
Abstract
Severe acute respiratory syndrome-coronavirus 2 (SARS-CoV-2), which causes coronavirus disease 2019 (COVID-19), threatens global public health. The world needs rapid development of new antivirals and vaccines to control the current pandemic and to control the spread of the variants. Among the proteins synthesized by the SARS-CoV-2 genome, main protease (Mpro also known as 3CLpro) is a primary drug target, due to its essential role in maturation of the viral polyproteins. In this study, we provide crystallographic evidence, along with some binding assay data, that three clinically approved anti hepatitis C virus drugs and two other drug-like compounds covalently bind to the Mpro Cys145 catalytic residue in the active site. Also, molecular docking studies can provide additional insight for the design of new antiviral inhibitors for SARS-CoV-2 using these drugs as lead compounds. One might consider derivatives of these lead compounds with higher affinity to the Mpro as potential COVID-19 therapeutics for further testing and possibly clinical trials.
Similar content being viewed by others
Introduction
In late December 2019, COVID-19 was reported in Hubei province, Wuhan, China, and it soon became a global pandemic affecting public health, social interactions, and economies1,2. The causative agent of COVID-19, SARS-CoV-2 is related to a group of positive-sense single-stranded RNA-containing coronaviruses which are pathogenic in vertebrates including humans3. Even though effective vaccines are now available, the need for the development of SARS-CoV-2-specific antivirals remains. The process of designing and developing new antiviral compounds can be lengthy. As an alternative, one approach is to search for existing approved drugs to repurpose. These repurposed drugs can then be minimally altered to increase their specificity to make effective SARS-CoV-2 antiviral therapeutics, thus expediting their approval for this new purpose.
The 30 kb single stranded RNA of the SARS-CoV-2 encodes four structural, sixteen non-structural (NS), and nine accessory proteins, 29 total, from fourteen open-reading frames (ORFs). Two of these ORFs, encode polyproteins (ORF1a and ORF1ab), that are proteolytically cleaved into sixteen non-structural proteins (NSP1-NSP16). The protein NSP3 is the papain-like protease (PLpro), and NSP5 is the main protease (Mpro); they are responsible for processing the polyproteins. NSP3 and NSP5 are part of a larger replicase-transcriptase complex which includes other NSPs such as NSP7 and NSP8 (primase complex), NSP12 (RNA-dependent RNA polymerase—RdRp), NSP13 (helicase-triphosphatase), NSP14 (exoribonuclease), NSP15 (endonuclease), and NSP10 and NSP16 (methyltransferases). NSP2, NSP9, and NSP11 have unknown functions. Proteins NSP4 and NSP6 make a complex with NSP34.
The structural proteins of the SARS-CoV-2 include spike (S), envelope (E), membrane (M), and nucleocapsid (N), which are involved in cell binding and fusion5, host recognition and virulence6, inhibiting interferon production7, and genome packaging8, respectively. The 3′ end of the genome expresses these four structural proteins as well as nine accessory proteins. The functions of most of the accessory proteins are unknown except for three of them, which are involved in inflammasome (Orf3a), as type I IFN antagonist (Orf6), and as suppressor of host antiviral response (Orf9b)4.
Almost all the SARS-CoV-2 proteins and their associated partner proteins, such as the type II transmembrane serine protease TMPRSS2, the adventitious partner of spike protein9, are potential targets for development of small molecule or biologic inhibitors. The first FDA-approved small molecule inhibitor for a SARS-CoV-2 protein was remdesivir, which was repurposed as an inhibitor of NSP12 (RdRp)10. Remdesivir was originally designed for combating Hepatitis C and subsequently the Ebola virus in 2014. Since NSP3 (PLpro) and NSP5 (Mpro) are part of a larger RNA polymerase, some suggest that small molecule inhibitors of NSP3 and NSP5 will have synergistic effects to inhibitors of RNA polymerase such as remdesivir11.
