
Abstract
Sizzled (Szl) is both a secreted frizzled related protein (sFRP) and a naturally occurring inhibitor of the zinc metalloproteinase bone morphogenetic protein-1 (BMP-1), a key regulator of extracellular matrix assembly and growth factor activation. Here we present a new crystal structure for Szl which differs from that previously reported by a large scale (90°) hinge rotation between its cysteine-rich and netrin-like domains. We also present results of a molecular docking analysis showing interactions likely to be involved in the inhibition of BMP-1 activity by Szl. When compared with known structures of BMP-1 in complex with small molecule inhibitors, this reveals features that may be helpful in the design of new inhibitors to prevent the excessive accumulation of extracellular matrix that is the hallmark of fibrotic diseases.
Similar content being viewed by others


Introduction
Bone morphogenetic protein-1 (BMP-1, EC 3.4.24.19) is a secreted multidomain Zn-metallopeptidase of the astacin M12A subfamily which plays a key role in extracellular matrix (ECM) deposition, growth factor activation and the generation of animal form1,2. As a member of the BMP-1/tolloid-like family (BTPs), BMP-1 controls ECM deposition by site-specific proteolysis of procollagens, proteoglycans, prolysyl oxidases and other matrix proteins; BTPs also control growth factor activity, particularly members of the TGF-β superfamily, by proteolytic cleavage of precursor forms or growth factor antagonists1. One such antagonist is chordin, whose cleavage by BTPs activates growth factors BMP-2, -4 and -7, thereby controlling the chordin morphogen gradient that specifies dorso-ventral patterning during embryonic development2,3,4. In most vertebrates (with the exception of mammals) this gradient is finely tuned by the presence of the protein Sizzled (Szl) which is a potent and specific inhibitor of BTPs (Ki = 1.5 nM)5.
Both Szl and the related protein Crescent (which also inhibits BTPs6) are members of the family of secreted frizzled related proteins (sFRPs) which are known to compete for binding of Wnt proteins to Frizzled protein receptors7,8, albeit that whether Szl itself is a Wnt antagonist remains controversial9,10,11,12. All sFRPs consist of an N-terminal cysteine-rich domain (CRD, also known as Fz domain) followed by a netrin-like domain (NTR), where binding to Wnts involves mainly the CRD domain10. In mammals, there are five sFRPs (sFRP1-5) for which sequence comparison and phylogenetic analysis reveal two subgroups7, sFRP1/2/5 and sFRP3/4. These sFRPs are also found in fish, amphibians, reptiles and birds, along with Szl and Crescent, which show sequence similarities closest to sFRP1/2/5. Among sFRP1-5, only sFRP2 has been reported to affect BTPs, either by inhibiting13,14 or enhancing15,16 activity, with others finding no effect at all5,17.
In view of their role in the excess ECM deposition that is the hallmark of fibrotic diseases, BMP-1 and other BTPs are recognized as targets for antifibrotic therapies18. Several small molecule hydroxamate-based inhibitors of BMP-1 have been designed and tested for efficacy in animal models, none of which have so far been approved for clinical use19,20,21,22,23. Understanding the molecular mechanism by which Szl inhibits BTP activity would therefore have implications for the development of new therapeutic strategies.
A first crystal structure of Szl from Xenopus laevis (PDB ID 5XGP) was recently described9. Here we report a new crystal structure that differs from that of Bu et al. by a 90° rotation of the CRD domain with respect to the NTR domain. We also present results of a molecular docking analysis of the interaction between Szl and the catalytic domain of BMP-1 (PDB ID 3EDH)24, following earlier studies5, that yield new insights into the key interactions involved.
Methods
Expression vectors and cloning
Native full-length Szl with a C-terminal 6xHistidine tag5 was expressed in pHLsec25 by cloning between the AgeI and KpnI restriction sites. To produce Se-Met labeled Szl, two more constructs were prepared, using the same restriction sites, with pURD26 and modified pHLsec (from the Oxford Protein Production Facility, OPPF) both of which include a rho-1D4 tag for enrichment of low expressing proteins. Primers used for sequencing were as follows:
-
pHLsec-forward: 5′-GCTGGTTATTGTGCTGTCTCATC-3′
-
pHLsec-reverse: 5′-CACCAGCCACCACCTTCTGATAG-3′
-
pURD-forward: 5′-GCTGGTTGTTGTGCTGTCTCATC-3′
-
pURD-reverse: 5′-GGAAGCAATAGCATGATACA-3′
Protein expression and purification
Native Szl was produced by transient transfection of HEK 293T cells25. Cells (obtained from “Cellulonet—SFR Biosciences” Lyon, France) were first grown in DMEM medium supplemented with 10% FBS in 10-step cell stacks (Corning, USA) at 37 °C with 5% CO2. When cells were at 80% cell confluency, the Szl plasmid was introduced using the transfection agent polyethylenimine (PEI) with a DNA:PEI ratio of 1:3 (w/w). Conditioned media containing the secreted Szl protein was collected 3- and 5-days post transfection. Protease inhibitors NEM (2 mM) and Pefabloc (0.2 mM) were added to the harvested medium followed by centrifugation at 10,000g for 15 min at 4 °C. The clear supernatant was then dialyzed against 50 mM Na phosphate buffer pH 8.0, 0.3 M NaCl and mixed with Ni–NTA resin (pre-equilibrated in the same buffer). After 2 h of incubation at 4 °C on a rotating shaker, BioRad columns were manually loaded then washed and eluted with a ~ 200–250 mM linear imidazole gradient. Szl eluate was then dialyzed against 20 mM MES pH 6.0, 0.5 M NaCl (buffer optimised by thermal shift assay at the CTPF Platform, Paris-Saclay), concentrated using a Centricon 70 spin column (Millipore) then further purified on a Superdex 75 10/300 Increase column (GE Healthcare). Typical yields of purified Szl were 1.5 mg/L of culture medium. Purified protein was then checked by SDS-PAGE and MALS-SEC.
