
Abstract
PILRA (rs1859788 A > G) has been suggested to be a protective variant for Alzheimer’s disease (AD) and is an entry co-receptor for herpes simplex virus-1. We conducted a nested case–control study of 360 1:1-matched AD subjects. Interactions between the PILRA-A allele, APOE risk variants (ε3/ε4 or ε4/ε4) and GM17 for AD risk were modelled. The associations were cross-validated using two independent whole-genome sequencing datasets. We found negative interactions between PILRA-A and GM17 (OR 0.72, 95% CI 0.52–1.00) and between PILRA-A and APOE risk variants (OR 0.56, 95% CI 0.32–0.98) in the discovery dataset. In the replication cohort, a joint effect of PILRA and PILRA × GM 17/17 was observed for the risk of developing AD (p .02). Here, we report a negative effect modification by PILRA on APOE and GM17 high-risk variants for future AD risk in two independent datasets. This highlights the complex genetics of AD.
Similar content being viewed by others

Introduction
The underlying cause of Alzheimer´s disease (AD) is considered to involve both genetic and environmental factors1. The major genetic risk allele for late-onset AD is the ε4 variant of the apolipoprotein E gene (APOE) on chromosome 192. Large genome-wide association studies (GWAS) have discovered several other risk loci for AD3,4, many of which are also associated with immune dysfunction in the central nervous system5. Using a candidate gene approach, a new potential risk variant for AD was identified in the immunoglobulin heavy chain G (IGHG) genes on chromosome 146. The risk allele of IGHG encodes the immunoglobulin (Ig) Ƴ marker (GM) 17 allotype, and homozygosity for GM17 was independently associated with a fourfold increased risk of AD. Interestingly, both APOE and GM17 might affect host susceptibility to herpes simplex virus 1 (HSV-1) infections6,7,8,9,10. Another gene implicated in AD predisposition is PILRA located on chromosome 711,12,13,14.
PILRA encodes the protein paired immunoglobulin-like type 2 receptor alpha (PILRA), an inhibitory surface receptor expressed by myeloid cells and other tissues including the nervous system15, which appears to regulate immune cells and inflammation16,17,18. Also, PILRA plays an important role in the life cycle of HSV-1, acting as an entry co-receptor for HSV-1 through the binding of viral glycoprotein B15. Transfection of PILRA enables the spreading of HSV-1 in normally resistant cell lines15. PILRA rs1859788 c.232A > G (p.Arg78Gly) is thought to be a functional variant in the region adjacent to its sialic binding pocket. This missense mutation (PILRA R78G), where glycine (G) coded by the G allele is substituted for arginine (R) coded by the A allele, is suggested to be a protective variant for AD11. The A allele of PILRA R78G attenuates infection through reduced binding for several of its ligands, including HSV-1 glycoprotein B11.
Environmental exposure to infectious pathogens like HSV-1 might contribute to the pathogenesis of AD19,20. HSV-1 infection in mouse models and 3D brain organoids has been shown to induce typical features of AD21,22. Epidemiological observations of an association between HSV-1 infection and increased AD risk have provided further support for the link in humans8,9,10,23,24,25,26. Antiviral drugs given in the event of a recurrent herpes infection seem to reduce this risk according to recent retrospective cohort studies27,28,29,30,31.
AD is probably a polygenic disorder involving multiple genes and their combined effects32. Different allelic combinations can explain, at least in part, why only a subset of those carrying HSV-1 develop AD, since the virus is highly prevalent33. The recent finding that the A allele of PILRA R78G might be a protective gene variant for AD needs to be further investigated11. The aim of this study was to ascertain if PILRA R78G was associated with the risk of subsequent AD independently, or, by modifying the effect of other known risk markers, such as APOEε4, GM17, and HSV-1, in a nested case–control study of 360 AD subjects and their matched controls from Northern Sweden Health and Disease Study (NSHDS). Also, the associations were validated using two independent whole-genome sequencing datasets from the National Institute of Mental Health (NIMH) and from the National Institute of Aging’s (NIA) Alzheimer’s disease Sequencing Project (ADSP): NIA ADSP.