The importance and efficacy of the non-structural NS3/4A protease inhibitors of Hepatitis C Virus (HCV) against SARS-CoV-2 has drawn much attention in the scientific community; many agree that they are effective inhibitors of SARS-CoV-212,13. The α-ketoamide-containing covalent inhibitors are among lead candidates for binding to cysteine proteases, especially Mpro14,15,16, because the two adjacent C=O groups create an especially powerful electrophile. Boceprevir was among the first Hepatitis C Virus antiviral agents to show inhibitory activity against Mpro and coronavirus17,18,19. Based on structural similarities of the HCV NS3/4A protease and Mpro, several authors suggest that a variety of NS3/4A inhibitors would be effective against SARS-CoV-2 main protease12,13. Also, a structural analysis of binding of α-ketoamide inhibitors from experiments at room temperature suggests that a high degree of flexibility of the Mpro active site facilitates the binding of these antivirals20,21.
In this study, we attempted to obtain direct X-ray crystallographic binding evidence on six HCV NS3/4A protease inhibitors against SARS-CoV-2 main protease. Based on an initial molecular docking study on boceprevir, and a broad knowledge-based literature search, we selected boceprevir, telaprevir, narlaprevir, asunaprevir, grazoprevir, and simeprevir (Table S1) for co-crystallization trials. Also, we selected some other covalent binders (Table S2), mostly related to the family of small molecule cysteine protease and cathepsin inhibitors, for co-crystallization against Mpro. Out of the other compounds incubated with Mpro, only VBY-825 and leupeptin produced crystals suitable for X-ray crystallography.
Materials and methods
Cloning, expression, and purification of Mpro
The codon-optimized synthetic gene of full length Mpro from SARS-CoV-2 was cloned into the pET29b vector carrying a C-terminal 6 × His-Tag sequence. The plasmid encoding Mpro was transformed into competent E. Coli BL21 (DE3) cells. Multiple colonies from the transformed plate were picked and incubated in the LB media containing 100 μg/ml kanamycin overnight at 37 °C. The cells were inoculated into 500 ml of auto induction ZYM 5052 medium and grown at 37 °C to an OD600 of about 0.6. The cells were then allowed to auto-induce22 overnight at 20 °C. The cells were harvested by centrifugation at 5000 rpm for 20 min, lysed at ice-cold temperature using bacterial protein extraction agent (B-PER, Thermo Fisher Scientific) in the presence of lysozyme and benzonase. The soluble and insoluble fractions were separated by centrifugation at 16,000 rpm for 25 min. The resulting cell-free supernatant was allowed to bind for 20 min at 20 °C with Ni–NTA agarose (thermo scientific) resin that had earlier been equilibrated with buffer A (40 mM Tris, 400 mM NaCl, 10 mM imidazole, pH 8.0). This mixture was then poured into a column and the resin was washed with 50 ml binding buffer. The protein was eluted using a step gradient with increasing concentration of imidazole (50, 100 and 250 mM). Fractions of the eluate were analyzed on 4–10% SDS–PAGE gel. Further purification was achieved by a size-exclusion (Superdex increase-200) column that had previously been equilibrated with a buffer containing 40 mM Hepes pH 7.4, 2 mM TCEP, and 300 mM NaCl. The histidine tag was cleaved by human rhinovirus (HRV) 3C protease (AcroBIOSYSTEMS) and further purified by reverse nickel-affinity chromatography. The purified protein was then dialyzed overnight at 4 °C against 30 mM Hepes pH 7.4, 200 mM NaCl, 1 mM TCEP, and concentrated to ~ 7 mg/ml and used for crystallization or stored at −80 °C.
Compounds
Boceprevir, Telaprevir, Narlaprevir, Grazoprevir, Asunaprevir, Simeprevir, Leupeptin, VBY-825, Balicatib, CA-074, Calpeptin, E-64, E-64c, JPM-OEt, LY-3000328, Odanacatib, CPI (Cysteine Protease Inhibitor), Aloxistatin, Cinanserin, MDL-28170, MG-101, ONO-5334, (±)Alliin, and Z-LVG-CHN2 were purchased from MedChemExpress, LLC. (www.medchemexpress.com). DiscoveryProbe™ FDA-approved Drug Library (23 plates of 96-well, 1971 compounds) were purchased from APExBIO (https://www.apexbt.com).
