
Abstract
Ladle furnace slag (LFS) can undergo hydration and carbonation reactions as cement. This article explores the effect of LFS hydration and carbonation reactions on cementitious substances at different temperatures and different LFS particle sizes, determining the effect of these varying conditions on the microstructure and formation mechanism of cementitious substances. The results show that in the early stages, C2S and C3S undergo hydration to generate C–S–H gel, which then undergoes decalcification and condensation to generate CaCO3 and Ca-deficient C–S–H gel; the hydration reaction and carbonation reaction promote and influence each other. The increase in temperature was found to hinder the formation of CaCO3 from Ca2+ and CO32−, thus reducing the efficiency of hydration carbonation. The increase in particle size was not conducive to the leaching of C2S and C3S to the surface of the reaction phase, which in turn reduced the degree of decalcification and polymerization of the C–S–H gel in the carbonation phase. It was concluded that the optimum LFS hydration and carbonation reactions were achieved at 20 °C and with a LFS particle sizes < 38 μm.
Similar content being viewed by others
Introduction
Ladle furnace slag (LFS) is an alkaline solid waste material with a high calcium content, that is produced as a by-product of secondary refining processes in the iron and steel industry1. The mineral phase components of LFS mainly contain calcium metasilicate (CaSiO3, CS), dicalcium silicate (Ca2SiO4, C2S), tricalcium silicate (Ca3SiO5, C3S) and free calcium oxide (f-CaO)2. LFS exhibits poor stability and poor compactness due to its high content of f-CaO. In addition, it exerts a filler effect that can increase the density of cementitious substances, as its mineral phase composition is similar to that of Ordinary Portland cement (OPC) and it contains the same oxides that are present in OPC, while also having the advantages of a high affinity to cement-based materials and highly abundant availability3. The addition of LFS to cement-based materials as a mineral additive can accelerate the hydration reaction and increase the formation of hydration products such as calcium silicate hydrate (C–S–H) gel4. As the most important hydration product of cement slurries, C–S–H gel is an important source of mechanical properties in cementitious materials and considerably influences the carbonation of LFS and the preparation of cement5.
The calcium-based substances in LFS exhibit good carbonation performance with a theoretical carbonation efficiency of 30–60%6. The poor stability and poor compactness of LFS can be mitigated by absorbing CO2 to produce stable calcium carbonate (CaCO3). Xu7 used CO2 to immobilize heavy metals in LFS and enhance its strength. Monkman8 established that the extractable CaO content of carbonated LFS was reduced by about 95% compared with uncarbonated LFS, while its strength is greatly improved when formed into mortar. Therefore, carbonation can not only effectively achieve CO2 storage but also improve the compressive strength of the final product. In the early stage of carbonation, the cementitious substances in LFS (C2S and C3S) undergo hydration reactions to form C–S–H gel. When comparing the changes of C–S–H gel during natural carbonation or accelerated carbonation, Auroy9 concluded that CaCO3 was more conducive to forming crystalline spherical aragonite with a relatively stable structure after accelerated carbonation. Therefore, hydration and carbonation reactions are particularly important in the process of LFS resource utilization.
Huijgen10 analyzed the controlling factors that affect the carbonation reaction, including particle size, temperature, solid–liquid ratio, stirring speed, and aeration rate and demonstrated that temperature and particle size are the two most important factors. Ashraf11 compared the carbonation process of CS, C2S, and C3S at different temperatures, reporting that the reactivity of CS samples was lower than that of C2S and C3S samples. Previous studies12,13 have reported that reducing the average particle size of slag can shorten the leaching channels from the metal ion core to the inner surface, thus promoting the utilization of slag and interactions between the slag-water–gas system. Suh14 reported that heating an anhydrous calcium silicate clinker to 200 °C resulted in a gradual increase in Q1 structure (SiO4 tetrahedra connected to a SiO4 tetrahedra) and Q2 structure (SiO4 tetrahedra connected to two SiO4 tetrahedra) peak intensities, according to deconvolution of the recorded 29Si solid-state nuclear magnetic resonance (NMR) spectra. Suh’s results show that the hydration reaction of anhydrous calcium silicate can be accelerated by increasing the reaction temperature within a certain range. The aforementioned studies have generally assessed the effects of hydration and carbonation reactions in isolation on mineral phase composition, with only a few studies having combined both reactions to analyze the change mechanism of the cementitious substances under varying temperature and particle size conditions.