Results
The descriptive statistics of the 360 AD cases and 360 matched controls from the discovery dataset (i.e. NSHDS) are presented in Table 1. The mean time to event was 9.6 ± 4.1 years (i.e. time between blood collection and AD diagnosis). The mean age of AD diagnosis was 70.8 ± 6.4 years.
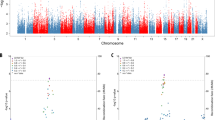
The PILRA R78-A allele was not associated with AD in the discovery dataset (crude Odds ratio (OR) 0.94, 95% confidence interval (CI) 0.74–1.21, p = 0.656; Table 2). The interactions terms were modelled using conditional logistic regression and additive coding for PILRA R78G-A and GM17 (see Methods). We found negative interactions between PILRA R78G-A x GM17 and PILRA R78G-A x APOE risk variants (ε3/ε4 or ε4/ε4) for the risk of AD (OR for the interaction 0.72, 95% CI 0.52–1.00 and 0.56, 95% CI 0.32–0.98 respectively; Table 3). The interaction term of PILRA R78G-A x anti-HSV-1 IgG seropositivity was not significant (Table 3). These interaction effects are also visualized in Fig. 1A–C where PILRA R78G is plotted against APOE, GM genotypes, and anti-HSV-1 IgG in separate groups.

Proportions of PILRA R78G genotype and anti-HSV IgM + respectively. A) Stratified by APOEε4 genotype and case–control status. B) Stratified by GM genotype and case–control status. C) Stratified by anti-HSV-1 IgG + and case–control status. D) Proportion of anti-HSV IgM + stratified by APOE risk variants and PILRA R78G genotype.
Table 4 shows the descriptive statistics of subjects with different PILRA R78G genotypes among cases and controls separately. The distribution of PILRA R78G genotype in cases and controls, stratified by APOE, GM17, and anti-HSV-1 IgG status is also presented in Fig. 1A–C. Controls with APOE risk variants, the GM17 allele and anti-HSV-1 IgG antibodies all seemed to have higher frequencies of PILRA A/A genotype compared to their cases (Fig. 1A–C). In contrast, subjects (cases and controls combined) carrying both PILRA R78G A/A and APOE risk variants had lower frequencies of detectable anti-HSV IgM antibodies compared to subjects with APOE risk variants and non-PILRA R78G A/A genotypes (Fig. 1D).
Next, we sought to assess the main or interaction effects of PILRA R78G in two AD whole-genome sequencing datasets with different study designs: a large family-based AD sample from NIMH and an AD case–control dataset from NIA ADSP (Table 5). The case–control sample from the NIA ADSP contained three subcohorts: a Non-Hispanic White cohort, an African-American cohort and a Hispanic cohort.
Using transmission family-based approaches, we saw an association of AD risk with PILRA R78G (p = 0.0495) and APOE rs429358 (ε4, p = 1.78 × 10−15) and rs7412 (ε2, p = 5.01 × 10−5) SNPs, but not with GM17 (rs1071803, p = 0.9). This method is used to evaluate both linkage and association with the phenotype of interest in family pedigrees. When including one of the following interaction terms: PILRA R78G × APOE risk variants or PILRA R78G × GM 17/17, we found that the family-based joint test for the main effect PILRA G78R and the interaction effect PILRA R78G × GM 17/17 was significant (p = 0.02, Table 6). However, none of the interaction terms in each of the two models was significant. Finally, in the non-Hispanic white subpopulation of the NIA ADSP dataset (n = 1669), PILRA R78G was not associated with AD (p = 0.94). The variant rs1071803, which codes for GM17, was missing in NIA ADSP and the interaction term PILRA R78G × APOE risk variants were not significant (p = 0.66 using additive coding and p = 0.27 using recessive coding).
Discussion
The key finding of our study is that the PILRA R78G-A allele negatively modifies the effect of APOE and GM17 high-risk variants on AD risk (OR for the GM17 interaction 0.72, 95% CI 0.52–1.00 and OR for the APOE interaction 0.56, 95% CI 0.31–0.98; Table 3 in the discovery cohort). The effect modification seems to be of increased strength in APOEε4 and GM17 homozygotes (Fig. 1A, B), revealing a potential dose-dependent pattern. Similarly, we found a significant joint effect of PILRA R78G and PILRA R78G × GM 17/17 for AD in the replication cohort. While having the PILRA R78G-A allele was associated with reduced risk of AD in the family cohort, this association was not replicated in the other two samples.
