27170 (2021).
Marcolongo, A. & Marzari, N. Ionic correlations and failure of Nernst-Einstein relation in solid-state electrolytes. Phys. Rev. Mater. 1, 025402 (2017).
France-Lanord, A. & Grossman, J. C. Correlations from ion pairing and the Nernst-Einstein equation. Phys. Rev. Lett. 122, 136001 (2019).
Kubo, R. Statistical-mechanical theory of irreversible processes. I. General theory and simple applications to magnetic and conduction problems. J. Phys. Soc. Jpn. 12, 570–586 (1957).
Van der Ven, A., Ceder, G., Asta, M. & Tepesch, P. First-principles theory of ionic diffusion with nondilute carriers. Phys. Rev. B 64, 184307 (2001).
Lawn, B. The thermal expansion of silver iodide and the cuprous halides. Acta Crystallogr. 17, 1341–1347 (1964).
Spencer, T., O’Dell, L., Moudrakovski, I. & Goward, G. Dynamics of Ag\(^+\) ions in RbAg\(_4\)I\(_5\) probed indirectly via \(^{87}\)Rb solid-state NMR. J. Phys. Chem. C 117, 9558–9565 (2013).
Burbano, J. C., Correa, H. & Lara, D. P. A new position in \(\alpha\)-\(\rm RbAg_4I_5\) at room temperature by molecular dynamics simulations. Mol. Simul. 46, 375–379 (2020).
Bradley, J. & Greene, P. Relationship of structure and ionic mobility in solid MAg\(_4\)I\(_5\). Trans. Faraday Soc. 63, 2516–2521 (1967).
Geller, S. Crystal structure of the solid electrolyte, RbAg\(_4\)I\(_5\). Science 157, 310–312 (1967).
Agrawal, R. C. DC polarisation: An experimental tool in the study of ionic conductors. Indian J. Pure Appl. Phys. 37, 294–301 (1999).
Meyer, B. & Vanderbilt, D. Ab-initio study of BaTiO\(_3\) and PbTiO\(_3\) surfaces in external electric fields. Phys. Rev. B 63, 205426 (2001).
Vivas, E. et al. Structural and transport properties for the superionic conductors AgI and RbAg\(_4\)I\(_5\) through molecular dynamic simulation. Ionics 27, 781–788 (2021).
Wood, B. C. & Marzari, N. Dynamical structure, bonding, and thermodynamics of the superionic sublattice in \(\alpha\)-AgI. Phys. Rev. Lett. 97, 166401 (2006).
Funke, K. AgI-type solid electrolytes. Prog. Solid State Chem. 11, 345–402 (1976).
Funke, K. & Hackenberg, R. Ionen-halleffekt in \(\alpha\)-AgJ. Berichte der Bunsengesellschaft für physikalische Chemie 76, 883–885 (1972).
Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 (1984).
Verlet, L. Computer, “experiments’’ on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 159, 98 (1967).
Liou, Y., Hudson, R., Wonnell, S. & Slifkin, L. Ionic Hall effect in crystals: Independent versus cooperative hopping in AgBr and \(\alpha\)-AgI. Phys. Rev. B 41, 10481 (1990).
Stuhrmann, C., Kreiterling, H. & Funke, K. Ionic hall effect measured in rubidium silver iodide. Solid State Ionics 154, 109–112 (2002).
Perrott, C. & Fletcher, N. H. Heat capacity of silver iodide. II. Theory. J. Chem. Phys. 48, 2681–2688 (1968).
Yokota, I. On the deviation from the Einstein relation observed for diffusion of Ag+ ions in \(\alpha\)-Ag2S and others. J. Phys. Soc. Jpn. 21, 420–423 (1966).
Kvist, A. & Tärneberg, R. Self-diffusion of silver ions in the cubic high temperature modification of silver iodide. Zeitschrift für Naturforschung A 25, 257–259 (1970).