For Szl-SeMet, both the pURD-1D4 and pHLSec-1D4 vectors containing Szl inserts were used to generate stable and transiently transfected 293T cell lines using the robotic cell culture facility at STRUBI, Oxford, UK. Briefly, HEK cells (293 T) were grown in normal DMEM medium containing 10% FBS in roller bottles (Greiner Bio-One; 250 mL × 12). When 80% confluent, cells were transfected with pURD-Szl-1D4 and pHLSec-szl-1D4 plasmids using PEI as transfection reagent with a DNA:PEI ratio of 1:2 (0.5 mg DNA/roller bottle) then further grown in DMEM containing 2% FBS for 24–48 h in a rolling incubator. For Se-Met labelling, first the culture medium was removed then cells were gently washed with PBS. Freshly prepared labelling medium (250 mL methionine-free DMEM, 2% FBS, l-glutamine and non-essential amino acids) supplemented with 0.5 mL of l-Se-methionine (SeMet, 20 mg/mL stock, Eburon Organics) was then added to each roller bottle to give a final concentration of 40 mg/L SeMet. Cells were incubated for 4 days then conditioned media was collected. To the 6 L of the harvested medium, protease inhibitors NEM (2 mM) and PMSF (0.2 mM) were added followed by centrifugation at 3000 rpm, 4 °C for 30 min. After filtering through 0.22 µm Millipore Express PLUS disposable units, medium was then loaded on a column of activated 1D4-agarose resin (manually prepared by coupling Rho-1D4 antibody to CNBr-activated Sepharose 4B (GE 17-0430-01 2.5 mL) under gravity flow overnight at 4 °C, followed by washing with 20 mM MES pH 6.0, 0.5 M NaCl. Szl-SeMet was eluted using the peptide TETSQVAPA (Genscript), at 400 µg/mL. For elution, 3 mL of the peptide solution was added to the resin with bound Szl-SeMet then shaken at 4 °C for 2 h. Three fractions of 1.5 mL each were collected in 20 mM MES pH 6.0, 0.5 M NaCl, then Szl-SeMet eluates were pooled and loaded onto a Superdex 200 10/300 Increase column (GE Healthcare) pre-equilibrated and eluted using the same buffer. Szl-SeMet eluates from peak fractions were then pooled and concentrated using Vivaspin concentrators (Sartorius) for setting up crystallization screens. The final yield of Szl-SeMet was 450 µg from 12 L of conditioned medium.
Small angle X-ray scattering
SAXS data were collected at B21 at the Diamond Light Source UK using samples in 20 mM MES pH 6.0, 0.5 M NaCl in the concentration range 3.5–9.5 mg/mL Data were collected at 15 °C in the q range 0.015–0.4 Å−1 using a Pilatus 2 M detector (3 min/180 frames per sample). After buffer subtraction, concentration-normalised intensity data were extrapolated to zero concentration using PRIMUS27 then P(r) analysis was carried out using GNOM28. Ab initio models were generated from the experimental data using DAMMIF29 then averaged and filtered using the DAMAVER suite30 to generate the final DAMFILT model.
Crystallization of native Szl and Szl-SeMet
Prior to crystallization, proteins were concentrated to 12 mg/mL using Vivaspin concentrators. Initial crystals of native Szl were grown employing the sitting-drop vapour-diffusion method using 96-well plates (Greiner) at 20 °C. A Mosquito Nanolitre Robot (STP Labtech) was used to set up crystallization screens. Crystals of Szl were grown in 0.1 M NH4OAc and 18% PEG 3350 after incubation for 2 weeks. Diffraction quality crystals were then obtained in optimized conditions using 0.2 M NH4OAc, 18% PEG 3350 as precipitant. These latter were flash frozen in liquid nitrogen with 17% glycerol added to the crystallization buffer as cryo-protectant. Crystals of native Szl were reproduced in hanging drops and used for X-ray data collection.
For Szl-SeMet crystallization, screening was carried out as for Szl. Limited protein (Oxford Protein Production Facility) and PEGRx (Hampton Research, USA) screens were used for crystallization assays employing a Cartesian Technologies pipetting robot to set up 100 nL drops with a protein:precipitant ratio of 1:1. Crystals of Szl-SeMet appeared in 24 h from a crystallization buffer containing 0.1 M Bicine pH 8.5, 15% PEG 1500. Further optimizations were carried out using PEG 1500 concentrations of 17–11.3% in 0.1 M Bicine pH 8.5 in decreasing steps of 3% and with a varying protein to precipitant ratio (1:1/1:2/1:3). Large crystals of Szl-SeMet were obtained with 17% PEG 1500 which grew to full size within 3 days. The quality of these latter deteriorated within 5 days. Szl-SeMet crystals were flash-frozen in liquid nitrogen using reservoir solution supplemented with 20% glycerol as cryo-protectant.
