
Abstract
Heart failure (HF) is the inability of the heart to pump blood sufficiently to meet the metabolic demands of the body. HF with reduced systolic function is characterized by cardiac hypertrophy, ventricular fibrosis and remodeling, and decreased cardiac contractility, leading to cardiac functional impairment and death. Transverse aortic constriction (TAC) is a well-established model for inducing hypertrophy and HF in rodents. Mice globally deficient in sirtuin 5 (SIRT5), a NAD+-dependent deacylase, are hypersensitive to cardiac stress and display increased mortality after TAC. Prior studies assessing SIRT5 functions in the heart have all employed loss-of-function approaches. In this study, we generated SIRT5 overexpressing (SIRT5OE) mice, and evaluated their response to chronic pressure overload using TAC. Compared to littermate controls, SIRT5OE mice were protected against adverse functional consequences of TAC, left ventricular dilation and impaired ejection fraction. Transcriptomic analysis revealed that SIRT5 suppresses key HF sequelae, including the metabolic switch from fatty acid oxidation to glycolysis, immune activation, and fibrotic signaling pathways. We conclude that SIRT5 is a limiting factor in the preservation of cardiac function in response to experimental pressure overload.
Similar content being viewed by others


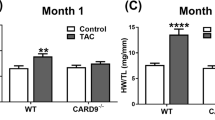
Introduction
Heart failure (HF) represents the end stage of a variety of cardiovascular pathologies, leading to insufficient oxygen delivery to meet the metabolic demands of the body. The incidence of HF is predicted to increase by 46% between 2012 and 2030 in the US, mainly due to an increase in population longevity coupled with improved survival rates following cardiovascular injury1. In 2018 alone, HF claimed 83,616 lives2. HF with systolic dysfunction is characterized by a progressive decline in contractile function and chronic hemodynamic overload, and by ventricular hypertrophy and remodeling, neurohormonal compensation mechanisms, and myocardial damage. Key cellular mechanisms thought to contribute to HF include myocyte death, reduced ability to maintain calcium homeostasis, and changes in the production and utilization of high-energy phosphates3. However, the molecular changes that lead to HF are still incompletely understood, and currently there are no mechanism-based treatments for HF.
Transverse aortic constriction (TAC) is a well-established experimental rodent model for pressure overload-induced HF. In TAC, the aorta is surgically narrowed, chronically increasing resistance to outflow from the left ventricle (LV)4. The resulting increased left ventricular burden initially induces concentric hypertrophy to compensate for TAC-induced elevated resistance. However, chronic pressure overload leads to contractile dysfunction and extensive remodeling, including fibrosis and ventricular dilation, resulting in functional decline. Over time, the deteriorating LV becomes unable to meet the hemodynamic needs of the body, resulting in progressive HF5.
Sirtuins are a family of NAD+-dependent deacylases playing major roles in cellular stress responses. In mammals, each of the seven sirtuins possesses a distinct combination of enzymatic activities, molecular targets, subcellular localization, and tissue expression6. Sirtuin 5 (SIRT5) possesses robust deacylase activity against lysine succinylation, malonylation, and glutarylation, negatively-charged lysine acyl modifications derived from their respective Coenzyme A (CoA) species7,8,9,10,11. SIRT5 target proteins reside in the mitochondrial matrix, cytosol, peroxisome, and nucleus; many function in metabolic pathways, including the citric acid cycle: e.g. pyruvate dehydrogenase and succinate dehydrogenase (SDH), and fatty acid oxidation: e.g. trifunctional enzyme subunit alpha (ECHA)8,12. Although many SIRT5 targets take part in key metabolic pathways, no striking metabolic phenotypes have been observed in Sirt5 knockout (SIRT5KO) or overexpressing (OE) mice under basal, unstressed conditions13,14,15.
Among normal tissues, a major site of SIRT5 function appears to be the heart. SIRT5KO mice display a modest, age-associated decrease in cardiac function, along with increased fibrosis and hypertrophy12. SIRT5KO mice are also hypersensitive to cardiac stress. In an ischemia–reperfusion (I/R) injury model, SIRT5KO mice show increased infarct size and impaired recovery16. In these studies, proteomic experiments identified ECHA and SDH, respectively, as the likely substrates most relevant to SIRT5’s cardioprotective activity. In response to TAC, whole-body SIRT5KOs developed more severe cardiac dysfunction, exacerbated hypertrophy, and increased fibrosis compared to wild-type (WT) littermates17. The authors of this study proposed that, during TAC, SIRT5 deficiency is associated with impaired electron flow in oxidative phosphorylation, elevated mitochondrial NADH levels, and impaired oxidative metabolism, with greater reliance on glycolysis. However, a subsequent study by the same group using a cardiomyocyte (CM)-specific SIRT5KO strain showed no differences between WT and SIRT5 mutants in response to TAC18.
In this study, we generated SIRT5 overexpressing (SIRT5OE) mice and evaluated their response to cardiac pressure overload. SIRT5OE mice displayed no obvious cardiac phenotype and very few gene expression changes in the absence of stress compared to WT littermates. However, SIRT5OE mice showed robust protection against TAC-induced LV dilation and subsequent functional decline. Transcriptomic analysis linked the protection conferred by SIRT5 overexpression to suppression of key transcriptional and signaling events accompanying ventricular hypertrophy and HF, including cardiac fibrosis, inflammatory cytokine signaling, and the metabolic switch from fatty acid oxidation to glycolysis. Our results suggest that SIRT5 levels are limiting in the context of the cardiac response to pressure overload.
Results
Generation and characterization of SIRT5-overexpressing mice
To investigate the effects of SIRT5 on cardiac function, we generated a transgenic SIRT5 overexpressing (SIRT5OE) mouse strain by inserting a LoxP-STOP-LoxP-SIRT5 cassette into the 3’UTR of the Col1A1 locus, using a well-characterized transgene insertion system (Fig. 1A)19. Constitutive, whole-body SIRT5 overexpression is driven by the CAGGS promoter (Supplemental Fig. 1A)20. SIRT5OE mice were born at normal Mendelian and sex ratios, and grossly indistinguishable from their WT littermates, with no obvious differences in weight gain with age in either sex (Supplemental Fig. 1B, Fig. 1B). We tested the molecular effects of SIRT5 overexpression on bulk protein acylation in the heart and found modest decreases in total lysine succinylation (Ksucc) and lysine malonylation (Kmal), which were significant for Kmal (Fig. 1C-E).