Protein crystallization
Mpro crystals were grown using either hanging drop or sitting drop vapor diffusion methods by manual and robotic methods. Crystallization conditions were 22% PEG 4000, 0.1 M Hepes pH 7.0, and 3% dimethyl sulfoxide (DMSO). Boceprevir, telaprevir and narlaprevir, were dissolved in 100% molecular biology grade DMSO and they were diluted to 1 mM solution using the crystallization well solution prior to the final drop setup with a 2:1, 1:1, and 1:2 ratio of Mpro (5.7–6.7 mg/ml) and the well solution containing the inhibitors at 1 mM concentration.
The first contact between the Mpro and HCV inhibitors was the crystallization drop. Mpro-inhibitor complex plate-shaped crystals appeared three to five days after the crystallization setup. Leupeptin-containing (0.6 mM in final drop) Mpro crystals were grown using a 1:0.75 ratio of precipitant:Mpro. For the VBY-825 Mpro complex, a final drop concentration of 1 mM VBY-825 and 4% DMSO was used in a 2:1 ratio of precipitant:Mpro due to the challenges posed by insolubility issues of VBY-825 in crystallization conditions.
Also, we used a High Throughput Acoustic Droplet Ejection (HT-ADE) method to grow Mpro-telaprevir and Mpro-boceprevir crystals. Crystallization drops were set-up with 40 nL of Mpro solution and 40 nL of the well solution containing 1 mM telaprevir or boceprevir followed by 2.5 nL of seed stock generated from apo Mpro crystals grown with conventional sitting drop vapor diffusion. HT-ADE co-crystallization attempts were also made for grazoprevir (1 mM), asunaprevir (1 mM), and simeprevir (0.5 mM), however no ligand was observed in electron density maps arising from these attempts.
For completeness we considered the following additional compounds as inhibitors for Mpro co-crystallization, which did not produce diffraction quality crystals: Balicatib, CA-074, Calpeptin, E-64, E-64c, JPM-OEt, LY-3000328, Odanacatib, CPI, Aloxistatin, Cinanserin, MDL-28170, MG-101, ONO-5334, (±)Alliin, and Z-LVG-CHN2.
For the FDA-approved drug library, we used an Opentrons high-precision OT-2 laboratory robot (https://opentrons.com/ot-2) to prepare the plates by dispensing 100 µL of 100% molecular biology grade DMSO to solubilize the ligands. We attempted the Mpro crystallization using 88 selected compounds (based on higher score hits calculated as binding affinity in kcal/mol from the molecular docking as well as their chemical reactivity towards cysteine) using the high throughput ADE method. In cases where sample identity could not be confirmed, or hits were duplicated (different salts of the same compounds), or the unavailability of the compounds, the hits were omitted from the crystallization trials. The final drop contained 40 nL Mpro, 40 nL precipitant, 2.5 nL ligand and 2.5 nL seeding materials. Most of the selected compounds in this category did not produce diffraction-quality crystals or the ligand did not bind to the Mpro.
Data collection, structure determination and refinement
All data were collected at the 17-ID-1 (AMX) and 17-ID-2 (FMX) beamlines at the NSLS-II, Brookhaven National Laboratory (BNL), Upton, NY, United States. The energy of the X-ray beam was 12.66 keV (0.979 Å) at 17-ID-2 and 13.5 keV (0.920 Å) at 17-ID-1 beamline. To collect data at 100 K, we used Oxford cryosystems 800, and to capture the diffraction images we used Eiger 16 M and Eiger 9 M pixel array detectors from Dectris. Diffraction images were indexed, integrated, and scaled using XDS-based FastDP23,24. The Matthews coefficient (VM) was calculated as 2.02 Å3 Da−1, which corresponds to one monomer per asymmetric unit with an estimated solvent content of 39% (e.g., PDB entry: 7K40). A summary of the data-collection statistics is shown in Tables S3 and S4.