Data collection, data reduction, phasing and structure determination
Diffraction data for native and Szl-SeMet, were collected at 100 K on beamlines I02 and I03 of the Diamond Light Source (Oxfordshire, UK). Data for Szl-SeMet crystals were collected by inverse beam SAD in two sweeps starting at 0–45° and 180° apart. Native Szl crystals diffracted X-rays to 1.9 Å while for Szl-SeMet the best crystal diffracted X-rays to 3.3 Å resolution. Data were processed using the Xia2 pipeline31 at Diamond combining XDS32 and POINTLESS from the CCP4 suite33. For phasing, four independent datasets from 4 different crystals were merged to enhance the anomalous signal. Heavy-atom coordinates of merged datasets for Szl-SeMet were located using Autosol from the PHENIX suite34 though with poor phasing power. Szl reflections were finally phased by combining a partial molecular replacement model obtained using the PDB coordinates of the cysteine rich domain (CRD) of mouse SFRP-3 (PDB ID 1IJX)35 and also the “search for model in map” module of the programme MOLREP36 employing the 1.95 Å native Szl dataset. Manual iterative rebuilding and fitting were done using COOT37 and model refinement was done with phenix.refine38 and Refmac 5.639 using a combination of TLS and restrained refinement. Model geometry and stereochemistry was examined using Molprobity40.
Molecular docking
Prior to docking, the protein preparation wizard PrepWizard (Schrödinger release 2018-3, Bioluminate, Schrödinger (suites2018-3/psp-v5.3), LLC, New York, NY, 2018) was used to optimize the crystal structures of Szl (determined in this study) and of the BMP-1 catalytic domain (PDB ID 3EDH)24. This included addition of hydrogen atoms and missing side-chains, filling missing loops, optimization of bond orders and hydrogen bonds and generation of ionized states. Water and other hetero-atoms were also removed. Restrained minimization of structures was carried out within an overall root mean square deviation (RMSD) of 0.3 from the initial energy minimized state. The PRIME structure prediction panel of the Schrödinger-Bioluminate suite was used for building the missing loop/linker (138DTLSKEYQYSYKE150) region connecting the CRD and NTR domains of Szl. Loops were further refined in PRIME to avoid any clashes. The crystal structure of Szl has two independent monomers (chains A and B) in the asymmetric unit, with the chain B structure being the most complete. Chain B was therefore further subjected to energy minimization and used for docking (with and without inter-domain linker loops) into the BMP-1 catalytic pocket.
Previously acquired data on key residues in Szl (Glu44, Asp92)5 and BMP-1 (Arg182)24 required for inhibition of BMP-1 activity were exploited to generate docking poses of the proteinase-inhibitor complex using the PIPER module of the Schrödinger-Bioluminate suite41, followed by refinement and energy minimization in PRIME. PIPER uses efficient Fast Fourier Transformation (FFT) in combination with pairwise interaction potentials using a docking code42 which greatly reduces the number of initial poses. PIPER also increases the number of near-native conformations in the initial selection of poses relative to other FFT-based docking programs. Alternatively, for docking analysis using more open structures of the BMP-1 catalytic domain in the region of the Cys65-Cys66 vicinal disulphide bond, previously generated from 3EDH by energy minimisation43, the ClusPro 2.0 server44 was used.
Structural analysis and figure rendering
Docked poses were submitted to the Protein Interactions Calculator (PIC) webserver (http://pic.mbu.iisc.ernet.in/) to map contacts at the protein complex interface45, as well as to the PDBePISA v1.52 server. Parameters such as numbers of hydrogen bonds, hydrophobic residues, aromatic and ionic interactions were considered to examine the strength of the docked poses. Detection of hinge regions was performed using the HingeProt server46. Prior to calculating SAXS profiles from the crystal structures using CRYSOL 3.047, the programmes Modeller48 and AllosMod-FoXS49 were used to complete missing regions including N- and C-termini and His-tags. Protein topology was determined using PDBsum50. All structure figures were generated using UCSF Chimera51 with the exception of Fig. 2, for which PyMol (Schrödinger, LLC) was used.
Accession code
Coordinates and structure factors have been deposited in the Protein Data Bank under accession code 7EL5.
Results
Preliminary analysis
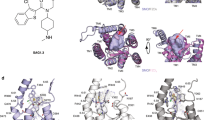
By SDS-PAGE, Szl showed a single band at ~ 30 kDa in non-reducing conditions. This was confirmed by both MALS-SEC and small angle X-ray scattering, the latter showing a linear Guinier plot indicative of a monodisperse population of molecules with radius of gyration (Rg) 24.8 Å, molecular mass 30.9 kDa and longest dimension 80 Å. Further analysis of the SAXS data revealed a bi-lobed structure consistent with the CRD and NTR domains (Fig. 1A).

Low resolution envelope of Szl determined by SAXS with superimposed crystal structures of 5XGP and the new structure reported here (PDB-ID 7EL5). (a) SAXS envelope only shown in side view (top), top view (middle) and side view rotated 90° about the vertical axis (bottom). (b) With superimposed full length ribbon diagram of the structure reported here (in green and including modelled inter-domain linker and N- and C-terminal ends). (c) As B but for 5XGP (in blue). (d) Both structures superimposed and aligned using their N-terminal CRD domains.
Crystal structure of Szl
Crystals from native and selenomethionine-labelled Szl belonged to space groups P212121 and P21 and diffracted X-rays to 1.95 and 3.32 Å, respectively (Table 1). The crystal structure was determined by combining SAD phasing using the Szl-SeMet data with a partial molecular replacement model from the high-resolution native data (1.95 Å) and refinement to the same resolution. Data collection, scaling and refinement statistics for native and Szl-SeMet are summarized in Table 1. Whereas Szl was found to be a monomer in solution by both MALS-SEC and SAXS, the crystal asymmetric unit contains two molecules A and B (Supplementary Fig. S1A).