Previous epidemiological studies have shown that HSV-1 is associated with increased AD risk in genetically predisposed individuals carrying the APOEε4 allele or other AD risk genes7,8,9,10,23. The finding that the PILRA R78G-A allele might modify the risk of AD in APOEε4 and GM17 carriers (Table 3) might further enhance our understanding of the complex gene-gene and gene-environment interactions for HSV1-associated AD risk.
PILRA R78G has previously been linked to both HSV-1 and AD11,15. The A allele of PILRA R78G causes a conformational change in its sialic binding pocket, which leads to impaired binding capacity for HSV-1 and other ligands11. This could make target cells less susceptible to HSV-1 infection through reduced HSV-1 cell fusion, and limit viral entry into neurons in the brain, thus offering some protection against HSV-1-associated AD. The effect of PILRA could also possibly be explained by fewer latently infected neurons in the periphery, which correlate with lower reactivation rates of HSV-134. Importantly, PILRA also function as an inhibitory regulator of microglia activation35, and reduced PILRA signaling in R78G-A allelic variants could result in the enhancement of microglial activity11. It is therefore possible that the decrease in AD risk associated with having the PILRA R78G-A allele might be attributed to more properly regulated microglia and possibly improved amyloid-β clearance36. However, the exact role of microglia in AD initiation and progression remains to be fully elucidated, and it might vary during the course of the disease.
In the discovery cohort, we observed a potential modifying effect of PILRA R78G A/A on the risk of having anti-HSV IgM antibodies (a marker of recent HSV reactivation) among carriers of APOE risk variants (Fig. 1D). Notably, we have previously shown that having APOE risk variants were associated with a higher prevalence of anti-HSV IgM antibodies in the NSHDS sample6, thus an association that seems to be negatively modified by PILRA. Figure 1C illustrates that PILRA R78G A/A homozygosity also could have a protective impact on the HSV-1 associated AD risk, although not statistically significant (Table 3). Herein, HSV-1 seropositive controls had a higher frequency of PILRA R78G A/A genotypes compared to HSV-1 seronegative controls.
The primary strength of this study is that controls, sampled from the same population, were closely matched on possible confounding and demographic variables. Another major strength is the prospective design, where blood specimens were obtained several years prior to the disease onset, making it possible to estimate future disease risk. Limitations include the observational nature of our study, as potential unaccounted confounding factors could influence the associations and that the AD diagnoses were clinical and not based on evidence of amyloid deposition or pathologic tau. A further limitation noticed was that only 5.3% of AD cases and 7.3% of controls were PILRA R78G A/A homozygotes (Table 1), suggesting that this genotype is not common in the studied population. The allele frequency of PILRA rs1859788 seems to vary globally, and is higher in the East Asian population37. This variation in allele frequency could possibly explain the lack of association between AD and PILRA R78G in the NSHDS and NIA ADSP material, which was indicated by another study11 and the family-based NIMH dataset.
Conclusion
Here, we report a negative effect modification by the PILRA R78G-A allele on APOE and GM17 risk variants for future AD risk in two independent datasets. This observation might provide further insight into the complex genetics of HSV1-associated AD.
Methods
Study design
Discovery dataset NSHDS
We used a nested case–control study design, where 360 subjects clinically diagnosed with AD were identified from the population-based Northern Sweden Health and Disease study (NSHDS)38. The NSHDS consists of three subcohorts: the Västerbotten Intervention Programme (VIP), the Mammography Screening Project (MA), and The Northern Sweden Monica Project (MO). Blood samples were previously drawn and stored in the Medical Biobank in Umeå, extracted for analysis on average 9.6 years before the AD diagnosis. Controls without neurodegenerative disorders were randomly selected from the NSHDS cohort and matched 1:1 by age, sampling dates, sex, and subcohort. The diagnostic procedure and selection of subjects have been described in a previous publication25.
NIMH family-based dataset and ADSP case–control dataset
The results were cross-validated using two independent whole-genome sequencing datasets, a family-based AD cohort from NIMH and an AD case–control sample from the NIA (ADSP).