We used Phaser25 (2.8.3) and Dimple26 (2.5.7) for molecular replacement and ligand search. We used Refmac27 (5.8.0) as implemented in CCP428 (7.1.0) and Phenix29,30,31 (1.19.2) for refinement, and Coot32 (0.9.4) for model building. Refinement statistics and model validation values are shown in Tables S3 and S4. All molecular-graphics figures were created using PyMOL (v.2.4.1; by Schrödinger) (https://pymol.org).
Molecular docking studies
We used AutoDock Vina (version 1.0) for docking studies (https://vina.scripps.edu)33 on three HCV NS3/4A protease inhibitors against SARS-CoV-2 Mpro. We defined the minimum arguments required to run the docking program including the receptor, ligand, and search space arguments. We used a random seeding for the ligand which start the docking with a random conformation of the ligand. Exhaustiveness of the global search and number of binding modes were defined as 8 and 20, respectively. We used the following Mpro PDB entries with all the water and ligand molecules removed for the preparation of the receptors: 6WNP for boceprevir, 7C7P for telaprevir, and 6XQT for narlaprevir. We used AutoDockTools version 1.5.6 (http://mgltools.scripps.edu/) for preparing the ligand and receptor files and to define the search space. Receptor and ligand molecules were treated as rigid and as flexible, respectively.
Also, for virtual screening using high throughput molecular docking , we used a modified script (File S1 and File S2) to employ AutoDock Vina on small molecule databases. The original shell script is available on the AutoDock Vina website (https://vina.scripps.edu). We used ZINC15, a free database of commercially available compounds for virtual screening, for building our small molecule databases (https://zinc15.docking.org/)34. We compiled three databases: “fda” subset with 1426 compounds, “in-trials” subset with 6848 compounds, and “in-vitro” subset with 161,758 compounds. These databases were used for molecular docking against some of the SARS-CoV-2 targets. We used both personal computers and the National Synchrotron Light Source II High Performance Computing (NSLS-II HPC) AMX and FMX nodes for virtual screening of Mpro against the “fda” subset which contains a list of FDA-approved drugs. The name of the compounds, their crystallization status with the Mpro and some of the docking results are included in Tables S5 and S6, respectively.
Microscale thermophoresis binding assays
Thermophoretic assays were carried out using a Microscale Thermophoresis Monolith NT.115 apparatus (NanoTemperTechnologies). We fluorescently labeled the target protein Mpro by coupling of Mpro lysine residues to N-hydroxysuccinimide of the dye NT647 (NanoTemper Technologies). We incubated Mpro and NT647 dye on ice in darkness for 30 min and separated fluorescently labeled Mpro from free dye by size-exclusion chromatography using a buffer composed of 30 mM sodium phosphate pH 8.0, 200 mM NaCl, 1 mM DTT. Labelling efficiency of Mpro was verified prior to performing the binding test and equilibrium dissociation constant (Kd) determination. For the initial binding test, 200 nM fluorescently labeled Mpro was incubated with 50 μM of ligands (telaprevir and narlaprevir) for 15 min prior to detection. For Kd determination, 200 nM fluorescently labeled Mpro was incubated with serial dilution of telaprevir or narlaprevir for 15 min, before loading of approximately 5 μL of the samples into capillaries. Thermophoretic measurements were performed using 40% MST power and 80% LED power at 25 °C.
Results
Mpro and HCV protease inhibitor complexes
Three Hepatitis C virus NS3/4A α-ketoamide protease inhibitors with a similar peptidomimetic scaffold (boceprevir, telaprevir, and narlaprevir) (Fig. S1A-C) can bind to the SARS-CoV-2 Mpro active site (Fig. 1). Electron densities show a high occupancy by these inhibitors, with very good shape complementarity to the Mpro active site. The six functional groups of the inhibitors (P1′, P1-P5) occupy the Mpro active subsites as they are available; boceprevir lacks the P5 functional group and its P1′ does not have a cyclopropyl substitution. The P1′ amide of all these inhibitors binds to the S1′ active subsite of the Mpro and their ketone group undergoes a nucleophilic attack by Cys145 of the enzyme to make a hemithioketal covalent linkage.