The new crystal structure described herein is somewhat similar to that reported earlier9, henceforth referred to as 5XGP, and resembles the number 8 with the N-terminal CRD domain tethered via a polypeptide linker and an interdomain S–S bridge to the C-terminal NTR domain to form an elongated shape of length 70 Å and width 30 Å (Supplementary Fig. S1B). The total buried surface area is 628 Å2. The structural fold of Szl is stabilized by eight disulphide bonds (seven intra-domain formed by cysteines 27–90, 37–83, 74–109, 98–136, 102–126, 159–231, 176–281 and one inter-domain formed by cysteines 115–156). In the crystal structure, the dimer interface is stabilized by a salt bridge (not present in 5XGP) between E229 (chain A) and H116 (chain B) and by an extensive set of non-bonded van der Waals interactions involving mainly Q223, L226, I227, N228, R275. No electron density was observed for the linker (residues 138–150) connecting the CRD to the NTR domain, probably due to high flexibility. Also lacking was electron density corresponding to the side-chains of residues 188–200 and 191–195 in chains A and B, respectively.
Topological features of the Szl CRD (Fig. 2, Supplementary Figs. S1, S2) were found to be highly conserved compared to the 5XGP structure starting with two short β-strands and thereafter consisting mainly of α-helices (α1–α4). The globular fold of the CRD domain is stabilized by five disulphide bridges. In contrast, the NTR domain displays mixed β-α-β topology, consisting of seven β-strands (β3–β9), flanked by helices α5–α8. This domain is mainly stabilized by hydrophobic contacts as reported for 5XGP. The solvent accessible surface areas of the two monomers of Szl chains A and B were 12,765 and 12,588 Å2, respectively, perhaps due to slight differences in the orientation of each molecule. Structurally, molecules A and B aligned well with a RMSD of 1.16 Å except in the long loop at the C-terminal end of the CRD preceding the missing linker region.

Overall fold and topology diagram of the new crystal structure of Szl. This structure (PDB-ID 7EL5), shows α-helices in orange (CRD) and salmon (NTR), β-sheets in purple (CRD) and teal (NTR) and unstructured areas and loops in green. Also shown are positions of S2′, S1′, S1 and S2 subsites in Szl referred to in the text, as well as positions of corresponding residues in Szl and interaction partners in BMP-1 (the latter in the blue box). The positions of subsites are indicated by yellow stars on the ribbon presentation.
Despite the above similarities when comparing individual CRD and NTR domains, striking differences were observed when the overall structure of Szl described herein was compared to 5XGP (RMSD 1.578 Å). When the NTR domains of the overall structures were aligned, there was no alignment between the corresponding CRD domains, or vice versa. Instead, there was a rotation about the long axis of one domain with respect to the other of approximately 90°, pivoting around the inter-domain disulphide bridge (Fig. 3, Supplementary Fig. S3). This was confirmed using HingeProt46 which revealed hinge residues close to and on the N-terminal side of Cys156. Even though there was a difference in the relative orientations of the CRD and NTR domains between 5XGP and the structure reported here, theoretical SAXS profiles for both structures (calculated using CRYSOL) were indistinguishable and consistent with the experimental SAXS data (Supplementary Fig. S4). However, difference Fourier maps (Supplementary Fig. S5) confirmed the orientation of the CRD domain with respect to the NTR domain and vice-versa of Szl studied herein.

Overlay of the two crystal structures of Szl. (a) Side views of the new structure described here “7EL5” (green) and 5XGP (blue) aligned using their NTR domains and shown as ribbon presentations. (b) Top view showing the relative rotation of the CRD domains by about 90° as indicated by the dotted lines and marker residues shown in orange (new structure) and magenta (5XGP). Disulphide bridges are depicted in yellow with the inter-domain bond labelled “SS”.
BMP-1cat/Szl complex
Several attempts were made to co-crystallize the complex between Szl and the BMP-1 catalytic domain (BMP-1cat), all of which were unsuccessful. Therefore, we made a theoretical 3D structural model of the complex by protein–protein in silico docking using the Schrödinger suite. To do so, the first step was to build the 13-residue loop comprising the linker in Szl (138DTLSKEYQYSYKE150) connecting the CRD and NTR domains (Supplementary Fig. S1B). As similar docking models/poses were obtained for the BMP-1 catalytic site both with and without the Szl CRD-NTR linker, for further analysis only the docked poses of Szl with linker were used. Analysis of the multiple poses obtained revealed that the interface between BMP-1cat and Szl was relatively stable having a free energy (ΔG) value of − 22 kcal/mol, occupying a total interactive surface area of 3626 Å2, and including a total of 59 hydrogen-bonds and 12 salt-bridges (Supplementary Table S1). The dynamics and stability of the theoretical BMP-1cat/Szl complex was further analyzed in a 200 ns molecular dynamics simulation run (Supplementary Methods, Supplementary Fig. S6 and Supplementary Table S2), and displayed a calculated binding free energy of ~ 10 kcal/mol being in agreement with the aforementioned calculated free energy.
Inspection of the docked poses of the BMP-1cat/Szl model complex showed that a long loop containing Szl Asp92, previously found to be essential for inhibitory activity5,52,53, fits neatly into the BMP-1cat active site cleft close to the catalytic zinc ion and the cysteine-rich loop containing the vicinal disulphide bond (Cys65-Cys66) (Supplementary Figs. S7, S8). This agrees with the previously established specificity of the BMP-1 S1’ pocket for substrates with Asp at the P1’ position1,54. This interface is stabilized by an extensive network of direct hydrogen-bonded interactions between Szl Asp92 and BMP-1 His103 and Cys66, the latter being a part of the vicinal disulphide-bridge that forms a flap at the entrance to the deep “V” shaped catalytic pocket of BMP-1. In addition, Szl Asp92 forms hydrogen bonds with BMP-1 Tyr152 and Asn128 (Supplementary Fig. S7b, Table 2) as well as charged/ionic interactions with BMP-1 His103 and His 105 at the S1′ site and with the catalytic Zn2+ at the active site.