SIRT5OE mice exhibit decreased mildly decreased cardiac lysine malonylation. (A) Immunoblot analysis of SIRT5 expression in heart samples presented in c-e. Original immunoblot images are presented in Supplemental Fig. 7A. (B) Weight (g) of WT and SIRT5OE male and female mice with age. (C-E) Representative immunoblot analysis of succinyl-lysine (Ksucc), and malonyl-lysine (Kmal) levels in WT and SIRTOE hearts, with quantification. Original immunoblot images are presented in Supplemental Fig. 7B-C.
SIRT5 overexpression protects against TAC-induced heart failure
To evaluate sensitivity to cardiac stress, male WT and SIRT5OE littermates between 4–8 months of age were randomly assigned to a sham or TAC operation (Fig. 2A)21. TAC surgically narrows the transverse aorta, generating a pressure gradient that reflects the severity of pressure overload on the heart. Sham animals undergo an identical surgical procedure but without aortic banding. Echocardiograms (echos) to assess cardiac structure and function were performed prior to surgery, and then repeated at 4-weeks, at which point tissue was collected for downstream analyses (Fig. 2A). WT and SIRT5OE sham animals did not exhibit any notable differences by echo, indicating that global SIRT5 overexpression does not alter cardiac structure or function under basal conditions (Table 1). Additionally, the magnitude of the aortic pressure gradient was comparable between WT and SIRT5OE TAC mice (Fig. 2B). Thus, any differences observed between genotypes following TAC were not due to discrepancies in the degree of cardiac outflow obstruction. Pressure overload first induces concentric cardiac hypertrophy, a temporary compensatory mechanism to relieve stress on the heart22. Echo-based measurement of the interventricular septum (IVS) and the posterior wall (PW) thickness during both systole and diastole showed comparable LV hypertrophy at four weeks after TAC in all mice irrespective of genotype (Fig. 2C-D, Table 1). Consistent with other studies, the TAC procedure itself was associated with a 15–18% mortality rate, with 3 mice and 4 mice dying in the WT and SIRT5OE groups respectively (Supplemental Table 1). Therefore, SIRT5 overexpression does not affect cardiac concentric hypertrophy in response to TAC.

SIRT5OE mice are protected against TAC-induced heart failure. Echocardiography was performed on WT sham (n = 16), WT TAC (n = 12), SIRT5OE sham (n = 10) and SIRT5OE TAC (n = 14) mice to measure changes in cardiac function four weeks post-surgery. (A) Depiction of groups, procedures, and timeline of surgery. (B) Aortic pressure gradient in mice after TAC. Echo measurements for (C) systolic interventricular septum (IVS) thickness; (D) systolic posterior wall thickness (PWT); (E) LV end-diastolic diameter; (F) left ventricle mass normalized to body weight; (G) fractional shortening; (H) ejection fraction. (I) Quantification of CM cell area, normalized to WT sham four weeks after surgery [WT sham (n = 4), WT TAC (n = 4), SIRT5OE sham (n = 4) and SIRT5OE TAC (n = 10)]. (J) Representative wheat germ agglutinin-stained cardiac sections of the indicated genotypes and treatments four weeks post-surgery. Scale bar = 50 um. (K) qRT-PCR for Acta1 (n = 3 for all groups); Nppa and Myh6 [WT sham (n = 5), WT TAC (n = 5), SIRT5OE sham (n = 3) and SIRT5OE TAC (n = 6)]; and Myh7 [WT sham (n = 5), WT TAC (n = 4), SIRT5OE sham (n = 3) and SIRT5OE TAC (n = 5)] expression normalized to GAPDH. Statistical significance was determined using Student’s t-test for 2-group analysis or two-way ANOVA followed by Sidak’s correction for multiple comparisons for 4-group analyses.
After prolonged pressure overload, concentric hypertrophy progresses to ventricular dilation and heart failure (HF)5. Four weeks after TAC, WT mice showed significantly increased LV diameter, indicating a transition from adaptive to maladaptive ventricular hypertrophy. In comparison, LV diameter did not significantly increase in SIRT5OE TAC mice (Fig. 2E, Table 1). The combination of ventricular hypertrophy and dilation was also reflected in the LV size normalized to body weight. Both genotypes showed increased normalized LV mass after TAC; however, this increase was blunted by SIRT5 overexpression (Fig. 2F). Ventricular dilation leads to reduced fractional shortening (FS) and impaired ejection fraction (EF), a measure of systolic function. Both were significantly reduced in response to chronic pressure overload in the WT mice but preserved in the SIRT5OE mice (Fig. 2G-H). These patterns were also reflected in CM size, with WT TAC mice showing a significant increase in CM area compared to both WT sham and SIRT5OE TAC groups (Fig. 2I-J).
To complement echo-based measurements, we assessed RNA levels of standard markers of cardiac hypertrophy and HF, skeletal muscle Actin (Acta1) and atrial natriuretic peptide (Nppa)23. As expected, expression of both genes increased in response to TAC, but this increase was blunted in SIRT5OE TAC mice compared to WT TAC (Fig. 2K). The failing myocardium also shifts contractile protein expression from myosin heavy chain isoform α (Myh6) to β (Myh7) (Fig. 2K)23. The decrease in Myh6 expression by TAC was attenuated in SIRT5OE mice compared to WT controls. Overall, based on these physiological and molecular assays, we conclude that SIRT5 overexpression protects against the transition from adaptive, concentric hypertrophy to maladaptive ventricular dilation and systolic dysfunction.
SIRT5 overexpression mitigates transcriptomic changes induced by TAC
To understand how SIRT5 overexpression protects against TAC-induced stress, we performed RNA-seq transcriptomic profiling on whole hearts from WT sham, WT TAC, SIRT5OE sham, and SIRT5OE TAC mice four weeks after surgery. Principal component analysis (PCA) showed that SIRT5 overexpression did not appreciably alter baseline gene expression in the heart, as sham mice clustered together regardless of genotype. In contrast, TAC induced a marked transcriptional response in both genotypes. Apart from one animal, SIRT5OE TAC mice clustered between sham animals and WT TAC animals, suggesting that SIRT5OE blunts the overall TAC transcriptional response (Fig. 3A). Hierarchical cluster analysis (HCA) of the top 30 genes ranked by variance generated a dendrogram with two major clades (Fig. 3B). Sirt5 transcript levels were elevated in the SIRT5OE samples but were not altered by TAC in either genotype (Fig. 3A, Supplemental Fig. 2A-B). Apart from one animal, all SIRT5OE mice clustered together with WT sham mice in one clade. The second clade contained all WT TAC mice and one SIRT5OE TAC animal. Genes in signaling pathways important to heart failure were evident in this unbiased HCA clustering (Fig. 3B). Consistent with the qRT-PCR data (Fig. 2K), atrial natriuretic peptide (Nppa), skeletal muscle Actin (Acta1), and myosin heavy chain isoform α (Myh6) increased with TAC but these increases were blunted in SIRT5OE TAC mice compared to WT TAC mice (Fig. 3B). Fibroblast remodeling of the extracellular matrix (ECM) and increased fibrosis is a key component of maladaptive remodeling in HF. Postn, a key marker of cardiac fibroblast activation and differentiation, was strongly induced in WT TAC samples, but trended less so in the SIRT5OE TAC mice (Fig. 3C)24,25. Markers of monocyte-derived macrophages that stimulate fibrosis, Spp1 and Thbs1, were also elevated in TAC samples, and this increase was significantly mitigated in the SIRT5OE mice (Fig. 3C)26.Thus, based on both PCA and HCA, SIRT5OE TAC mice are generally more transcriptionally similar to WT sham mice than WT TAC mice.