Active subsites of the Mpro binding pocket (S1′–S5), ligand functional groups or positions (P1′–P5), and hydrogen bonding interactions of the HCV NS3/4A inhibitors with Mpro. (A) Electron density of the boceprevir complex at 1.35 Å resolution (PDB entry: 7K40). Boceprevir lacks the P1′ and P5 functional groups and therefore the associated enzyme subsites are not occupied. (B) Hydrogen bonding interactions of boceprevir complex. One structural water molecule facilitates the binding of the amine group of the α-ketoamide moiety. (C) electron density of the telaprevir complex at 1.48 Å resolution (PDB entry: 7K6D). (D) Hydrogen bonding interactions of the telaprevir complex. One structural water molecule facilitates the binding of the pyrazine (P5) moiety. (E) Electron density of the narlaprevir complex at 1.79 Å resolution (PDB entry: 7JYC). (F) Hydrogen bonding interactions of the narlaprevir complex. Ligands are shown as ball-and-stick representation. Electron densities (2Fo–Fc, 1 rmsd) are shown as purple mesh. All distances are in Å. (C) reproduced with permission of the International Union of Crystallography, Acta Cryst. (2021), A77, C194 (https://doi.org/10.1107/S0108767321094885).
In all three inhibitors, the hemithioketal oxygen makes a strong and short distanced hydrogen bond (Low-Barrier H-Bond: LBHB35) to the His41 as follows: 2.51 Å in boceprevir, 2.49 Å in telaprevir, 2.45 Å in narlaprevir. Our observations are consistent with the longer hydrophobic substitutions at P1′ and P1 being able to render an electron-donation propensity which facilitates short hydrogen bond formation. However, it is in contrast with other detailed studies others reported20 (2.4 Å in boceprevir, 2.5 Å in telaprevir, 2.8 Å in narlaprevir). We speculate that the difference could be attributed to the data-collection temperature (100 K in this study vs. the previous study at 293 K) or different protonation states of the hemithioketal oxygen and His41 side chain.
The hydrogen bonding patterns (Fig. 1) of these peptidomimetic inhibitors with Mpro are similar, and depend in part on the presence of their specific functional groups (P1′, P1–P5). Some noticeable differences in the H-bonding patterns exist including involvement of a structural water for binding of the amide group of boceprevir (P1′) to the Thr26 main chain, the interaction of telaprevir’s P4 and P5 carbonyls with Gln189 and Gln192 side chain and main chain (via H2O), respectively, and no water-mediated interaction for narlaprevir binding. Telaprevir is the only inhibitor that interacts with Gln189.
Microscale thermophoresis binding assays
The equilibrium dissociation constants (Kd) between Mpro-telaprevir (23 ± 4 μM) (Fig. 2A) and Mpro-narlaprevir (12 ± 3 μM) (Fig. 2B) were determined by thermophoretic experiments in which the drugs were titrated against fluorescently labeled Mpro. Our measured dissociation-constant values are consistent with the reported IC50, EC50, or binding constants within the margin of error i.e., in the low µM binding range for these inhibitors (Table 1)13,17,20,36,37,38. For boceprevir, binding experiments were not feasible due to the poor solubility of the ligand.

The equilibrium dissociation constants between (A) Mpro-telaprevir (23 ± 4 μM) and (B) Mpro-narlaprevir (12 ± 3 μM) were determined by thermophoretic experiments titrated against fluorescently labeled Mpro. Dissociation curves were fit to the data to calculate the Kds. Data shown are the mean ± SD, n = 3. For boceprevir, the binding experiment was not successful due to the precipitation of the ligand in the assay buffer (30 mM sodium phosphate pH 8.0, 200 mM NaCl, 1 mM DTT). The precipitation led to turbidity and a non-uniform fluorescent signal.
Mpro and other inhibitor complexes
In the case of the VBY-825 molecule (Fig. 3A–C), which has the same α-ketoamide functional group as the other three HCV inhibitors, the binding pattern of the ketone is like that of the HCV inhibitors. However, the shape complementarity of the rest of the molecule, in terms of binding to the active site as well as the occupancy level, seemed sub-optimal. We speculate that the geometry of the molecule and its low solubility under the crystallization conditions may contribute to the partial occupancy at the active site.