In this study, changes in the hydration reaction of C2S and C3S in LFS and the carbonation reaction of C–S–H gel when applying different temperatures and LFS particle sizes, were explored using solid-state 29Si NMR technology. The samples were characterized by scanning electron microscopy (SEM), thermogravimetry (TG), differential thermogravimetry (DTG), X-ray diffraction (XRD) and Brunner–Emmet–Teller (BET) analysis of nitrogen adsorption–desorption isotherms. Peak Fit software was used to analyze the changes in the cementitious substances during the hydration and carbonation stages and revealed their mechanism of microstructure formation. The present study aimed to understand the effect of different temperatures and particle sizes on cementitious substances subjected to hydration and carbonation reactions, providing theoretical guidance for the application of LFS and LFS-based cement materials.
Materials and methods
Experimental materials
The LFS used in this study was taken from a steel plant in Xinjiang (China), with the chemical composition and morphology of the material shown in Table 1 and Fig. 1, respectively. XRD analysis showed that the main mineral phase components of LFS include CS, C2S, C3S and CaAl2Si2O8. The solid LFS material was pulverized using a jaw crusher (BB200, Retsch, Germany) and then ground with a vibrating disc grinder (RS200, Retsch, Germany). Varying particle sizes were obtained by passing the ground sample through 180 μm, 96 μm, 48 μm, and 38 μm pore size sieves before experiments.

Micrograph of the original LFS material.
Experimental process
A three-phase reaction tank was employed to investigate the influence of particle size and temperature on the carbonation of LFS. Slurries of LFS fractions containing each respective particle size range, were prepared with a solid–liquid ratio of 1:5, for use in experiments at a CO2 ventilation rate of 500 mL/min, with continual stirring at 700 rad/min. Hydration and carbonation reactions were considered to be complete when the mass remained constant. To determine the influence of particle size, slurries with LFS particles sizes of 96–180 μm, 48–96 μm, 38–48 μm and < 38 μm were used at a reaction temperature of 20 ℃. To analyze the influence of temperature, samples with a particle size < 38 μm were reacted at 20 ℃, 40 ℃, 60 °C and 80 °C. At the end of the experiment, the solids in the slurry were separated from the liquid and dried at 105 °C for 8 h to obtain carbonation products for analysis.
Characterization methods
X-ray diffraction analysis was performed to identify carbonation products using an X-ray powder diffractometer (D8 Advance, Bruker, Germany). For all materials, two samples were analyzed under the following experimental conditions: Cu Kα radiation (λ = 0.154 nm), scanning range of 10°–70° and a scanning rate of 2.0°/min. The mineral phase composition was determined via thermal stability analysis (TG-DTG) using a simultaneous thermogravimetric analyzer (STA 7300, Hitachi, Japan) with 10 mg of sample analyzed at a temperature range from ambient temperature to 900 °C at a heating rate of 10 °C/min in a single measurement, as variations in outcome were expected to be small15. A scanning electron microscope (LEO 1430VP, Carl Zeiss, Germany) was used at an accelerating voltage of 15 kV to analyze the micro-morphology of samples, which were sprayed with gold before the test to stabilize images. Analysis of the microstructure changes during C2S and C3S carbonation was carried out via 29Si NMR analysis (600 M spectrometer, Agilent, US), with a 4-mm ZrO2 rotor at a rotation speed of 8 kHz and TMS solution as a 29Si chemical shift calibration reference.