Transcriptomic analysis of heart tissue four weeks after TAC identifies a dampened transcriptional response to pressure overload in SIRT5OE mice. (A) Principal component analysis (PCA) of RNA-sequencing data of the four groups of mice [WT sham (n = 4), WT TAC (n = 4), SIRT5OE sham (n = 3) and SIRT5OE TAC (n = 4)]. (B) Hierarchical clustering analysis (HCA) of the RNA-seq data based on the top 30 genes, determined by greatest variance from the mean. Genes with higher expression compared to the mean skew red; genes with lower expression compared to the mean skew blue. (C) Postn, Spp1, and Thbs1 mRNA levels measured by RNA-seq. (D) Venn Diagram comparing exclusive and overlapping differentially expressed genes in the WT (sham vs TAC) mice and the SIRT5OE (sham vs TAC) mice. (E–G) Top five positively and negatively enriched Gene Ontology (GO) pathways from the indicated comparisons, determined by gene set enrichment analysis (GSEA). NES: normalized enrichment score. All listed pathways were significantly enriched with a false discovery rate (FDR) < 0.0001.
We defined the criteria for differentially expressed genes (DEGs) between two groups as a false discovery rate (FDR) < 0.05 and fold-change of |1|. Strikingly, there were only 8 DEGs, including SIRT5, between sham WT and SIRT5OE mice, consistent with the observation that SIRT5 overexpression does not significantly alter baseline cardiac parameters. Following TAC, the number of DEGs increased dramatically in both genotypes. There were 2,606 unique DEGs between WT sham and TAC hearts, but only 177 in SIRT5OE sham and SIRT5OE TAC. There were 870 DEGs in common between the WT and SIRT5OE sham to TAC comparisons, representing a core group of genes that respond to chronic pressure overload across both genotypes (Fig. 3D). The lower number of DEGs in SIRT5OE mice after TAC suggests that transcriptional responses induced by pressure overload are partially mitigated by SIRT5 overexpression.
Unbiased analysis of gene signatures enriched by the DEGs of each group was performed using Ingenuity Pathway Analysis (IPA) and Gene Set Enrichment Analysis (GSEA)27,28. The top 5 most positively and negatively enriched Gene Ontology (GO) pathways based on normalized enrichment score (NES) were extracellular matrix (ECM) remodeling and metabolic pathways, respectively in sham to TAC comparisons of both genotypes, albeit to different magnitudes (Fig. 3E-F). The most enriched GO terms between TAC samples revealed that SIRT5OE TAC hearts show reduced activation of the immune system and suppressed ECM organization while maintaining expression of respiratory genes compared to WT TAC hearts (Fig. 3G). IPA analysis of these samples showed similar patterns of pathway enrichment (Supplemental Fig. 2C-E). In summary, based on PCA, HCA, and function enrichment analysis, transcriptomic analysis supports the conclusion that SIRT5 overexpression protects against pressure overload-induced HF partially through the suppression of molecular signaling pathways responsible for HF progression.
Pressure overload induces similar metabolic changes in both WT and SIRT5OE hearts
Under normal conditions, the heart utilizes fatty acids as its main substrate for ATP generation. The failing heart increases glycolysis, without a concomitant increase in glucose oxidation, leading to overall decreased mitochondrial oxidative metabolism, resulting in an energy deficit29. Gene ontology analysis of the RNA-seq data revealed that WT TAC mice showed significant downregulation of cellular respiration and mitochondrial matrix composition pathways (Fig. 3E). Similarly, SIRT5OE mice post-TAC also showed reduced expression of genes in various catabolic processes, including fatty acid and small molecule metabolite breakdown (Fig. 3F). Comparison of the WT and SIRT5OE TAC transcriptomes revealed up-regulation of hallmark glycolytic genes in the WT TAC samples. Conversely, genes for fatty acid metabolism and components of the respiratory electron transport chain (ETC) were enriched in the SIRT5OE TAC samples (Fig. 4A). Overall, SIRT5OE TAC hearts exhibits an intermediate metabolic state between healthy (sham) hearts and the failing, WT TAC hearts 4 weeks after TAC.

WT TAC mice exhibit a metabolic shift by RNA-seq, but are functionally comparable to SIRT5OE TAC mice four weeks post-surgery. (A) GSEA of SIRT5OE TAC compared to WT TAC RNA-seq samples. NES: normalized enrichment score. FDR: false discovery rate. (B-C) Immunoblot analysis of electron transport chain subunits and quantification of CV-ATP5A. Original immunoblot images are presented in Supplemental Fig. 8A. (D) Agilent/Seahorse analysis of electron flow through the ETC in mitochondria isolated from WT (n = 5) and SIRT5OE (n = 6) hearts 4 weeks after TAC. y-axis indicates oxygen consumption rate (OCR) in pmol/min. (E) qPCR of mitochondrially encoded (mtDNA) genes 16S and ND1, normalized to HK2, encoded by the nuclear genome (nuDNA). (F) Sod2 mRNA levels measured by RNA-seq. (G-H) SOD2 immunoblot from heart lysates and quantification. Original immunoblot images are presented in Supplemental Fig. 9A.
To investigate whether RNA expression differences in these metabolic pathways manifested at the protein level, we immunoblotted for components of the ETC, and found similar reductions in expression in both TAC groups (Fig. 4B-C, Supplemental Fig. 3A). We then directly assessed ETC activity using mitochondria isolated from hearts four weeks post-TAC using the Agilent/Seahorse XFe96 analyzer. Both groups exhibited similar rates of election flow (Fig. 4D).