Jordan, P. & Poclon, M. Détermination des coefficients de diffusion de l’argent et de l’iode dans les trois modifications de l’iodure d’argent par la méthode des échanges isotopiques hétérogènes. Helv. Phys. Acta 30, 33 (1957).
Arzigian, J. S. & Lazarus, D. Silver diffusion and isotope effect in silver rubidium iodide. Phys. Rev. B 23, 640 (1981).
Bentle, G. Silver diffusion in a high-conductivity solid electrolyte. J. Appl. Phys. 39, 4036–4038 (1968).
Okazaki, H. & Tachibana, F. A computer simulation of the Haven ratio in \(\alpha\)-AgI. Solid State Ionics 9, 1427–1432 (1983).
Slifkin, L. The ionic Hall effect in crystals. In Diffusion in Materials (eds Laskar, A. et al.) 1–17 (Kluwer Academic Publishers, 1990).
Funke, K., Banhatti, R., Brückner, S., Cramer, C. & Wilmer, D. Dynamics of mobile ions in crystals, glasses and melts, described by the concept of mismatch and relaxation. Solid State Ionics 154, 65–74 (2002).
Agrawal, R., Kathal, K. & Gupta, R. Estimation of energies of Ag\(^+\) ion formation and migration using transient ionic current (TIC) technique. Solid State Ionics 74, 137–140 (1994).
Monty, C. About ionic conductivity/diffusion relationships in yttria doped zirconia. Ionics 8, 461–469 (2002).
Raleigh, D. O. Ionic conductivity of single-crystal and polycrystalline RbAg\(_4\)I\(_5\). J. Appl. Phys. 41, 1876–1877 (1970).
Guo, Y.-G., Lee, J.-S. & Maier, J. Preparation and characterization of AgI nanoparticles with controlled size, morphology and crystal structure. Solid State Ionics 177, 2467–2471 (2006).
Guo, Y.-G., Lee, J.-S., Hu, Y.-S. & Maier, J. AgI nanoplates in unusual 7h/9r structures: Highly ionically conducting polytype heterostructures. J. Electrochem. Soc. 154, K51 (2007).
Suci, W. G. et al. Review of various sulfide electrolyte types for solid-state lithium-ion batteries. Open Eng. 12, 409–423 (2022).
Soler, J. M. et al. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 14, 2745–2779 (2002).
Ceperley, D. M. & Alder, B. J. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 45, 566–569 (1980).
Troullier, N. & Martins, J. L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 43, 1993–2006 (1991).
Sánchez-Portal, D., Ordejón, P., Artacho, E. & Soler, J. M. Density-functional method for very large systems with LCAO basis sets. Int. J. Quantum Chem. 65, 453–461 (1997).
Sánchez-Portal, D., Junquera, J., Paz, Ó. & Artacho, E. Numerical atomic orbitals for linear-scaling calculations. Phys. Rev. B 64, 235111 (2001).
Methfessel, M. XBS version Nov. http://www.ccl.net/chemistry/resources/software/X-WINDOW/xbs/index.shtml (1995).
Jmol version 14.6.4, http://jmol.sourceforge.net/.
Acknowledgements
This research/project is supported by the Ministry of Education, Singapore, under its Research Centre of Excellence award to the Institute for Functional Intelligent Materials (I-FIM, project No. EDUNC-33-18-279-V12). The computational work was supported by the Centre of Advanced 2D Materials, funded by the National Research Foundation, Prime Ministers Office, Singapore, under its Medium-Sized Centre Programme.
Author information
Authors and Affiliations
Contributions
A.C. and S.N. designed the method and performed the calculations. A.H.C.N. defined the problem statement and supervised the project. All authors contributed to the interpretation of results and writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Carvalho, A., Negi, S. & Neto, A.H.C. Direct calculation of the ionic mobility in superionic conductors. Sci Rep 12, 19930 (2022). https://doi.org/10.1038/s41598-022-21561-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-21561-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