Genotyping in NSHDS
Samples were genotyped for APOE (rs429358 and rs7412) and PILRA R78G (rs1859788) using Illumina genome-wide array Human-OmniExpress24 (deCODE genetics, Reykjavik, Iceland)9. QPCR-based genotyping assays11,39 were employed for confirmation of inconclusive sequences. A custom design TaqMan genotyping assay was employed for genotyping of the GM3 and17 alleles (i.e. to determine GM 3/3, GM 3/17 and GM 17/17 genotypes)6.
WGS analysis in NIMH and ADSP
Whole genome sequencing in the National Institute of Mental Health (NIMH) AD cohort and AD diagnoses are described elsewhere40,41. Variant calls in vcf format for the National Institute of Aging’s (NIA) Alzheimer’s disease sequencing project (ADSP) cohort were obtained from the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS) under accession number: NG00067. The NIA ADSP dataset was divided into three subcohorts: Non-Hispanic White, African-American and Hispanic based on derived principal components. In order to derive more recent admixture principal components were calculated based on 100,000 rare variants using a modified genetic relationship matrix based on the Jaccard index42. Outliers based on principal components were excluded.
Serology—NSHDS
Enzyme-linked immunosorbent assays were used for the detection of anti-HSV IgG, anti-HSV-1 IgG, and anti-HSV IgM as previously described25.
Statistical analyses
Variables for APOE, GM, and PILRA R78G genotypes
We used additive coding for GM17 and PILRA R78G, as having 0, 1 or 2 copies of the minor allele (i.e. the PILRA R78G-A or GM17 alleles). The APOE variable was dichotomized as having high risk variants (ε3/ε4 or ε4/ε4) compared to ε3/ε4 and ε4/ε4 non-carriers. The rationale for dichotomizing APOE is that the effect of APOEε4 on AD risk is not additive, and the APOE locus is not bi-allelic.
APOE, GM, PILRA R78G, HSV-1, and the risk of AD in NSHDS
Associations between the risk of AD and the PILRA R78G-A allele were assessed by conditional logistic regression models. Interaction models were fitted for PILRA R78G-A and AD with interaction terms for PILRA R78G-A x APOE risk variants, PILRA R78G-A x GM17 and PILRA R78G-A x anti-HSV-1 IgG seropositivity. Each interaction term was modeled separately to estimate the effect modification by the PILRA R78G-A allele on AD risk per these factors.
The gene variables contained missing data ranging from n = 3 to 10 (APOE: n = 3 cases and n = 7 controls, PILRA R78G: n = 8 cases and n = 6 controls, GM: n = 10 cases and n = 8 controls). Subjects with missing values were omitted from the statistical analyses. This strategy was chosen since data can be assumed to be missing completely at random due to their blood samples containing insufficient amounts of DNA.
Statistical analyses were performed using R version 4.1.3. A two-tailed p-value < 0.05 was considered significant. The codes are available as supplementary files (Supplementary file 1: discovery cohort and Supplementary file 2: replication cohorts).
APOE, GM, PILRA R78G, and the risk of AD in NIMH and ADSP
PLINK243 (www.cog-genomics.org/plink/2.0/) was used to pre-process and extract variants of interest. In the NIMH cohort, we used a robust gene-by-environment test44, which is based on the family-based association test (FBAT)45, a generalization of the transmission disequilibrium test. We used the function “fbatge” from the “fbati” package in R. In the case–control cohort, we used PLINK2 and R to perform logistic regression with covariates (Age, Sex, Sequencing center, and first 5 principal components to adjust for the population structure) and the corresponding interaction term. If not mentioned otherwise, we considered an additive model for PILRA G78R and considered the following interaction terms: PILRA R78G A/A x APOE risk variants, PILRA R78G A/A x GM 17/17, PILRA R78G A/A x APOE, or GM 17 risk variants. Information on anti-HSV-1 IgG seropositivity was not available in WGS cohorts.
Ethical approval
The study was performed in accordance with the Declaration of Helsinki and was approved by the Regional Ethical Review Board in Umeå, Sweden (diary no. 09-190 M and 2017/18-31). All participants provided informed consent for long-term storage of blood specimens and for research on the stored samples.