With comparable protein expression levels of ETC components, and electron flow, we then asked whether differences in total mitochondrial content might contribute to the discrepancies in LV function between the two TAC groups at four weeks post-surgery. Mitochondrial mass was quantified using two distinct approaches. First, qPCR was performed for mitochondrial protein-encoding genes 16S and Nd1, and normalized to Hk2. Then, each sample was normalized to WT sham (Fig. 4E)30. Second, we assessed mRNA and protein levels of the mitochondrial protein SOD2 (Fig. 4F-H). In both assays, TAC resulted in a notable decrease in mitochondrial content, particularly in WT mice. However, only modest differences were evident between genotypes in the TAC groups. From these data, overall we conclude that while WT and SIRT5OE TAC hearts show transcriptional differences related to electron transport and other metabolic pathways, these expression differences do not robustly manifest at the level of electron transport chain activity, levels of respiratory complexes proteins, or mitochondrial content.
To test whether the cardio-protective properties of SIRT5 overexpression might be associated with alterations in the metabolome, we performed unbiased metabolomics on cardiac samples across all groups at four weeks post-surgery using LC/MS-based mass spectrometry. PCA showed minimal variance between samples on the first two principal components (Fig. 5A). TAC was the main influence on sample clustering, with minimal contribution by genotype. Analysis of metabolite differences between genotypes also supported this conclusion, with only 6 and 4 metabolites significantly altered in WT sham to SIRT5OE sham and WT TAC to SIRT5OE TAC comparisons, respectively. In contrast, the WT sham to TAC comparison showed the greatest number of significantly different metabolites (44), followed by the SIRT5OE sham to TAC comparison (25).

TAC alters the cardiac metabolomic landscape. (A) PCA of the metabolomics data from hearts four weeks post-surgery across all groups [WT sham (n = 10), WT TAC (n = 6), SIRT5OE sham (n = 6) and SIRT5OE TAC (n = 4)]. (B) Succinate levels. (C-D) Immunoblot analysis of Ksucc in sham and TAC mice, with quantification. Statistical significance was determined using two-way ANOVA followed by Sidak’s correction for multiple comparisons for 4-group analyses. Original immunoblot images are presented in Supplemental Fig. 10A.
Increased glucose usage and glycolytic flux are commonly observed during HF31. Levels of glycolytic metabolites did not show any significant changes between groups (Supplemental Fig. 4A-B). In contrast, several TCA cycle metabolites were significantly affected by TAC (Supplemental Table 2, Supplemental Fig. 4C-D). Succinate levels exhibited an interesting pattern, showing decreased levels with both TAC and SIRT5 overexpression (Fig. 5B). Since succinate is a substrate of SDH, a known SIRT5 target, we directly measured SDH activity but found no differences between the groups (Supplemental Fig. 5A). This result was consistent with electron flow data, where no differences in complex II/SDH activity were observed between mitochondria isolated from TAC hearts (Fig. 4C). Succinate is also linked to SIRT5 through SIRT5’s desuccinylase activity, as succinyl-CoA donates the succinyl group used to generate the succinyl-lysine modification (Supplemental Fig. 5B). Ksucc levels were lower in both SIRT5OE and TAC samples (Fig. 5C-D).
To complement these data, we obtained heart tissue samples from human HF patients. Similar to the mouse hearts, Ksucc levels trended lower in the human failing hearts compared to the control samples (p = 0.07) (Supplemental Fig. 5C-D). Therefore, cardiac stress leads to decreased levels of Ksucc, independent of SIRT5 genotype, in both mice and humans. Taken together, transcriptomic and metabolomic analyses reflect a primarily TAC-driven, rather than genotype-associated, shift in the metabolic landscape in both genotypes following TAC.
Cardiac fibrosis is suppressed in the SIRT5OE mice after TAC.
To further investigate the underpinnings of the improved response of SIRT5OE mice to TAC, we extracted a list of pathways IPA categorized as “cardiovascular disease-related pathways” (Fig. 6A). These pathways play critical roles in the development and progression of pathologic hypertrophy and HF, and all were more significantly enriched in WT mice with TAC compared to SIRT5OE mice. Fibrosis-related pathways (i.e., “Hepatic fibrosis signaling pathway”, “Apelin cardiac fibroblast signaling pathway”, “TGF-β signaling”) were readily apparent (Figs. 6A and 3E-G). Hepatic Fibrosis Signaling was the most significant pathway identified in the IPA analysis, and includes signaling occurring during fibroblast activation, an essential process during adaptive hypertrophy by which resident fibroblasts differentiate into myofibroblasts (Fig. 6A). However, sustained myofibroblast activity results in excessive cardiac remodeling and is also a major contributor to LV dysfunction32.

SIRT5OE protects against TAC-induced fibrosis four weeks after surgery. (A) Heat map of gene ontology analysis of WT (sham vs TAC) and SIRT5OE (sham vs TAC) mapping to cardiovascular disease related pathways using IPA. Blue color scale represents -log(p-value) of each pathway. (B) GSEA of SIRT5OE TAC compared to WT TAC RNA-seq samples. NES: normalized enrichment score. FDR: false discovery rate. (C) Volcano plot highlighting significant genes in from the GO: collagen containing extracellular matrix gene list. (D) Quantification of the amount of fibrosis in each heart sample, expressed in percentage [WT sham (n = 3), WT TAC (n = 4), SIRT5OE sham (n = 4) and SIRT5OE TAC (n = 10)]. Two-way ANOVA interaction term: p-value = 0.0012. (E) Representative images of heart sections stained with Picrosirius red for collagen. (F) Summary figure of effects of SIRT5 overexpression during pressure overload.
GSEA confirmed that WT TAC samples were significantly enriched with genes upregulated by TGFβ1, a cytokine central to the fibrotic activation response (Fig. 6B). TGFβ1 triggers expression of a fibrotic gene program that stimulates proliferation, cell migration, and increased secretion various macromolecules that restructure the ECM and promote wound healing33. ECM-related genes were highly enriched in WT TAC samples, including ECM components (Col1a1, Col4a2, Fbn1), ECM modifying genes (Timp1, Adamts4, Anxa2, Plod3), and fibrotic signaling cytokines (Tgfβ1, Gdf15, Icam1) (Fig. 6B-C)33,34. mRNA levels of these genes were significantly lower in the SIRT5OE hearts after TAC. Thus, SIRT5 overexpression suppresses pressure overload-induced fibrosis signaling. These differences in mRNA expression were also reflected in tissue histology. Staining for collagen using picrosirius red showed a dramatic increase in ECM accumulation in WT TAC but not SIRT5OE TAC tissue (Fig. 6D-E). Thus, gene ontology and histologic analyses show that SIRT5 overexpression blunts TAC-induced cardiac fibrosis.