Results and discussion
XRD analysis of the hydration and carbonation reaction
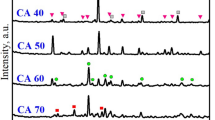
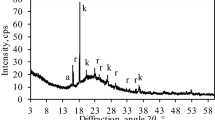
Figure 2a shows the XRD patterns of LFS samples (< 38 μm) hydrated and carbonated at different temperatures, while Fig. 2b shows the XRD patterns of LFS samples of different particle sizes after reaction at 20 ℃. These results confirm that the LFS solid waste material is composed mainly of silicate minerals, such as CS, C2S, and C3S, with additional amounts of calcium hydroxide Ca(OH)2 and anorthite (CaAl2Si2O8). After the hydration and carbonation reaction processes, the diffraction peaks of the calcium silicate phases decreased or disappeared. Simultaneously, three diffraction peaks appeared at around 2θ = 28.5°, 31.2°, and 61.2°, which were assigned to C–S–H gel16,17,18,19, indicating that the calcium silicate phases formed C–S–H gel during the hydration and carbonation reaction, and LFS has potential pozzolanic activity, Si and Al will also react with Ca(OH)2 to form C–S–H gel. As C2S and C3S are known to undergo hydration reactions20, C2S and C3S present on the surface of LFS particles can undergo hydration to form C–S–H gel in the early stage of the reaction. As the reaction progresses, the C–S–H gel releases Ca2+ to maintain the required alkaline environment21, resulting in decalcification of the C–S–H gel. CO2 gas was continuously passed through the solution to generate H2CO3 of weak acidity, which is extremely unstable and rapidly ionize in solution to generate HCO3− and CO32−. The free Ca2+ in the solution combines with CO32− to form stable CaCO3. C–S–H gel will eventually provide more Ca2+ than the present Ca(OH)2 when cement paste is exposed to high concentrations of CO222, Finally, Ca2+ in CaCO3 mainly originates from the C–S–H gel hydration product and since the presence of moisture is a necessary condition to initiate the carbonation reaction23,24, hydration will occur prior to the carbonation reaction. The XRD patterns in Fig. 2 indicate the formation of large amounts of CaCO3 in the reaction at different temperatures and different LFS particle sizes, indicating that the main product after carbonation was CaCO3. As CS cannot undergo hydration11,25,26, the CS present on the surface of LFS particles does not form C–S–H gel in the early stage of the reaction, but instead directly combines with CO32− in solution to form CaCO3. In addition, the diffraction peaks for CaAl2Si2O8 did not change significantly following the hydration and carbonation reactions, indicating that the anorthite structure was relatively stable and did not participate in the reaction process, which is consistent with the results reported by Ashraf27. With the increase in reaction temperature, the CaCO3 diffraction peaks gradually decreased, which may be due to the faster movement of CO2 molecules and expansion of the materials volume, resulting in an increase in the distance between the molecules of CO2 and H2O, weakening the intermolecular forces and hindering the generation of CO32-28. In addition, the increase in temperature also caused the solubility of CO2 to gradually decrease29, ultimately leading to a reduction in carbonation efficiency. With the decrease in particle size, the diffraction peaks of the calcium silicate phases and Ca(OH)2 gradually decreased, which might be explained by the simultaneous increase in surface area enhancing the contact area between particles and the solution, increasing the rate of reaction occurrence. However, a considerable amount of calcium silicates and Ca(OH)2 did not participate in the hydration and carbonation reactions in the largest LFS particle size, resulting in an incomplete reaction.

XRD patterns of original LFS and LFS samples after hydration and carbonation: (a) at different reaction temperatures, (b) at different particle sizes.
TG-DTG analysis of the hydration and carbonation reaction
Figure 3a shows the TG-DTG diagrams of LFS samples (< 38 μm) after hydration and carbonation at different temperatures, while Fig. 3b shows the TG-DTG diagrams of LFS samples of different particle sizes after hydration and carbonation reactions were performed at 20 ℃. As shown in Fig. 3, the mass losses were divided into three distinct temperature ranges: (1) 100–300 °C, which was attributed to dehydration of the C–S–H gel and the Ca-deficient C–S–H gel30; (2) 300–500 °C, which was attributed to the dehydration of Ca(OH)231; (3) 500–850 °C, which was attributed to the decarbonization of CaCO332. In terms of dry weight, the mass loss over the entire temperature range was referred to as the total mass loss. The mass of CaCO3 can be obtained from the CO2 content generated by CaCO3 decarbonization33. The weight-loss peak appearing in the range of 300–500 °C before the hydration and carbonation reactions may be caused by the formation of Ca(OH)2 due to the combination of calcium-based components of LFS with atmospheric water under natural conditions. Two weight-loss peaks appeared in the range of 100–300 °C and 500–850 °C after the hydration and carbonation reactions, indicating that the calcium based components of LFS underwent hydration and carbonation reactions successively, resulting in C–S–H gel, Ca-deficient C–S–H and CaCO3 being generated.