Apart from hydrogen-bonded and charged/ionic interactions involving catalytic site and S1′ residues, the docking also revealed significant interactions at the S2, S1, and S2′ subsites, all of which are distant from the catalytic site cleft and hence different from interaction sites normally expected for substrates (Fig. 2, Supplementary Fig. S7b and S8). The majority of these interactions involve charged/polar residues including Arg8(BMP-1)-Glu44(Szl), Glu7(BMP-1)-Ser43(Szl) at the S1 subsite near the hydrophobic pocket formed by the vicinal disulphide-bridge, while at the S2′ subsite BMP-1 Glu133 forms a salt bridge with Szl Lys26. Moreover, additional salt bridges/H-bonds at the S2 subsite involving Arg72(BMP-1) with Asp138(Szl), Arg61(BMP-1) with Glu133/Asp134(Szl) and Tyr60(BMP-1) with Glu133/Asp134 (Szl) further strengthen the positioning of Szl. The interface of the BMP-1cat-Szl theoretical complex is also stabilized by numerous face-to-face π–π interactions involving Tyr55(BMP-1)-Tyr144(Szl), Tyr127(BMP-1)-Phe94(Szl), Tyr68(BMP-1)-Tyr42(Szl) and hydrophobic contacts for Tyr68(BMP-1)-Val89(Szl), Val69(BMP-1)-Leu91(Szl), and Trp102(BMP-1)-Leu91(Szl) (Table 2, Supplementary Fig. S9). Tyr68 of BMP-1 further ensures tight binding of inhibitor residues through hydrophobic contacts at the hydrophobic surface below the S1 site (Table 2, Supplementary Fig. S7b), a feature also seen for the binding of recently reported reverse hydroxamate inhibitors to BMP-122.
Discussion
The ~ 90° rotation in the relative orientations of the Szl CRD and NTR domains, comparing the results reported here to those presented earlier9, was unexpected. This was particularly so in view of the inter-domain S–S bridge present in Szl, in addition to the interdomain linker, which together might be thought to constrain such movements. As described here however, there is clear evidence for hinge-bending about the inter-domain S–S bridge and linker region. To explore this further, we superimposed each crystal structure onto the sock-like SAXS low-resolution envelope. As shown in Fig. 1, both the 5XGP structure and the structure presented here fitted neatly within this shape, with the CRD domains fitting best in the thin “foot” region and the wider NTR domains in the cylindrical “leg” region. When viewed from the top (Fig. 1 middle), the relative rotations of the NTR domains are seen. Since hinge-like rotations between domains did not affect the SAXS scattering curves at the resolution of the data (~ 20 Å; Supplementary Fig. S4), all curves being additive for different shapes, it is tempting to speculate that the circular outline of the “leg” region (viewed from above) might reflect the range of conformations present in solution due to hinge movements. Such large conformational changes involving domain movements are tightly related to function in numerous proteins55,56,57,58. In this regard, the question of whether Szl plays a role in the inhibition of Wnt signalling remains controversial10,11. In the previously reported crystal structure of Szl9 (5XGP), it was suggested that the position of the NTR domain might prevent Wnt binding due to the tip of the extended hydrophobic palmitoleic acid (PAM) group colliding with charged residues His251 and Lys253 of the NTR domain (Supplementary Fig. S10). In contrast, with Szl 7EL5, because of the hinge rotation, the tip of the PAM group fits into the hydrophobic Leu225, Leu226 and Ile227 region of NTR, thus favouring PAM binding.
Szl residues Asp92, Phe94 and Glu44 were previously shown by site-directed mutagenesis to play key roles in the inhibition of BMP-1 activity5. The results presented here suggest that this list should be extended to Lys26, Glu133, Asp134 and Asp138 which are also important in anchoring Szl to BMP-1 as judged from the docking studies. Lys26 was previously suspected of being involved in binding to Szl, though its substitution by Ala was found to have little effect on inhibitory activity5. This underlines the prominent role played by cooperative interactions in stabilizing the complex. In addition, the flexible nature of particularly Asp92 (interaction with site S1′ in BMP-1) and E133 (interaction with site S2) in conjunction with residues interacting with sites S1 and S2′ located in secondary structure elements that are more or less superposable between the two Szl structures, points to a recognition/binding mode requiring alternating rigid anchoring points and flexible regions. Also, when superimposing the Danio rerio Sizzled 3D structure model as generated by Alpha-Fold259 (hereafter “AF2”, publicly available on www.uniprot.org), the rotation of the NTR domain relative to the CRD domain is similar to what we have observed for 7EL5 (Supplementary Fig. S3), with residues corresponding to Asp92 and Glu133 also showing a certain flexibility. We note that D. rerio Szl displays 52% strict sequence identity with X. laevis Szl and also inhibits BTPs60. While Szl binds to BMP-1 via a loop tipped by Asp92 that penetrates into the active site cleft (Supplementary Figs. S7 and S8), with its tip aligned in the direction of an authentic substrate61, Szl itself is not cleaved by BMP-1. This is despite the fact that Asp92 is by far the most commonly found P1′ residue bound to the S1′ site for all known BMP-1 substrates62. Interestingly, an Asp-tipped loop also plays a key role in the inhibition of another astacin family metalloproteinase, meprin β, by the endogenous protein fetuin-B63. Also, when superimposing the full-length three-dimensional structure of proBMP-1 as generated by Alpha-Fold259 onto the complex reported herein, it can be noted that whereas most of the BMP-1 propeptide is unstructured, the stretch containing the so-called “aspartate switch” is structured and buried in the BMP-1 catalytic domain (Supplementary Fig. S11). Intriguingly, this switch which is common to all astacin metalloproteinases co-localizes with the Asp-92 loop in Szl from our model complex, with the Asp residue at the tips in both model structures. Moreover, both loops are rich in hydrophobic residues including Phe52 from the proBMP-1 AF2 model which is located close to the critical Phe94 in Szl. Finally, we note that Asp92 in Szl is too far away (> 9 Å) to interact with Arg182 in BMP-1, formerly called Arg175 and thought to be key for binding to substrates with Asp in the P1′ position24. This may partially account for Szl not being cleaved by BMP-1, in addition to it binding to BMP-1 mostly by straddling the catalytic site cleft.