Discussion
Taken together, these data demonstrate that SIRT5 is a limiting factor in maintaining cardiac structure and function during chronic pressure overload. Supra-physiological SIRT5 expression protects against HF-associated phenotypes, in particular systolic dysfunction and fibrosis (Fig. 6F). Elevated SIRT5 levels preserved cardiac contractile function after 4 weeks of pressure overload, at which point WT mice had developed systolic dysfunction, characterized by decreased EF, ventricular dilation, remodeling and fibrosis. Consistently, RNA-seq analysis revealed that key cardiac disease-related signaling pathways, including TGFβ, IL6, Renin-Angiotensin, and NFAT, were blunted in SIRT5OE TAC mice. These data demonstrate a cardioprotective role of SIRT5 in response to stress and are consistent with several previous studies showing that SIRT5KO mice are more susceptible to damage and dysfunction after cardiac stress12,16,17,18.
At least three non-mutually exclusive models may explain how SIRT5 confers protection against the deleterious consequences of TAC-induced pressure overload. First, SIRT5-mediated regulation of specific metabolic pathways is an attractive model, as metabolic dysfunction is both a driver and a consequence of hypertrophy and HF29. A variety of studies have shown that SIRT5-dependent regulation of its targets can be highly context-dependent35. Studies conducted using SIRT5KO mice and cell culture models have indicated that SIRT5 promotes fatty acid oxidation by desuccinylating ECHA12. The heart shifts from fatty acid oxidation to increased glucose consumption upon hypertrophy and failure29. In our studies, WT TAC mice exhibited transcriptional downregulation of genes involved in fatty acid catabolism and oxidative phosphorylation, with concurrent upregulation of glycolytic genes, compared to SIRT5OE TAC mice. However, these transcriptional signatures were not reflected in changes in ETC protein expression, electron flow, or total mitochondrial content.
In an ischemia–reperfusion model, SIRT5KOs exhibited a larger infarct size, and inhibition of SDH reduced this infarct size in both WT and SIRT5KO mice16. Furthermore, succinate injection is sufficient to induce cardiomyocyte hypertrophy36. Our metabolomics analysis revealed a borderline significant elevation (p = 0.08) in succinate levels in the WT sham hearts compared to SIRT5OE sham hearts (Fig. 5B). Although SIRT5 regulates SDH activity in a cell- and context-dependent fashion8,35,37, we were unable to detect changes in SDH activity in our SIRT5OE mice. Nevertheless, overexpression of SIRT5 might protect against hypertrophy and HF through regulating levels of succinate, and/or other key metabolites.
ROS management may represent another target of SIRT5 in the context of SIRT5-mediated cardioprotection. Increased ROS production due to pathological stimuli, including cytokine signaling or mechanical stretching, can stimulate cardiomyocyte hypertrophy, ventricular dilation, and contractile dysfunction38. Rat myoblasts treated with H2O2 show decreased SIRT5 levels and induction of apoptosis; in contrast, we did not observe changes in SIRT5 transcript or protein levels after TAC (Supplemental Fig. 2A-B). In rat myoblasts, SIRT5 overexpression protects against H2O2-driven apoptosis through interactions with BCL-XL, an anti-apoptotic member of the Bcl-2 family39. In other cell types, SIRT5 reduces cellular ROS levels via desuccinylation-mediated activation of SOD1 and promotes NADPH production by desuccinylation and deglutarylation of IDH2 and G6PD, respectively35. Elevated cellular ROS induces expression of a specific set of genes to detoxify ROS, an effect mediated in part through the NRF2 transcription factor40. NRF2-mediated oxidative stress response was significantly enriched in both WT and SIRT5OE RNA-seq comparisons, but blunted in the latter group (Fig. 6A). Future studies are needed to determine whether this effect is a driver or a consequence of attenuated cardiac dysfunction in SIRT5OE mice.
Second, immune-related pathways were among the most significantly altered by SIRT5OE in the context of TAC (Fig. 3F). Early immune infiltration after TAC is necessary for the transition from ventricular hypertrophy to dilation during pressure overload41,42. Blocking this inflammatory response alleviates late LV remodeling and dysfunction, cardiac fibrosis, and T cell expansion43,44,45. Similarly, in our studies, both genotypes developed significant hypertrophy after 4 weeks of TAC, but SIRT5OE mice were protected against subsequent ventricular dilation and dysfunction (Fig. 2C-H). Although current information on SIRT5 function in the immune system is incomplete, SIRT5 might protect against TAC by dampening the pro-inflammatory response in macrophages, where SIRT5 has been shown to desuccinylate and activate PKM2, blocking LPS-driven macrophage activation46. Likewise, malonylation of GAPDH promotes TNFα translation and macrophage activation, although SIRT5’s role in this process is uncharacterized47. Consistently, single-cell RNA-seq data from TAC mice reveals that SIRT5 is expressed in macrophage and T-cell populations, in addition to cardiomyocytes and other cell types in the heart (Supplemental Fig. 6A-B)45. In this regard, a pair of studies from the Hirschey group showed that while constitutive whole-body SIRT5KO mice displayed increased mortality after TAC, cardiomyocyte-specific SIRT5KOs and controls responded to TAC similarly, raising the possibility of cardiomyocyte-non-autonomous functions of SIRT5 in response to TAC17,18.
Third, SIRT5 might directly regulate cardiac fibrosis, a process initiated and sustained through a variety of signaling and mechanical factors, including inflammatory cytokines (e.g., TGFβ, Angiotensin II, TNFα), neuroendocrine factors, and ventricular wall stretch33. These factors signal cardiac fibroblasts to activate, migrate, proliferate, and differentiate into myofibroblasts. In addition to ECM secretion and remodeling, activated fibroblasts exert protective effects through suppressing proinflammatory phenotypes and even promote the hypertrophic response through secretion of critical cytokines48,49. However, chronic myofibroblast activity inhibits oxygen and nutrient delivery, resulting in cardiomyocyte atrophy and death, ultimately leading to ventricular dilation and dysfunction32,33. The fibrotic response following TAC represents one of the most pronounced differences between WT and SIRT5OE mice. SIRT5 may play a role in regulating cytokine-mediated activation of fibrosis, in immune cells where cytokines originate, in triggering fibroblast activation, and/or in myofibroblasts themselves. Further studies are needed to determine (1) how long SIRT5 overexpression protects mice from dilation and contractile failure after TAC and (2) in which cell and tissue type(s) SIRT5 functions to confer cardioprotection.
In summary, we have shown that increased expression of the deacylase SIRT5 is sufficient to confer improved maintenance of cardiac function and decreased fibrosis during pressure overload. Sirtuin activity is amenable to enhancement by allosteric activators and NAD + precursors35. Intriguingly, we found that NADH levels and NAD+/NADH ratios were impacted by SIRT5 overexpression, those most of these differences failed to achieve statistical significance (Supplemental Fig. 4E, Supplemental Table 2)50,51. Overall, our results hint that SIRT5 might represent an attractive novel target in cardiac fibrosis, a condition that afflicts millions in the US and worldwide, but which currently lacks mechanism-based therapies.