Active subsites of the Mpro binding pocket (S1′–S5), ligand functional groups (P1, P1′, P1′′, & P2), and hydrogen bonding interactions of the novel covalent inhibitor VBY-825 with Mpro. Two different conformations of VBY-825 are shown. (A) Partial electron density of the VBY-825 Mpro complex at 1.70 Å resolution and 0.5 rmsd (PDB entry: 7MNG). VBY-825 lacks the P3–P5 functional groups and therefore the associated enzyme subsites are not occupied. Binding of a DMSO molecule to the S1 site forces the VBY-825 to bind in a tilted conformation to Cys145 (B) Hydrogen bonding interactions of VBY-825 Mpro complex shown for conformer A. Structural water molecules may facilitates the binding of the highly polarized fluorine groups of the trifluoromethyl moiety. A charge-assisted H-bond (CHAB)35 between H and F is possible, albeit not optimal. (C) A detailed 2D LigPlot+53 diagram of all the molecular interactions between conformer A of the VBY-825 and Mpro is shown. Hydrogen bonds are shown as green dotted lines. Spoked arcs represent nonbonded contacts such as hydrophobic interactions. The solid purple line between Cys145 and VBY-825 represents a covalent binding. (D) An overlay (0.28 Å r.m.s.d.) of the apo (black) (PDB: 7K3T) and VBY-825 bound (yellow) (PDB: 7MNG) Mpro. The comparative changes in Cα distances are shown as green dotted lines with values. Binding of the VBY-825 displaces the P2 helix by 1.1 Å (red arrows) and predominantly displaces the side chains of Gln189 and Met165. However, the conformational changes are less significant in comparison with other HCV NS3/4A inhibitor-Mpro complexes20.
In the complex with VBY-825, the S3-S5 binding sub-sites of Mpro are vacant and its cyclopropylmethanesulfonyl moiety (P1′′) binds close to the S1′ sub-site. The bulky trifluoroethyl 4-fluorophenyl seems to be very mobile and binds with a low occupancy to the S2 subsite in two different conformations. The solvent DMSO, required to solubilize the VBY-825 ligand is at a concentration of 4% in the crystallization drop. DMSO binds to the S1 sub-site, forcing the VBY-825 covalent bond to Cys145 into a slightly sub-optimal geometry, and the short ethyl group (P1) of VBY-825 cannot fully displace the DMSO molecule (Fig. 3A).
As shown in Fig. 4, the aldehyde group of the microbial peptide leupeptin stereo-specifically reacts with Cys145 to form a hemithioacetal as an S enantiomer and is hydrogen bonded to the main chain nitrogen of the Cys145. This observation is consistent with room temperature studies reported20. New in this study is the finding that the arginine moiety of the leupeptin seems to make a weak hydrogen bond with the Glu166 side chain. Also, two structural waters facilitate the binding of leupeptin to the Gln189 and Asn142 side chains (Fig. 4B). Leupeptin makes three additional hydrogen bonds to the Mpro main chain.

Active subsites of the Mpro binding pocket (S1′–S5), ligand functional groups (P1–P4), and hydrogen bonding interactions of the covalent inhibitor leupeptin with Mpro. (A) electron density of the leupeptin Mpro complex at 2.3 Å resolution and 0.5 rmsd (PDB entry: 7MRR). Leupeptin lacks the P5 functional group and therefore the associated enzyme subsite is not occupied. (B) hydrogen bonding interactions of leupeptin Mpro complex shown. Two structural water molecules shown facilitate the binding of leupeptin.
Molecular docking studies
Molecular docking is often used to study and predict the binding modes of a ligand to a receptor molecule and is widely used for drug discovery. Even though the binding mode observed by X-ray crystallography is often among the most favored modes of binding, the most favorable binding-energy mode may not necessarily be observed in X-ray crystallographic studies12.