Data availability
Discovery cohort, NSHDS: The dataset generated and analyzed during the current study is uploaded as a supplementary file. Additional information is available from the authors upon reasonable request, and after review and with permission from The Biobank Research Unit at Umeå University. Replication cohorts: The NIMH WGS dataset analyzed during the current study was funded by Cure Alzheimer's Fund, a non-profit organization, and is available from the authors on reasonable request. The NIA ADSP WGS dataset is available from DSS NIAGADS (https://dss.niagads.org/) under accession number: NG00067. Data used in preparation of this article were in part obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
References
Gatz, M. et al. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174. https://doi.org/10.1001/archpsyc.63.2.168 (2006).
Farrer, L. A. et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer disease meta analysis consortium. Jama 278, 1349–1356 (1997).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413. https://doi.org/10.1038/s41588-018-0311-9 (2019).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436. https://doi.org/10.1038/s41588-022-01024-z (2022).
Lambert, J. C. et al. Implication of the immune system in Alzheimer’s disease: Evidence from genome-wide pathway analysis. J. Alzheimers Dis. 20, 1107–1118. https://doi.org/10.3233/jad-2010-100018 (2010).
Pandey, J. P. et al. An Ig γ marker genotype is a strong risk factor for Alzheimer disease, independent of apolipoprotein E ε4 genotype. J. Immunol. (Baltimore, Md. 1950) 205, 1318–1322. https://doi.org/10.4049/jimmunol.2000351 (2020).
Itzhaki, R. F. et al. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet (London, England) 349, 241–244. https://doi.org/10.1016/s0140-6736(96)10149-5 (1997).
Lovheim, H. et al. Herpes simplex virus, APOEvarepsilon4, and cognitive decline in old age: Results from the Betula cohort study. J. Alzheimers Dis. 67, 211–220. https://doi.org/10.3233/jad-171162 (2019).
Lopatko Lindman, K. et al. A genetic signature including apolipoprotein Eε4 potentiates the risk of herpes simplex-associated Alzheimer’s disease. Alzheimers Dement. (N Y) 5, 697–704. https://doi.org/10.1016/j.trci.2019.09.014 (2019).
Linard, M. et al. Interaction between APOE4 and herpes simplex virus type 1 in Alzheimer’s disease. Alzheimer’s Dement. 16, 200–208. https://doi.org/10.1002/alz.12008 (2020).
Rathore, N. et al. Paired Immunoglobulin-like type 2 receptor alpha G78R variant alters ligand binding and confers protection to Alzheimer’s disease. PLoS Genet. 14, e1007427. https://doi.org/10.1371/journal.pgen.1007427 (2018).
Patel, T. et al. Whole-exome sequencing of the BDR cohort: Evidence to support the role of the PILRA gene in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 44, 506–521. https://doi.org/10.1111/nan.12452 (2018).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. https://doi.org/10.1038/ng.2802 (2013).
Park, Y. H. et al. Association of blood-based transcriptional risk scores with biomarkers for Alzheimer disease. Neurol. Genet. 6, e517. https://doi.org/10.1212/nxg.0000000000000517 (2020).
Satoh, T. et al. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132, 935–944. https://doi.org/10.1016/j.cell.2008.01.043 (2008).
Shiratori, I., Ogasawara, K., Saito, T., Lanier, L. L. & Arase, H. Activation of natural killer cells and dendritic cells upon recognition of a novel CD99-like ligand by paired immunoglobulin-like type 2 receptor. J. Exp. Med. 199, 525–533. https://doi.org/10.1084/jem.20031885 (2004).
Wang, J., Shiratori, I., Uehori, J., Ikawa, M. & Arase, H. Neutrophil infiltration during inflammation is regulated by PILRα via modulation of integrin activation. Nat. Immunol. 14, 34–40. https://doi.org/10.1038/ni.2456 (2013).
Kohyama, M. et al. Monocyte infiltration into obese and fibrilized tissues is regulated by PILRα. Eur. J. Immunol. 46, 1214–1223. https://doi.org/10.1002/eji.201545897 (2016).
Eimer, W. A. et al. Alzheimer’s disease-associated beta-amyloid is rapidly seeded by Herpesviridae to protect against brain infection. Neuron 99, 56-63.e53. https://doi.org/10.1016/j.neuron.2018.06.030 (2018).