A comparison of the docked poses of the modelled BMP-1cat/Szl complex was also carried out with docking performed using Szl and an open conformation of BMP-1 generated by energy minimization43. Overall fitting and positioning of the Szl inhibitory loop near the BMP-1 catalytic site appeared to be preserved as reflected in the RMSD of 0.9 Å for aligned C-α’s. However, subtle changes/shifts were observed for residues involved in interactions close to the catalytic site (Supplementary Fig. S12a). A total shift of 2 Å was seen for the Asp92 side-chains of Szl which appears to be hydrogen-bonded to Tyr152 of BMP-1 close to the catalytic site (Supplementary Fig. S7b). This was not surprising in that Asp92 is located in a flexible loop (interacting with site S1′ in BMP-1) as observed when comparing the Szl structure determined here with that of 5XGP9 and in which a distance of 5.5 Å between the Cα atoms is found (Supplementary Fig. S3). Similar repositioning was seen in the cysteine rich loop containing the vicinal disulphide-bridge. To accommodate the interaction with Szl residues Lys26, Glu44 and Glu133, subtle repositioning of S2′, S1 and S2 BMP-1 subsite residues Glu133, Arg8 and Arg61, respectively, was observed indicating a flexible binding mode in the presence of the Szl inhibitor. Again, the flexible binding mode was in agreement with observations seen near the S2 site in BMP-1 when comparing our Szl structure (7EL5) with that of 5XGP; these revealed a shift of 9.2 Å between the Cα atoms of E133 in the respective structures (Supplementary Fig. S3). Concerning residues interacting with BMP-1 residues in sites S1 and S2′, as expected these are less flexible being located within more structured parts of Szl.
The catalytic subsites of BMP-1 and other Zn-dependent endopeptidases are now well established based on the crystal structures and modelling of several protease/peptide-inhibitor complexes22,64,65. Here we show that key residues in Szl occupy one of these subsites (S1′) as well as other subsites S2′, S1, S2 that span the catalytic site cleft (Fig. 1, Supplementary Figs. S7 and S8). In this regard, it has recently been shown22 that the binding to BMP-1 of small molecule hydroxamate (PDB IDs 6BTP, 6BTQ) and reverse hydroxamate (PDB IDs 6BSL, 6BSM) inhibitors can be associated with important movements in key residues of BMP-1, particularly its vicinal disulphide-bridge (Cys65–Cys66), as demonstrated most clearly with reverse hydroxamate compound 22 (PDB ID 6BSM, Supplementary Fig. S12b). These observations confirm the previously suspected flexibility of this region24,43. Regarding the role of the vicinal disulphide bridge in the BMP-1cat/Szl complex, Supplementary Fig. S12a shows that movements in this region similar to those observed with small molecule inhibitors may lead to changes in the relative orientations of BMP-1cat and Szl, particularly near subsite S2. Also, Szl and compound 22 (EVV) in the 6BSM crystal structure share common binding sites to BMP-1, such that conformational changes in BMP-1 residues Arg61 and Arg72 allow them to interact with both ligands (Supplementary Fig. S12b). Such conformational repositioning of subsite residues has previously been reported for other Zn-dependent endopeptidases66. This conformational flexibility may also contribute to the potencies of different BMP-1 inhibitors, as reflected in their Ki values of 6.8 pM for reverse hydroxamate compound 2222 and 1.5 nM for Szl5, both of which are lower than for hydroxamate-based inhibitors19.
In conclusion, this study reveals new structural insights into the flexibility between the two globular domains, CRD and NTR, of Sizzled. Despite its interdomain disulphide bridge, not present in other SFRPs except Crescent, Szl has remarkable freedom of movement about its CRD and NTR domains. Our studies have moreover shed light on the possible mechanism of inhibition of BMP-1 activity by the secreted frizzled related protein Sizzled. Interestingly, its likely mechanism of binding to BMP-1 has features in common with recently developed reverse hydroxamate inhibitors. These results may contribute to the development of future treatments to prevent the excess accumulation of extracellular matrix that is the hallmark of fibrotic diseases.
Data availability
Coordinates and structure factors have been deposited in the Protein Data Bank under accession code 7EL5. Other data are available from the corresponding author upon reasonable request.
References
Vadon-Le Goff, S., Hulmes, D. J. & Moali, C. BMP-1/tolloid-like proteinases synchronize matrix assembly with growth factor activation to promote morphogenesis and tissue remodeling. Matrix Biol. 44–46, 14–23 (2015).
De Robertis, E. M., Moriyama, Y. & Colozza, G. Generation of animal form by the Chordin/Tolloid/BMP gradient: 100 years after D’Arcy Thompson. Dev. Growth Differ. 59, 580–592 (2017).
Scott, I. C. et al. Homologues of twisted gastrulation are extracellular cofactors in antagonism of BMP signalling. Nature 410, 475–478 (2001).
Sun, M. et al. The expression of twisted gastrulation in postnatal mouse brain and functional implications. Neuroscience 169, 920–931 (2010).
Bijakowski, C. et al. Sizzled is unique among secreted frizzled-related proteins for its ability to specifically inhibit bone morphogenetic protein-1 (BMP-1)/tolloid-like proteinases. J. Biol. Chem. 287, 33581–33593 (2012).