Our molecular docking studies show that AutoDock Vina could predict binding modes of the HCV NS3/4A inhibitors to Mpro that accurately reflect published X-ray crystal structures by others and in this study (Fig. S2). Establishing the accuracy of the modeled binding modes and the calculation of the binding affinities are necessary for comparing them with the newly designed derivative such as L551. The affinity of the binding was calculated as − 7.7, − 6.7, and − 8.3 kcal/mol for boceprevir, telaprevir, and narlaprevir, respectively. Since AutoDock Vina does not simulate the covalent binding of these inhibitors to their target, there is a slight coordinate shift between the X-ray structures and the docking binding modes (poses). The shift is more prominent near the Cys145 where covalent binding to α-ketoamide occurs. Also, the calculations of the binding affinity in these cases do not consider the affinity of covalent binding.
Discussion
We have investigated the interaction between drugs already approved for treatment of hepatitis C with the main protease of SARS-CoV-2, and observe their binding in X-ray crystal structures. The existence of these drugs and others suggests a short path to creation of effective curative new antivirals that can help quell the current pandemic.
Mpro-inhibitor complexes
A major component in all the HCV NS3/NS4 inhibitor drugs (Table S1) is a pyrrolidine/proline-like ring moiety (a cyclopentane in case of simeprevir) which acts as a molecular scaffold for three to four other substitutions. X-ray crystallographic structures of the known complexes of these antiviral agents with Mpro (Table S1) show that the stereochemistry of covalent binding to the active site Cys145 usually places this ring in the S2 binding pocket of the Mpro near the hydrophobic residues of Met165 and Met49. Examination of the S2 binding pocket reveals that it can only accommodate a maximum combined substitution of three to four carbon or equivalent atoms at both positions 3 and 4 of the pyrrolidine ring. Boceprevir, telaprevir and narlaprevir are the only compounds that have small enough ring substitutions to fit into the S2 binding pocket of Mpro.
During our studies, we attempted to co-crystallize Mpro with grazoprevir, asunaprevir, and simeprevir as selected inhibitors without any success. The reason these compounds were not found to bind to the active site was apparently the presence of a large substitution at position 3 or 4 of the pyrrolidine ring. With similar reasoning we anticipated that no Mpro complex will be observed using any other HCV NS3/NS4 inhibitors that have similar large substitutions in position 3 or 4 (see Table S1); therefore, we omitted this class of compounds from our crystallographic studies. Molecular docking studies on some of these untested compounds may seem to indicate a binding to the main protease active site11. Based on our observations, the in-silico predictions do not necessarily translate into observable binding in X-ray crystallographic studies. However, possibly removal of the large substitutions at positions 3 or 4 of the pyrrolidine ring on these untested compounds may show binding. The same reasoning might be true for variants of these untested compounds with a different stereochemistry which avoids positioning the larger substitution into the S2 binding pocket.
VBY-825 is a powerful cathepsin-specific (B, L, S, and V cathepsins) cysteine protease inhibitor39 with potent anti-tumor activity. Binding of VBY-825 to Mpro is particularly interesting due to its ability to inhibit cathepsins, making it a dual action inhibitor since host cell cathepsins are involved in SARS-CoV cell entry40,41. There is no information available on whether VBY-825 can inhibit furin42 and TMPRSS2 which are other host cell proteases facilitating SARS-CoV-2 cell entry41. VBY-825 binding to Mpro is mainly due to the highly electrophilic α-ketoamide functional group, resembling the other HCV NS3/4A inhibitors, and to the hydrophobic interactions (fluorophenyl moiety). Also, binding of VBY-825 to Mpro induces some conformational changes around the active site even though it does not occupy the S3-S5 subsites. However, the induced conformational changes at S3-S5 seem to be minimal (e.g., P4 β-hairpin flap20). The P5 loop seems to be relatively mobile and the bulky fluorinated functional groups of VBY-825 exert a movement of 1.1 Å on the P2 helix (Fig. 3D). The r.m.s.d. between the apo (PDB: 7K3T) and VBY-825 bound (PDB: 7MNG) Mpro structures is 0.28 Å.