Methods
Generation of SIRT5OE mice
Mouse ES cell clone 5-11C was derived from V6.5 ES cells52. ES cells were cultured in high-glucose Dulbecco’s minimal essential medium supplemented with 15% fetal bovine serum, 1 μM 2-mercaptoethanol, 4 mM glutamine, 50 IU of penicillin per ml, 50 ug of streptomycin per ml, and 1,000 U of recombinant leukemia inhibitory factor (ESGRO; Millipore) on mitotically inactivated feeder cells as described53. Chromosome counting indicated that 90% of twenty counted chromosome spreads contained an euploid chromosome number. ES cells were microinjected into 96 blastocysts obtained from the mating of albino mice (C57BL/6BrdCrHsd-Tyrc, Envigo). Of twenty-one pups born, twenty were ES cell-mouse chimeras and eighteen of these were male chimeras with at least 70% contribution from 5-11C ES cells, as judged by coat color contribution. Sox2-Cre (JAX 008,454) were mated with mice containing the SIRT5OE cassette to delete the loxP-STOP-loxP site to generate a global SIRT5OE transgenic mouse.
Immunoblotting
Mice were anesthetized with isoflurane and immediately sacrificed by cervical dislocation. Heart tissue samples were flash frozen, pulverized, and resuspended in Laemmli sample butter (62.5 mM Tris pH6.8, 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 1% bromophenol blue). Samples were sonicated for 30 s on ice, then spun down at 4 °C for 15 min at 15,000xg, and the supernatant was collected for protein quantification using a DC Protein Assay (Bio-Rad #5,000,112). Samples were boiled, resolved by SDS-PAGE, and transferred to PVDF membrane overnight at 4 °C using a Bio-Rad Criterion system. Membranes were stained with Ponceau S to assess protein loading, and then blocked using 5% milk in TBST (TBS containing 0.1% Tween-20) at room temperature. Primary antibodies were diluted in 5% BSA in TBST and incubated with the blot overnight at 4 °C on a rocker. Secondary incubation was performed at room temperature for 1 h using either mouse or rabbit secondary antibodies (Jackson ImmunoResearch 115–035-062 or 111–035-045) diluted 1:10,000 in 5% milk in TBST. For detection, blots were immersed in a western chemiluminescent HRP substrate (Millipore P90720) and imaged using the GE ImageQuant LAS 4000. Quantification was performed using ImageJ. Primary antibodies used in this study are listed in the Supplemental Methods.
TAC and echocardiography
Detailed TAC and echocardiography procedures are detailed in the Supplemental Methods.
Mouse Study approval
All mice were housed at the Biomedical Science Research Building (UM). All vertebrate animal experiments were approved by and performed in accordance with the regulations of the University of Michigan Institutional Animal Care & Use Committee (IACUC). All authors complied with ARRIVE guidelines and all methods are reported in accordance with ARRIVE guidelines.
Human heart tissue procurement
Ventricular myocardial tissue from patients with advanced heart failure at the University of Michigan was collected at the time of cardiac transplantation following obtaining of written informed consent. Nonfailing ventricular myocardial tissue was collected from unmatched donor hearts from the University of Michigan. Co-morbidities in the donors that may have precluded use of their hearts for cardiac transplantation included age, hypertension, diabetes, minor coronary artery disease, alcohol or tobacco use. Prior to tissue retrieval, all hearts were perfused with ice-cold cardioplegia. Samples from each heart were snap frozen in liquid N2 at the time of arrival and stored at − 80 °C. This study was approved by the Institutional Review Boards of the University of Michigan Medical School (IRBMED) and performed in accordance with all relevant guidelines and regulations.
Mitochondrial isolation
Mitochondrial isolation was performed as previously described54. In brief, mice were anesthetized with isoflurane and immediately sacrificed by cervical dislocation. After cardiac harvest, ventricles were minced in ice-cold MIB (210 mM D-Mannitol, 70 mM sucrose, 5 mM MgCl2, 10 mM KH2PO4, 5 mM HEPES, 1 mM EGTA, 0.2% fatty acid-free BSA), mechanically homogenized using a Fisher Scientific PowerGen 125, and centrifuged at 800×g then 8000×g for 10 min at 4 °C. The final pellets, containing mitochondria, were resuspended in MAS (220 mM D-Mannitol, 70 mM sucrose, 5 mM MgCl2, 10 mM KH2PO4, 2 mM HEPES, 1 mM EGTA, 0.1% fatty acid-free BSA). Mitochondria were quantified using DC quantification assy. Both MIB and MAS pH were adjusted to 7.2 with KOH and stored at 4 °C before use.
Agilent/Seahorse assay
To assess the functionality of isolated mitochondria from WT and SIRT5OE hearts, the electron flow assay was performed using the XFe96 Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA) per the manufacturer’s instructions. Specific methods used in this study are in Supplemental Methods.
Metabolomics sample preparation
The metabolomics MS/MS system, liquid chromatography, and mass spectrometry protocols used are detailed in the Supplemental Methods. Mice were anesthetized with isoflurane and immediately sacrificed by cervical dislocation. Heart tissue samples were immediately flash frozen, pulverized to powder, vortexed, and centrifuged for 10 min at 13,000 × g, 4 °C. Then, 1 ml of the supernatant was aspirated from each tube, transferred to a tightly capped sample tube, and stored at − 80 °C until analysis.
Metabolomics data analysis
The QqQ data were pre-processed with Agilent MassHunter Workstation QqQ Quantitative Analysis Software (B0700). Each metabolite abundance level in each sample was divided by the median of all abundance levels across all samples for proper comparisons, statistical analyses, and visualizations among metabolites. A total of 230 metabolites were measured. The statistical significance test was done by a two-tailed t-test with a significance threshold level of 0.05.
SDH activity assay
Succinate dehydrogenase activity was measured from flash frozen and powderized sham and TAC hearts using a kit (ab228560) according to manufacturer’s instructions. Around 20 mg of heart was diluted in assay buffer at 1:20 ratio, homogenized using a dounce homogenizer, and 2.5 mg final, diluted tissue was used in the assay.