TG-DTG diagrams of the original LFS material and LFS samples after hydration and carbonation: (a) at different temperatures, (b) at different particle sizes.
Table 2 lists the CaCO3 and total mass losses measured by TG analysis at different reaction temperatures or with different LFS particle sizes, showing that the mass of CaCO3 decreased with increasing temperature or LFS particle size. The content of CaCO3, C2S, and C3S was calculated using the reference intensity ratio (RIR) method. Results show that, as the temperature decreased, the C2S and C3S content reduced, indicating that increasing amounts of C2S and C3S participated in the hydration reaction, while the C–S–H gel continuously participated in the carbonation reaction, further promoting the hydration reaction. Therefore, a reaction temperature of 20 °C was more advantageous to the occurrence of the carbonation reaction. With the increase in particle size, the content of C2S and C3S increased, possibly because C2S and C3S in LFS require longer leaching channels to participate in the hydration reaction. Furthermore, this also negatively affected the later carbonation reaction. Therefore, larger particle sizes were not conducive to the subsequent occurrence of the carbonation reaction.
SEM–EDS analysis of the hydration and carbonation reaction
Figure 4 shows the SEM images of LFS after hydration and carbonation at different temperatures and when using samples of different particle sizes. Fig. 5 shows a schematic diagram of the microstructure mechanism of hydration and carbonation of a slag particle and Table 3 lists the average elemental content of these areas according to energy-dispersive X-ray spectroscopy (EDS). As shown in Fig. 4, after hydration and carbonation, a large amount of C–S–H gel was formed, exhibiting rough surfaces and a coagulated or fibrous form (indicated by the red area in Fig. 4), as well as CaCO3 crystals (such as calcite) with a cubic structure (indicated by the blue area in Fig. 4)34,35. As the reaction temperature increased, more voids became visible in the structure. As the LFS particle size increased, the amount of calcite gradually decreased and the LFS surface became covered by the C–S–H gel and Ca-deficient C–S–H gel (indicated by the green area in Fig. 4). When the LFS particle size was 96–180 μm, the hydration product Ca(OH)2 was present in the form of hexagonal flakes (indicated by the yellow area in Fig. 4). These results could be attributed to the cementitious properties of C2S and C3S, which cause the hydration reaction to form C–S–H gel and Ca(OH)2. During carbonation, the C–S–H gel is decalcified to form Ca-deficient C–S–H gel and CaCO3. Furthermore, Ca(OH)2 directly reacts with CO32− in solution to form CaCO3. As illustrated in Fig. 5, the carbonation products CaCO3 and Ca-deficient C–S–H gel adhere to the surface of the reaction phase and the entire structure becomes dense, inhibiting the diffusion of Ca2+ into solution, which eventually prevents further progression of the reactions. According to the results shown in Table 3, the reaction products were mainly composed of C, O, Al, Si and Ca. The content of Ca gradually increased with increases in temperature or particle size, indicating that the decalcification degree of the C–S–H gel gradually decreased and that the concentration of Ca2+ ions leached into the solution decreased, further reducing the formation of CaCO3. Therefore, increased temperature or particle size was detrimental to the hydration and carbonation reactions.

SEM images of LFS materials following hydration and carbonation under varying conditions: reaction temperatures of: (a) 20 ℃, (b) 40 ℃, (c) 60 ℃, (d) 80 ℃; LFS particle sizes of: (e) < 38 μm, (f) 38–48 μm, (g) 48–96 μm, (h) 96–180 μm.