Kumar, D. K. et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 8, 340ra372. https://doi.org/10.1126/scitranslmed.aaf1059 (2016).
De Chiara, G. et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 15, e1007617. https://doi.org/10.1371/journal.ppat.1007617 (2019).
Cairns, D. M. et al. A 3D human brain-like tissue model of herpes-induced Alzheimer’s disease. Sci. Adv. 6, 8828. https://doi.org/10.1126/sciadv.aay8828 (2020).
Steel, A. J. & Eslick, G. D. Herpes viruses increase the risk of Alzheimer’s disease: A meta-analysis. J. Alzheimers Dis. 47, 351–364. https://doi.org/10.3233/jad-140822 (2015).
Lovheim, H., Gilthorpe, J., Adolfsson, R., Nilsson, L. G. & Elgh, F. Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimer’s Dement. 11, 593–599. https://doi.org/10.1016/j.jalz.2014.04.522 (2015).
Lovheim, H. et al. Herpes simplex infection and the risk of Alzheimer’s disease: A nested case-control study. Alzheimer’s Dement. 11, 587–592. https://doi.org/10.1016/j.jalz.2014.07.157 (2015).
Letenneur, L. et al. Seropositivity to herpes simplex virus antibodies and risk of Alzheimer’s disease: A population-based cohort study. PLoS One 3, e3637. https://doi.org/10.1371/journal.pone.0003637 (2008).
Tzeng, N. S. et al. Anti-herpetic medications and reduced risk of dementia in patients with herpes simplex virus infections-a nationwide, population-based cohort study in Taiwan. Neurotherapeutics 15, 417–429. https://doi.org/10.1007/s13311-018-0611-x (2018).
Bae, S. et al. Association of herpes zoster with dementia and effect of antiviral therapy on dementia: A population-based cohort study. Eur. Arch. Psychiatry Clin. Neurosci. https://doi.org/10.1007/s00406-020-01157-4 (2020).
Lopatko Lindman, K. et al. Herpesvirus infections, antiviral treatment, and the risk of dementia-a registry-based cohort study in Sweden. Alzheimers Dement. (N Y) 7, e12119. https://doi.org/10.1002/trc2.12119 (2021).
Hemmingsson, E. S. et al. Antiviral treatment associated with reduced risk of clinical Alzheimer’s disease-A nested case-control study. Alzheimers Dement. (N Y) 7, e12187. https://doi.org/10.1002/trc2.12187 (2021).
Chen, V. C. et al. Herpes zoster and dementia: A Nationwide population-based cohort study. J. Clin. Psychiatry 79, 5. https://doi.org/10.4088/JCP.16m11312 (2018).
Escott-Price, V. et al. Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain 138, 3673–3684. https://doi.org/10.1093/brain/awv268 (2015).
Olsson, J., Kok, E., Adolfsson, R., Lovheim, H. & Elgh, F. Herpes virus seroepidemiology in the adult Swedish population. Immun. Ageing 14, 10. https://doi.org/10.1186/s12979-017-0093-4 (2017).
Hoshino, Y., Pesnicak, L., Cohen, J. I. & Straus, S. E. Rates of reactivation of latent herpes simplex virus from mouse trigeminal ganglia ex vivo correlate directly with viral load and inversely with number of infiltrating CD8+ T cells. J. Virol. 81, 8157–8164. https://doi.org/10.1128/jvi.00474-07 (2007).
Tato, C. M. et al. The myeloid receptor PILRβ mediates the balance of inflammatory responses through regulation of IL-27 production. PLoS One 7, e31680. https://doi.org/10.1371/journal.pone.0031680 (2012).
Cai, Z., Hussain, M. D. & Yan, L. J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 124, 307–321. https://doi.org/10.3109/00207454.2013.833510 (2014).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74. https://doi.org/10.1038/nature15393 (2015).
Hallmans, G. et al. Cardiovascular disease and diabetes in the Northern Sweden Health and Disease Study Cohort - evaluation of risk factors and their interactions. Scand. J. Public Health Suppl. 61, 18–24. https://doi.org/10.1080/14034950310001432 (2003).