Ploper, D., Lee, H. X. & De Robertis, E. M. Dorsal-ventral patterning: Crescent is a dorsally secreted Frizzled-related protein that competitively inhibits Tolloid proteases. Dev. Biol. 352, 317–328 (2011).
Bovolenta, P., Esteve, P., Ruiz, J. M., Cisneros, E. & Lopez-Rios, J. Beyond Wnt inhibition: New functions of secreted Frizzled-related proteins in development and disease. J. Cell Sci. 121, 737–746 (2008).
Mii, Y. & Taira, M. Secreted Wnt ‘inhibitors’ are not just inhibitors: Regulation of extracellular Wnt by secreted Frizzled-related proteins. Dev. Growth Differ. 53, 911–923 (2011).
Bu, Q. et al. The crystal structure of full-length Sizzled from Xenopus laevis yields insights into Wnt-antagonistic function of secreted Frizzled-related proteins. J. Biol. Chem. 292, 16055–16069 (2017).
Janda, C. Y., Waghray, D., Levin, A. M., Thomas, C. & Garcia, K. C. Structural basis of Wnt recognition by frizzled. Science 336, 59–64 (2012).
Kawano, Y. & Kypta, R. Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 116, 2627–2634 (2003).
Collavin, L. & Kirschner, M. W. The secreted Frizzled-related protein sizzled functions as a negative feedback regulator of extreme ventral mesoderm. Development 130, 805–816 (2003).
Lee, H. X., Ambrosio, A. L., Reversade, B. & De Robertis, E. M. Embryonic dorsal-ventral signaling: Secreted frizzled-related proteins as inhibitors of tolloid proteinases. Cell 124, 147–159 (2006).
He, W. et al. Exogenously administered secreted frizzled related protein 2 (Sfrp2) reduces fibrosis and improves cardiac function in a rat model of myocardial infarction. Proc. Natl. Acad. Sci. U.S.A. 107, 21110–21115 (2010).
Kobayashi, K. et al. Secreted Frizzled-related protein 2 is a procollagen C proteinase enhancer with a role in fibrosis associated with myocardial infarction. Nat. Cell Biol. 11, 46–55 (2009).
Zhu, Q. et al. Synergistic effect of PCPE1 and sFRP2 on the processing of procollagens via BMP1. FEBS Lett. 593, 119–127 (2019).
von Marschall, Z. & Fisher, L. W. Dentin sialophosphoprotein (DSPP) is cleaved into its two natural dentin matrix products by three isoforms of bone morphogenetic protein-1 (BMP1). Matrix Biol. 29, 295–303 (2010).
Turtle, E. D. & Ho, W. B. Inhibition of procollagen C-proteinase: Fibrosis and beyond. Expert Opin. Ther. Pat. 14, 1185–1197 (2004).
Fish, P. V. et al. Potent and selective nonpeptidic inhibitors of procollagen C-proteinase. J. Med. Chem. 50, 3442–3456 (2007).
Bailey, S. et al. Succinyl hydroxamates as potent and selective non-peptidic inhibitors of procollagen C-proteinase: Design, synthesis, and evaluation as topically applied, dermal anti-scarring agents. Bioorg. Med. Chem. Lett. 18, 6562–6567 (2008).
Turtle, E. et al. Design and synthesis of procollagen C-proteinase inhibitors. Bioorg. Med. Chem. Lett. 22, 7397–7401 (2012).
Kallander, L. S. et al. Reverse hydroxamate inhibitors of bone morphogenetic protein 1. ACS Med. Chem. Lett. 9, 736–740 (2018).
Talantikite, M. et al. Inhibitors of BMP-1/tolloid-like proteinases: Efficacy, selectivity and cellular toxicity. FEBS Open Bio 8, 2011–2021 (2018).
MacSweeney, A. et al. Structural basis for the substrate specificity of bone morphogenetic protein 1/tolloid-like metalloproteases. J. Mol. Biol. 384, 228–239 (2008).
Aricescu, A. R., Lu, W. & Jones, E. Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. Sect. D. Biol. Crystallogr. 62, 1243–1250 (2006).
Zhao, Y., Malinauskas, T., Harlos, K. & Jones, E. Y. Structural insights into the inhibition of Wnt signaling by cancer antigen 5T4/Wnt-activated inhibitory factor 1. Structure 22, 612–620 (2014).
Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J. & Svergun, D. I. PRIMUS—A Windows-PC based system for small-angle scattering data analysis. J. Appl. Cryst. 36, 1277–1282 (2003).
Svergun, D. I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 25, 495–503 (1992).
Franke, D. & Svergun, D. I. DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Cryst. 42, 342–346 (2009).
Volkov, V. V. & Svergun, D. I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Cryst. 36, 860–864 (2003).
Winter, G., Lobley, C. M. C. & Prince, S. M. Decision making in xia2. Acta Crystallogr. Sect. D Biol. Crystallogr. 69, 1260–1273 (2013).
Kabsch, W. X. D. S. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 125–132 (2010).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 235–242 (2011).
Adams, P. D. et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 213–221 (2010).
Dann, C. E. et al. Insights into Wnt binding and signalling from the structures of two Frizzled cysteine-rich domains. Nature 412, 86–90 (2001).
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 22–25 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D. Biol. Crystallogr. 60, 2126–2132 (2004).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 68, 352–367 (2012).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 355–367 (2011).
Chen, V. B. et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 12–21 (2010).
Kozakov, D., Brenke, R., Comeau, S. R. & Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Proteins Struct. Funct. Genet. 65, 392–406 (2006).
Vajda, S. et al. New additions to the ClusPro server motivated by CAPRI. Proteins Struct. Funct. Bioinform. 85, 435–444 (2017).
Pulido, D. et al. Structural basis for the acceleration of procollagen processing by procollagen C-proteinase enhancer-1. Structure 26, 1384–1392 (2018).