Schematic diagram of the microstructure mechanism of hydration and carbonation of LFS.
Microstructure of the cementitious substances in the hydration and carbonation reaction
Table 4 shows the 29Si NMR chemical shift range of Qn structural units in silicates36, while Fig. 6 shows the 29Si NMR spectra following hydration and carbonation of LFS at different temperatures or when using different LFS particle sizes. In NMR analysis, Q0 represents isolated SiO4 tetrahedra, Q1 often appears at the end group of a straight chain, Q2 mostly appears as a middle group of a straight chain, Q3 has a double-stranded polymer structure or a layered structure and Q4 represents SiO4 tetrahedra connected to four SiO4 tetrahedra, in a three-dimensional network structure37. As shown in Fig. 6a, the main peak of uncarbonated LFS was Q0, with a few Q1 peaks appearing around -79 ppm, which may be due to the formation of the C–S–H gel by C2S and C3S hydration under natural conditions. As shown in Fig. 6b–i, after the hydration and carbonation reactions, Q2, Q3, and Q4 peaks were present.

29Si NMR spectra of original LFS: (a), and LFS samples after hydration and carbonation: (b) at 20 ℃, (c) at 40 ℃, (d) at 60 ℃, (e) at 80 ℃, (f) at < 38 μm, (g) at 38–48 μm, (h) at 48–96 μm, (i) at 96–180 μm.
Table 5 lists the calculated deconvolution results from 29Si NMR analysis. No Q3 and Q4 peaks were found to be present before the hydration and carbonation reactions, indicating that it is more difficult to complete C2S and C3S hydration and carbonation reactions under natural conditions. After the hydration and carbonation reactions, the content of Q0 gradually decreased or even disappeared, while the Q2 peak appeared, indicating that at this point C2S and C3S began to participate in the reaction due to the action of the hydration reaction, resulting in the formation of a C–S–H gel with longer chain lengths. Since the Si atom radius is smaller than the Ca atom radius, the bond length of the Si–O bond is shorter than that of the Ca–O bond, resulting in the bond energy of the Si–O bond being greater than that of the Ca–O bond. Furthermore, Si exhibits weak non-metal properties, making it difficult to form ions alone in compounds. Therefore, as the reaction time increased, the Ca–O bonds in the C–S–H gel were gradually broken under the action of the carbonation reaction, while the decalcification of the C–S–H gel results in Ca2+ combining with CO32− in solution to form CaCO3. After decalcification, the C–S–H gel was negatively charged due to the loss of Ca2+. To maintain the charge balance, H+ in solution is adsorbed by the C–S–H gel and combines with the broken Si–O– to form –Si–OH via protonation, which subsequently undergoes a condensation reaction with the adjacent –Si–OH. This condensation increases the mean silicate chain length and forms bridges between neighboring regions, thus pulling them closer together and causing shrinkage. This results in the formation of Q3 and Q4 structures of Ca-deficient C–S–H gel with longer chain lengths and a higher degree of polymerization. CS is composed of chains of SiO4 tetrahedra (Q2), which can directly form CaCO3 during the carbonation process11,38. Equations (1), (2), and (3) were used to express the degree of decalcification (Ld), the degree of polymerization (Pol) of the C–S–H gel, and the degree of hydration (H) of C2S and C3S, respectively5,39, as follows:
While H reflects the degree of C2S and C3S hydration, Ld reflects the degree of separation of Ca2+ in the C–S–H gel and Pol reflects the degree of re-polymerization of the SiO4 tetrahedra after decalcification of the C–S–H gel. According to the results shown in Table 5, the ratio of Ca-deficient C–S–H gel to the remaining C–S–H gel did not change significantly in the temperature range of 40–80 °C and with LFS particle sizes from 38 to 180 μm, with the decalcification degree of the C–S–H gel remaining low. When the reaction conditions included a temperature of 20 °C and LFS particle sizes of < 38 μm, Ld was significantly increased to 13, indicating that more Ca2+ leached into the solution, leading to more carbonation in the rapid carbonation phase. Figure 7 shows H and Pol at different reaction temperatures and with different particle sizes, indicating that, as the reaction temperature increased, both parameters exhibited a decreasing trend. This may have occurred as the reaction products cover the reaction phase surface, preventing further contact between water and the reaction phase, which ultimately inhibits the process of hydration. Furthermore, increasing the reaction temperature reduces the solubility of CO2, thereby inhibiting the carbonation reaction. The observed trend in Pol was negatively correlated with the LFS particle size, which may be due to the decrease in particle size causing the –Si–OH groups in Ca-deficient C–S–H to connect and form bridges more easily with adjacent Si–OH groups, with this polymerization accelerating the carbonation reaction. Furthermore, the released Ca2+ combines with CO32− in solution to form CaCO3, which leads to a decrease in Ca2+ concentration. According to Kurdowski's theory, a low concentration of Ca will accelerate the hydration reaction of C2S and C3S and generate more C–S–H gel and Ca-deficient C–S–H gel to participate in the carbonation reaction. Therefore, the carbonation reaction promotes the hydration reaction.