Calero, O., Hortigüela, R., Bullido, M. J. & Calero, M. Apolipoprotein E genotyping method by real time PCR, a fast and cost-effective alternative to the TaqMan and FRET assays. J. Neurosci. Methods 183, 238–240. https://doi.org/10.1016/j.jneumeth.2009.06.033 (2009).
Prokopenko, D. et al. Identification of novel Alzheimer’s disease loci using sex-specific family-based association analysis of whole-genome sequence data. Sci. Rep. 10, 5029. https://doi.org/10.1038/s41598-020-61883-6 (2020).
Blacker, D. et al. ApoE-4 and age at onset of Alzheimer’s disease: the NIMH genetics initiative. Neurology 48, 139–147. https://doi.org/10.1212/wnl.48.1.139 (1997).
Prokopenko, D. et al. Utilizing the Jaccard index to reveal population stratification in sequencing data: A simulation study and an application to the 1000 genomes project. Bioinformatics 32, 1366–1372. https://doi.org/10.1093/bioinformatics/btv752 (2016).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7. https://doi.org/10.1186/s13742-015-0047-8 (2015).
Hoffmann, T. J., Lange, C., Vansteelandt, S. & Laird, N. M. Gene-environment interaction tests for dichotomous traits in trios and sibships. Genet. Epidemiol. 33, 691–699. https://doi.org/10.1002/gepi.20421 (2009).
Laird, N. M., Horvath, S. & Xu, X. Implementing a unified approach to family-based tests of association. Genet. Epidemiol. 19(Suppl 1), S36-42. https://doi.org/10.1002/1098-2272(2000)19:1+%3c::Aid-gepi6%3e3.0.Co;2-m (2000).
Acknowledgements
NSHDS: This study was conducted in the context of the CHANCES project, which was funded in the FP7 framework program of DG RESEARCH in the European Commission. The study was further supported financially by grants from Region Västerbotten, the Kempe Foundation, Wallenberg Centre for Molecular Medicine in Umeå, the Swedish Medical Association, the Swedish Dementia Association, the Swedish Alzheimer Fund and the Umeå University Foundation for Medical Research. The authors wish to express their gratitude to Emma Honkala for laboratory assistance. Part of the genotype analysis was provided by deCODE genetics, Reykjavik Iceland. During the last 36 months B.W has received grants from The Swedish Brain Foundation and S.E has received project grants from The National Health and Medical Research Council (App1157607) outside of the submitted work.
NIMH and NIA ADSP cohorts: This work was supported by Cure Alzheimer’s Fund. During the last 36 months D.P has received support from the Women's Alzheimer's Movement outside of the submitted work. The computations in this paper were partially run on the FASRC Cannon cluster supported by the FAS Division of Science Research Computing Group at Harvard University. The funding body has no role in the design of the study or collection, analysis, or interpretation of data, nor in writing the manuscript. Please refer to the Supplementary Note for full acknowledgements.
Funding
Open access funding provided by Umea University.
Author information
Authors and Affiliations
Contributions
All authors contributed to and approved the final version. R.T., G.H., S.E., F.E. and H.L. designed and initialized the study. F.E. and J.O. were responsible for laboratory testing. K.L.L., D.P. and H.L. performed the statistical analyses. K.L.L., C.J. and H.L. wrote the first draft. All authors were involved in interpretation of data and critically reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
All authors declare no competing interests directly related to this project. R.T reported the following interests, unrelated to the submitted work: Financial Interest only: Amylyx / React Neuro / Cognitive Clarity / DRADS Capital / Verge Genomics / Cognoptix / Genomind / Interaxon/ Neurogenetics. Paid Consultant and Financial Interest: MarvelBiome / AZTherapies / Promis / Cerevance / Chromadex / Jefferson Pharmaceuticals / Annovis Bio/ TrialSight. Paid Consultant only: Takeda / FujiFilm / CAMP4 / Sarepta / Neurona.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lopatko Lindman, K., Jonsson, C., Weidung, B. et al. PILRA polymorphism modifies the effect of APOE4 and GM17 on Alzheimer’s disease risk. Sci Rep 12, 13264 (2022). https://doi.org/10.1038/s41598-022-17058-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-17058-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