Tissue preparation for RNA extraction
Heart tissue samples were flash frozen, pulverized, resuspended in TRIzol and homogenized using a power homogenizer (Fisher Scientific PowerGen 125). RNA was extracted using RNeasy Mini Kit (Qiagen 741–4) per the manufacturer’s instructions.
qRT-PCR
cDNA was generated from RNA using SuperScript III Reverse Transcriptase (Invitrogen 18,080,044) per the manufacturer’s instructions. TaqMan Gene Expression Assays (Applied Biosystems) for single-tube assays were used for all qRT-PCR experiments. Reactions were carried out as per the manufacturer’s methods, using TaqMan Gene Expression Assays as listed with the associated TaqMan Assay ID: Myh6 (Mm00440359_m1), Acta1 (Mm00808218_g1), Nppa (Mm01255747_g1), Myh7 (Mm00600555_m1).
RNA-sequencing bioinformatic analysis
RNA libraries were prepared using a standard Illumina protocol. Base-calling was performed on an Illumina HiSeq4000. Raw fastq files were analyzed using FastQC for quality control. Transcripts were aligned to mm ensemble cDNA release 101 using kallisto (v0.46.0) and counted using Tximport (v1.18.0)55,56,57. DESeq2 was used for calculating differential expression58. Genes with a false discovery rate (FDR) of less than 0.05 and a log2-fold change greater that |1| were designated as differentially expressed between groups. Pathway analysis was performed using Ingenuity Pathway Analysis (IPA) and gene set enrichment analysis (GSEA)27,28.
Cardiomyocyte size quantification
Sections were deparaffinized in xylene for three washes for 5 min each and rehydrated in 100% ethanol, then two 10-min washes each with 95% ethanol, followed by two 5-min washes in PBS, and staining with 10 μg/mL wheat germ agglutinin conjugated to Alexa Flour 488 (Invitrogen) diluted in PBS for one hour at room temperature. Sections were washed 3× with PBS, mounted with Prolong Gold with DAPI (Invitrogen), and imaged on Zeiss Confocal at 20× magnification. Three images of the left ventricle were taken of each cardiac section and cell surface area quantified on ImageJ from 200 cells per sample.
Fibrosis staining
Heart tissue was paraffin embedded, sectioned, and stained with picrosirius red at the University of Michigan Dentistry Histology Core. Paraffin embedded heart section were de-paraffinized in xylene, rehydrated in 100% EtOH, 95% EtOH, and 70% EtOH and dH20. Sections were then fixed in 10% buffered formalin and stained in picric acid solution with 0.1% Sirius red, dehydrated in 80% EtOH, 95% EtOH, then 100% EtOH, cleared in xylene, and finally mounted with Permount. For each heart, eight images of left ventricle sections, 500 μm × 400 μm, were acquired under polarized bright-filed light for fibrosis detection. Collagen area fraction (CAF) of the total areas, 500 μm × 400 μm, were measured by ImageJ using the equation ((collagen area)/(total area) = CAF). 4 images with the highest CAF were used for statistical analysis.
Statistics
Statistical analysis were conducted using Graphpad Prism, version 8. Multiple-group comparisons were analyzed using two-way ANOVA followed by Sidak’s correction for multiple comparisons. For analysis of two groups, Welch’s t-test was used. A p-value of less than 0.05 was considered statistically significant. Scatter plots include median and interquartile range markers. Significance markers: (*) p < 0.05, (**) p < 0.01, (***) p < 0.001, (****) p < 0.0001.
Data availability
Both the raw and analyzed data generated from this study are openly available at GEO (GSE198926).
References
Heidenreich, P. A. et al. Forecasting the impact of heart failure in the United States. Circulation Hear Fail 6, 606–619 (2013).
Virani, S. S. et al. Heart disease and stroke statistics—2021 update: a report from the american heart association. Circulation 143, e254–e743 (2021).
Miranda, D., Lewis, G. D. & Fifer, M. A. Pathophysiology of Heart Disease: A Collaborative Project of Medical Students and Faculty. (Wolters Kluwer, 2016).
Rockman, H. A. et al. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc National Acad Sci 88, 8277–8281 (1991).
Heineke, J. & Molkentin, J. D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Bio 7, 589–600 (2006).
Stein, A. B., Giblin, W., Guo, A. H. & Lombard, D. B. Introductory review on sirtuins in biology, aging, and disease. 155–173 (2018) doi:https://doi.org/10.1016/b978-0-12-813499-3.00012-5.
Du, J. et al. Sirt5 Is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 (2011).
Park, J. et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell 50, 919–930 (2013).
Rardin, M. J. et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab 18, 920–933 (2013).
Tan, M. et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab 19, 605–617 (2014).
Nishida, Y. et al. SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol Cell 59, 321–332 (2015).
Sadhukhan, S. et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc National Acad Sci 113, 4320–4325 (2016).
Lombard, D. B. et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation▿ †. Mol Cell Biol 27, 8807–8814 (2007).
Ogura, M. et al. Overexpression of SIRT5 confirms its involvement in deacetylation and activation of carbamoyl phosphate synthetase 1. Biochem Bioph Res Co 393, 73–78 (2010).
Bentley, N. L. et al. Protein hypoacylation induced by Sirt5 overexpression has minimal metabolic effect in mice. Biochem Bioph Res Co 503, 1349–1355 (2018).
Boylston, J. A. et al. Characterization of the cardiac succinylome and its role in ischemia–reperfusion injury. J Mol Cell Cardiol 88, 73–81 (2015).
Hershberger, K. A. et al. Sirtuin 5 is required for mouse survival in response to cardiac pressure overload. J Biol Chem 292, 19767–19781 (2017).
Hershberger, K. A. et al. Ablation of Sirtuin5 in the postnatal mouse heart results in protein succinylation and normal survival in response to chronic pressure overload. J Biol Chem 293, 10630–10645 (2018).
Beard, C., Hochedlinger, K., Plath, K., Wutz, A. & Jaenisch, R. Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis 44, 23–28 (2006).
Firestein, R. et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE 3, e2020 (2008).
Du, X.-J. Gender modulates cardiac phenotype development in genetically modified mice. Cardiovasc Res 63, 510–519 (2004).
Nakamura, A. et al. LV systolic performance improves with development of hypertrophy after transverse aortic constriction in mice. Am J Physiol Heart C 281, H1104–H1112 (2001).
Berthiaume, J. M. et al. Cardiovascular Pathology (Fourth Edition). 271–339 (2016) doi:https://doi.org/10.1016/b978-0-12-420219-1.00008-2.
Moore-Morris, T. et al. Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J Clin Invest 124, 2921–2934 (2014).
Frangogiannis, N. G. Cardiac fibrosis. Cardiovasc Res https://doi.org/10.1093/cvr/cvaa324 (2020).
Revelo, X. S. et al. Cardiac resident macrophages prevent fibrosis and stimulate angiogenesis. Circ Res 129, 1086–1101 (2021).