Kozakov, D. et al. The ClusPro web server for protein–protein docking. Nat. Protoc. 12, 255–278 (2017).
Tina, K. G., Bhadra, R. & Srinivasan, N. P. I. C. Protein interactions calculator. Nucleic Acids Res. 35, W473–W476 (2007).
Emekli, U., Schneidman-Duhovny, D., Wolfson, H. J., Nussinov, R. & Haliloglu, T. HingeProt: Automated prediction of hinges in protein structures. Proteins Struct. Funct. Genet. 70, 1219–1227 (2008).
Franke, D. et al. ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 50, 1212–1225 (2017).
Sali, A. & Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993).
Guttman, M., Weinkam, P., Sali, A. & Lee, K. K. All-atom ensemble modeling to analyze small-angle X-ray scattering of glycosylated proteins. Structure 21, 321–331 (2013).
Laskowski, R. A., Jabłońska, J., Pravda, L., Vařeková, R. S. & Thornton, J. M. PDBsum: Structural summaries of PDB entries. Protein Sci. 27, 129–134 (2018).
Pettersen, E. F. et al. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Yabe, T. et al. Ogon/Secreted Frizzled functions as a negative feedback regulator of Bmp signaling. Development 130, 2705–2716 (2003).
Martyn, U. & Schulte-Merker, S. The ventralized ogon mutant phenotype is caused by a mutation in the zebrafish homologue of Sizzled, a secreted Frizzled-related protein. Dev. Biol. 260, 58–67 (2003).
Schechter, I. & Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27, 157–162 (1967).
Bernstein, B. E., Michels, P. A. M. & Hol, W. G. J. Synergistic effects of substrate-induced conformational changes in phosphoglycerate kinase activation. Nature 385, 275–278 (1997).
Pijning, T. & Vujičic̈-Žagar, A., Kralj, S., Dijkhuizen, L. & Dijkstra, B. W.,. Flexibility of truncated and full-length glucansucrase GTF180 enzymes from Lactobacillus reuteri 180. FEBS J. 281, 2159–2171 (2014).
Ballut, L. et al. Active site coupling in Plasmodium falciparum GMP synthetase is triggered by domain rotation. Nat. Commun. 6, 8930 (2015).
Orellana, L. Large-scale conformational changes and protein function: Breaking the in silico barrier. Front. Mol. Biosci. 6, 117 (2019).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Muraoka, M. O. et al. Sizzled controls dorso-ventral polarity by repressing cleavage of the Chordin protein. Nat. Cell Biol. 8, 329–338 (2006).
Guevara, T. et al. Proenzyme structure and activation of astacin metallopeptidase. J. Biol. Chem. 285, 13958–13965 (2010).
Rawlings, N. D. et al. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46, D624–D632 (2018).
Eckhard, U. et al. The crystal structure of a 250-kDa heterotetrameric particle explains inhibition of sheddase meprin β by endogenous fetuin-B. Proc. Natl. Acad. Sci. U.S.A. 118, e2023839118 (2021).
Dive, V., Chang, C. F., Yiotakis, A. & Sturrock, E. D. Inhibition of zinc metallopeptidases in cardiovascular disease—from unity to trinity, or duality?. Curr. Pharm. Des. 15, 3606–3621 (2009).
Garrigue-Antar, L., Barker, C. & Kadler, K. E. Identification of amino acid residues in bone morphogenetic protein-1 important for procollagen C-proteinase activity. J. Biol. Chem. 276, 26237–26242 (2001).
Schiering, N. et al. Structure of neprilysin in complex with the active metabolite of sacubitril. Sci. Rep. 6, 1–5 (2016).
Acknowledgements
This research was funded by the French National Research Agency (project ‘TOLLREG’ awarded to CM and NA), FP7 PCUBE (ID: 227764), FP7 BioStruct-X FP7 (ID: 283570), CNRS and the Université Claude Bernard Lyon 1. US is currently supported by a Ramalingaswami Re-entry fellowship, Department of Biotechnology, Government of India. The Wellcome Centre for Human Genetics is supported by the Wellcome Trust (Grant 090532/Z/09/Z), Cancer Research UK (Grant C375/A17721) and the UK Medical Research Council (Grant MR/M000141/1). Technical support from staff on beamlines MX and FIP (European Synchrotron Radiation Facility), PX3 (Swiss Light Source, Switzerland), Proxima 1 (Soleil, France) and B21, I02 and I03 (Diamond Light Source, UK; proposal mx8423) is gratefully acknowledged. We also acknowledge assistance from the Protein Science Facility, SFR Biosciences Lyon (UAR3444/CNRS, US8/INSERM, Ecole Normale Supérieure de Lyon). Finally, we are grateful to Katalin Phimister of the Schrödinger technical support team for help with refinement of the docked poses.
Author information
Authors and Affiliations
Contributions
U.S., N.M., J.M.B. and C.B. carried out the production, purification and crystallization of native Szl. U.S., D.J.S.H. and Y.Z. prepared, expressed and purified SeMet-Szl. U.S., K.H. and N.A. collected and processed the X-ray data and determined the structures. U.S. carried out docking analysis of the BMP-1cat/Szl complex. U.S., S.V.L.G., D.J.S.H. and N.A. analysed the data. S.V.L.G., C.M., D.J.S.H. and N.A. supervised the study. U.S., D.J.S.H. and N.A. drafted the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sharma, U., Vadon-Le Goff, S., Harlos, K. et al. Dynamics of the secreted frizzled related protein Sizzled and potential implications for binding to bone morphogenetic protein-1 (BMP-1). Sci Rep 12, 14850 (2022). https://doi.org/10.1038/s41598-022-18795-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-18795-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