Hydration and polymerization degree of C2S and C3S at different reaction temperatures or with different LFS particle sizes.
Conclusions
-
1.
According to the obtained XRD, TG-DTG and SEM results, the hydration and carbonation reaction processes start with the hydration of C2S and C3S to form C–S–H gel. To maintain the required alkaline environment, C–S–H gel undergoes decalcification to form Ca-deficient C–S–H gel and Ca2+, with the Ca2+ and CO32− in solution forming structurally stable CaCO3. The Ca-deficient C–S–H gel undergoes condensation with adjacent –Si–OH groups to maintain charge equilibrium, resulting in a Ca-deficient C–S–H gel structure with a higher degree of polymerization of Q3 and Q4 units. Although increasing the temperature is beneficial to the conversion of Ca2+ in C2S and C3S, it hinders the combination of Ca2+ with CO32− and thus, inhibits the hydration carbonation efficiency. Increasing the particle size was not conducive to the hydration reaction of C2S and C3S, which in turn affects the subsequent carbonation reaction. Therefore, the hydration and carbonation reactions influence and promote each other.
-
2.
Deconvolution of the 29Si NMR spectra revealed that the highest degree of hydration of C2S and C3S, the largest C–S–H gel decalcification effect and the highest degree of polymerization were achieved at a temperature of 20 °C, indicating that within the investigated temperature range the hydration carbonation reaction was optimal at this temperature. Moreover, the hydration of C2S and C3S and the polymerization of C–S–H gel decreased gradually with increasing temperature, indicating that the increase in temperature was not favorable for the hydration and carbonation reactions.
-
3.
Deconvolution of the 29Si NMR spectra further indicated that the highest degree of hydration of C2S and C3S and the highest level of decalcification and polymerization of the C–S–H gel were achieved when the LFS particle size was smaller than 38 μm, indicating that the hydration and carbonation reaction was optimal at this particle size. The polymerization degree of the C–S–H gel decreased gradually with increasing particle size, showing that larger particle sizes were not conducive to the hydration and carbonation reactions.
-
4.
In future research, the effect of cementitious substances on the properties of LFS-based cement materials will be investigated.
Data availability
The datasets generated during and/or analyzed during the current study are available on resonable request, by contacting the corresponding author; Yuanrong Yi(yyrhyw@163.com).
References
Jiang, Y., Ling, T. C., Shi, C. & Pan, S. Y. Characteristics of steel slags and their use in cement and concrete—A review. Resour. Conserv. Recycl. 136, 187–197 (2018).
Yi, Y.-R. et al. Accelerated carbonation of ladle furnace slag and characterization of its mineral phase. Constr. Build. Mater. 276, 122235 (2021).
Adolfsson, D., Menad, N., Viggh, E. & Björkman, B. Steelmaking slags as raw material for sulphoaluminate belite cement. Adv. Cem. Res. 19, 147–156 (2007).
Araos Henríquez, P., Aponte, D., Ibáñez-Insa, J. & Barra Bizinotto, M. Ladle furnace slag as a partial replacement of Portland cement. Constr. Build. Mater. 289, 123106 (2021).