Krämer, A., Green, J., Pollard, J. & Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530 (2014).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. P Natl Acad Sci Usa 102, 15545–15550 (2005).
Bertero, E. & Maack, C. Metabolic remodelling in heart failure. Nat Rev Cardiol 15, 457–470 (2018).
Quiros, P. M., Goyal, A., Jha, P. & Auwerx, J. Analysis of mtDNA/nDNA ratio in mice. Curr Protoc Mouse Biology 7, 47–54 (2017).
Zhou, B. & Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest 128, 3716–3726 (2018).
Tallquist, M. D. & Molkentin, J. D. Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol 14, 484–491 (2017).
Gibb, A. A., Lazaropoulos, M. P. & Elrod, J. W. Myofibroblasts and fibrosis. Circ Res 127, 427–447 (2020).
Khalil, H. et al. Fibroblast-specific TGF-β–Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest 127, 3770–3783 (2017).
Kumar, S. & Lombard, D. B. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathobiology. Crit Rev Biochem Mol 53, 1–24 (2018).
Aguiar, C. J. et al. Succinate causes pathological cardiomyocyte hypertrophy through GPR91 activation. Cell Commun Signal 12, 78 (2014).
Zhang, Y. et al. Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J Biol Chem 292, 10239–10249 (2017).
Pol, A. van der, Gilst, W. H. van, Voors, A. A. & Meer, P. van der. Treating oxidative stress in heart failure: past, present and future. Eur J Heart Fail 21, 425–435 (2019).
Liu, B. et al. SIRT5: a safeguard against oxidative stress-induced apoptosis in cardiomyocytes. Cell Physiol Biochem 32, 1050–1059 (2013).
Sies, H. & Jones, D. P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Bio 21, 363–383 (2020).
Epelman, S., Liu, P. P. & Mann, D. L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol 15, 117–129 (2015).
Patel, B. et al. CCR2+ monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. Jacc Basic Transl Sci 3, 230–244 (2018).
Laroumanie, F. et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 129, 2111–2124 (2014).
Martini, E. et al. T cell costimulation blockade blunts age-related heart failure. Circ Res https://doi.org/10.1161/circresaha.119.316530 (2020).
Ren, Z. et al. Single-cell reconstruction of progression trajectory reveals intervention principles in pathological cardiac hypertrophy. Circulation 141, 1704–1719 (2020).
Wang, F. et al. SIRT5 desuccinylates and activates pyruvate kinase M2 to block macrophage IL-1β production and to prevent DSS-induced colitis in mice. Cell Rep. 19, 2331–2344 (2017).
Galván-Peña, S. et al. Malonylation of GAPDH is an inflammatory signal in macrophages. Nat Commun 10, 338 (2019).
Xiang, F.-L., Fang, M. & Yutzey, K. E. Loss of β-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat Commun 8, 712 (2017).
Russo, I. et al. Protective effects of activated myofibroblasts in the pressure-overloaded myocardium are mediated through smad-dependent activation of a matrix-preserving program. Circ Res 124, 1214–1227 (2019).
Matasic, D. S., Brenner, C. & London, B. Emerging potential benefits of modulating NAD+ metabolism in cardiovascular disease. Am J Physiol-heart C 314, H839–H852 (2018).
Yoshino, J., Baur, J. A. & Imai, S. NAD+ intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab 27, 513–528 (2017).
Pittman, D. L. et al. Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol Cell 1, 697–705 (1998).
Hughes, E. D. & Saunders, T. L. Advanced Protocols for Animal Transgenesis, An ISTT Manual. 291–325 (2011) doi:https://doi.org/10.1007/978-3-642-20792-1_14.
Rogers, G. W. et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS ONE 6, e21746 (2011).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527 (2016).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research 4, 1521 (2016).
Howe, K. L. et al. Ensembl 2021. Nucleic Acids Res 49, gkaa942- (2020).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014).
Acknowledgements
The authors would like to thank the University of Michigan Transgenic Animal Model Core for assistance with generation of the SIRT5OE mouse strain; Steven Whitesall, Kimber Converso-Baran, and Dr. Daniel Michele (Physiology and Phenotyping Core) for TAC and echo services and helpful discussions; and the Dentistry Histology Core. Dr. Heiko Bugger is also acknowledged for helpful discussions. We would also like to thank Dr. Daniel Goldstein and the other members of the Guo dissertation committee, and members of the Lombard Lab for useful feedback. Figure 2A and 6F was created with BioRender.com. This project was supported by R01GM101171, R21ES032305 and the Glenn Foundation for Medical Research (GFMR) (DBL); T32AG000114 and T32GM113900 (AHG); R25GM086262 (RKB); R37AG028730 and GFMR (DAS); R01CA248160, R01CA244931 and UMCCC Core Grant: P30CA046592 (CAL); NIHR00HL136695 (MJB); and American Heart Association Predoctoral Fellowship #825996 (RSG). ABS was funded by the University of Michigan Frankel Cardiovascular Center, the University of Michigan McKay Grant, and the Taubman Emerging Scholars Program. Metabolomics studies performed at the University of Michigan were supported by DK097153, the Charles Woodson Research Fund, and the UM Pediatric Brain Tumor Initiative. NJD was supported by the Frankel Cardiovascular Center Summer Undergraduate Fellowship Program.
Author information
Authors and Affiliations
Contributions
A.H.G., R.B., M.E.S., S.K., A.A., L.Z., R.S.G., N.J.D., M.W.M., S.M.D., M.J.B., C.A.L., A.B.S., and D.B.L. performed experiments and/or analyzed data. S.M. and D.A.S. provided the SIRT5OE mouse ES cells. A.H.G., A.B.S., and D.B.L. interpreted data. A.H.G. and D.B.L. wrote the manuscript. A.H.G. made the figures. D.B.L. and A.B.S. supervised overall design and study interpretation.
Corresponding author
Ethics declarations
Competing interests
DAS is a consultant to MetroBiotech, a company developing NAD + boosters to treat rare diseases; a complete list of DAS activities is at https://sinclair.hms.harvard.edu/david-sinclairs-affiliations. CAL has received consulting fees from Astellas Pharmaceuticals and is an inventor on patents pertaining to Kras regulated metabolic pathways, redox control pathways in pancreatic cancer, and targeting the GOT1 pathway as a therapeutic approach. DBL reports ownership of the equivalent in voting stock or share of ABBV and GILD. All other authors do not have any conflict of interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, A.H., Baliira, R., Skinner, M.E. et al. Sirtuin 5 levels are limiting in preserving cardiac function and suppressing fibrosis in response to pressure overload. Sci Rep 12, 12258 (2022). https://doi.org/10.1038/s41598-022-16506-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-16506-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