Gaitero, J. J., Campillo, I. & Guerrero, A. Reduction of the calcium leaching rate of cement paste by addition of silica nanoparticles. Cem. Concr. Res. 38, 1112–1118 (2008).
Pan, S. Y., Liu, H. L., Chang, E. E., Kim, H. & Chiang, P. C. Multiple model approach to evaluation of accelerated carbonation for steelmaking slag in a slurry reactor. Chemosphere 154, 63–71 (2016).
Xu, B. & Yi, Y. Treatment of ladle furnace slag by carbonation: Carbon dioxide sequestration, heavy metal immobilization, and strength enhancement. Chemosphere 287, 132–274 (2021).
Monkman, S., Shao, Y. & Shi, C. Carbonated ladle slag fines for carbon uptake and sand substitute. J. Mater. Civ. Eng. 21, 657–665 (2009).
Auroy, M. et al. Comparison between natural and accelerated carbonation (3% CO2): Impact on mineralogy, microstructure, water retention and cracking. Cem. Concr. Res. 109, 64–80 (2018).
Wouter, et al. Mineral CO2 sequestration by steel slag carbonation. Environ. Sci. Technol. 39, 9676–9682 (2005).
Ashraf, W. & Olek, J. Carbonation behavior of hydraulic and non-hydraulic calcium silicates: potential of utilizing low-lime calcium silicates in cement-based materials. J. Mater. Sci. 51, 6173–6191 (2016).
Reddy, K. R. et al. Effect of basic oxygen furnace slag particle size on sequestration of carbon dioxide from landfill gas. Waste Manage. Res. 37, 469–477 (2019).
Zhang, T., Yu, Q., Wei, J., Li, J. & Zhang, P. Preparation of high performance blended cements and reclamation of iron concentrate from basic oxygen furnace steel slag. Resour. Conserv. Recycl. 56, 48–55 (2011).
Suh, H., A et al. Influences of rehydration conditions on the mechanical and atomic structural recovery characteristics of Portland cement paste exposed to elevated temperatures. Construct. Build. Mater. 235, (2019).
Liu, W., Li, Y. Q., Tang, L. P. & Dong, Z. J. XRD and 29Si MAS NMR study on carbonated cement paste under accelerated carbonation using different concentration of CO2. Mater. Today Commun. 19, 464–470 (2019).
Song, S., Sohn, D., Jennings, H. M. & Mason, T. O. Hydration of alkali-activated ground granulated blast furnace slag. J. Mater. Sci. 35, 249–257 (2000).
Khan, M., Kayali, O. & Troitzsch, U. Effect of NaOH activation on sulphate resistance of GGBFS and binary blend pastes. Cement Concr. Compos. 81, 49–58 (2017).
Maddalena, R., Li, K., Chater, P. A., Michalik, S. & Hamilton, A. Direct synthesis of a solid calcium-silicate-hydrate (C-S-H). Constr. Build. Mater. 223, 554–565 (2019).
Yang, K. H. & Song, J. K. Workability loss and compressive strength development of cementless mortars activated by combination of sodium silicate and sodium hydroxide. J. Mater. Civ. Eng. 21, 119–127 (2009).
Li, Q., Hurt, A. P. & Coleman, N. J. The application of 29Si NMR spectroscopy to the analysis of calcium silicate-based cement using biodentine as an example. J. Funct. Biomater. 10, (2019).
Ma, H. & Li, Z. in The Twenty-Fourth KKCNN Symposium on Civil Engineering.
Chen, J. J., Thomas, J. J. & Jennings, H. M. Decalcification shrinkage of cement paste. Cem. Concr. Res. 36, 801–809 (2006).
Fang, Y. & Chang, J. Microstructure changes of waste hydrated cement paste induced by accelerated carbonation. Constr. Build. Mater. 76, 360–365 (2015).
Reddy, K. R., Gopakumar, A., Chetri, J. K., Kumar, G. & Grubb, D. G. Sequestration of landfill gas emissions using basic oxygen furnace slag: effects of moisture content and humid gas flow conditions. J. Environ. Eng. 145, (2019).
Ashraf, W., Olek, J. & Atakan, V. in 14th International Congress on the Chemistry of Cement (ICCC 2015).
Jang, J. G. & Lee, H. K. Microstructural densification and CO2 uptake promoted by the carbonation curing of belite-rich Portland cement. Cem. Concr. Res. 82, 50–57 (2016).
Ashraf, W., Olek, J. & Sahu, S. Phase evolution and strength development during carbonation of low-lime calcium silicate cement (CSC). Constr. Build. Mater. 210, 473–482 (2019).
Pan, S.-Y., Chang, E. E. & Chiang, P.-C. CO2 capture by accelerated carbonation of alkaline wastes: A review on its principles and applications. Aerosol Air Qual. Res. 12, 770–791 (2012).
Chang, E. E., Chen, C.-H., Chen, Y.-H., Pan, S.-Y. & Chiang, P.-C. Performance evaluation for carbonation of steel-making slags in a slurry reactor. J. Hazard. Mater. 186, 558–564 (2011).
Haha, M. B., Lothenbach, B., Le Saout, G. & Winnefeld, F. Influence of slag chemistry on the hydration of alkali-activated blast-furnace slag—Part I: Effect of MgO. Cem. Concr. Res. 41, 955–963 (2011).
Mo, L., Zhang, F. & Deng, M. Mechanical performance and microstructure of the calcium carbonate binders produced by carbonating steel slag paste under CO2 curing. Cem. Concr. Res. 88, 217–226 (2016).
Wang, D. & Chang, J. Comparison on accelerated carbonation of β-C2S, Ca(OH)2, and C4AF: Reaction degree, multi-properties, and products. Constr. Build. Mater. 224, 336–347 (2019).
Santos, A. et al. Larnite powders and larnite/silica aerogel composites as effective agents for CO2 sequestration by carbonation. J. Hazard. Mater. 168, 1397–1403 (2009).
Chang, J., Fang, Y., Shang, X. & Wang, J. Effect of accelerated carbonation on microstructure of calcium silicate hydrate. J. Chin. Ceram. Soc. 43, 1055–1060 (2015).
De Beer, M., Doucet, F. J., Maree, J. P. & Liebenberg, L. Synthesis of high-purity precipitated calcium carbonate during the process of recovery of elemental sulphur from gypsum waste. Waste Manage. 46, 619–627 (2015).
Lippmaa, E., Maegi, M., Samoson, A., Engelhardt, G. & Grimmer, A. R. Structural studies of silicates by solid-state high-resolution 29Si NMR. J. Am. Chem. Soc. 102, 4889–4893 (1980).
Feng, C., Wang, X. & Li, D. Application progress of solid 29Si, 27Al NMR in the research of cement-based materials. He Jishu/Nuclear Tech. 37, 010502–010505 (2014).
Hansen, M. R., Jakobsen, H. J. & Skibsted, J. 29Si chemical shift anisotropies in calcium silicates from high-field 29Si MAS NMR spectroscopy. Inorg. Chem. 42, 2368–2377 (2003).
Bosque, I. F. S. E. D., Martı´n-Pastor, M., Martı´nez-Ramı´rez, S. & TeresaBlanco-Varela, M. A. Effect of temperature on C3S and C3S+ nanosilica hydration and C-S–H structure. J. Am. Ceram. Soc. 96, 957–965 (2013).
Acknowledgements
This study was funded by the National Natural Science Foundation of China (51868072). The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
Y.Y. and W.M. contributed equally. Data curation: W.M.; methodology: A.S., Y.C.; project administration: M.F.; software: Z.M.; validation: S.B.; visualization: Y.L.; writing-original draft: Y.Y.; writing-review and editing: Y.Y., W.M.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yi, Y., Ma, W., Sidike, A. et al. Synergistic effect of hydration and carbonation of ladle furnace aslag on cementitious substances. Sci Rep 12, 14526 (2022). https://doi.org/10.1038/s41598-022-18215-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-18215-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