Leupeptin43 (a natural microbial peptide and protease inhibitor) forms a S-hemithioacetal with Cys145 through its aldehyde group. The PDB entry 7NEV44 for the leupeptin-Mpro complex shows two binding modes (R and S) in terms of the stereochemistry while 7MRR (this study) shows the S-enantiomer binding mode consistent with the reported room temperature studies (PDB entry 6XCH)20. As described in detail in room temperature studies20, the binding of leupeptin resembles the metastable tetrahedral intermediate of a protease, which eventually leads to the production of acyl intermediates.
Significance of the Mpro inhibitors to human health
Successful introduction of the effective SARS-CoV-2 vaccines and antibodies for treatment of COVID-19 patients, does not necessarily reduce the significance and the urgency of developing small-molecule inhibitor drugs, because the virus continually mutates45. The likelihood of the mutations is high for structural proteins such as spike protein. Certain mutations in the spike protein may reduce the effectiveness of the vaccines and antibodies as the mutations occur46.
Non-structural proteins on the other hand, are less prone to mutations. A mutation in the NSP5 or Mpro active site may subsequently require multiple concerted mutations in its substrates. The likelihood of multiple concerted Mpro mutations accumulating fast enough to produce a resistant virus is much lower than mutations of a structural proteins such as the spike protein. For this reason, the inhibitors of the Mpro, will likely be a more stable and effective solution in the long run. The same reasoning is true for other NSPs such as NSP3 or PLpro. Also, small molecule inhibitors are a better choice than vaccines for infected individuals and may work synergistically with neutralizing antibodies and remdesivir.
The rationale behind new drug design
A detailed analysis of the Mpro active subsites in terms of their hydrophilicity in relation to the functional groups of the inhibitors14,15,20 suggests that an improvement in binding is possible. The P1 functional group occupying the S1 subsite is the most noticeable group for a change. The P1 chemical group in all the three HCV inhibitors is a cyclic/linear hydrophobic chain which occupies a fully hydrophilic S1 subsite. Conceivably, replacing the current P1 groups with a functional group capable of making a hydrogen bond with both side chains of the His163 and Glu166 will make these inhibitors more specific for a more efficient binding to SARS-CoV-2 Mpro. Numerous PDB structures (e.g., 7JT747, 7LYH, 7LYI, 7CB748) suggest that a pyrrolidone functional group, among others, is one of the best fits in binding to the S1 subsite. This change may significantly improve the binding of these inhibitors without significantly changing their ADME49 (Absorption, Distribution, Metabolism, and Excretion) properties and safety margins as FDA-approved drugs.
This approach would shorten safety testing and evaluation times, helping to fulfil the urgent need for new antiviral drugs. We proposed that three compounds, with a design based on the HCV NS3/4A inhibitors (L551, L737, and L751) (Fig. S1D-F) may bind more tightly to the Mpro. While we were evaluating the viability of chemical synthesis and subsequent testing of these three new compounds for x-ray crystallography and binding studies, an experimental new drug by Pfizer (PF-07321332) (Fig. S1G) entered clinical trial50,51,52 (PDB IDs: 7SI9, 7VH8, 7RFS, 7RFW). The design of PF-07321332 was based on PF-00835231 (structurally similar to GC-37618,54); however, the final new drug is ultimately a pyrrolidone derivative of a boceprevir-like compound with a nitrile as an active functional group instead of α-ketoamide, which corroborates our hypothesis that pyrrolidone derivatives of HCV NS3/4A inhibitors are potentially good lead compounds for the design of new Mpro inhibitors. Also, the recently published structures of the Mpro with BBH-1, BBH-2, and NBH-2 shows that this approach can succeed55.
The Mpro S2 subsite is hydrophobic, and all the three inhibitors seem to be able to use this site for efficient binding using their hydrophobic P2 groups. The S3 and S5 binding subsites are shallow, and they lie at the surface of the Mpro. Telaprevir can efficiently use both subsites, especially S5, by making a hydrogen bond to Gln189. The S4 subsite is amphiphilic, and therefore an amphiphilic P4 functional group might be a better choice for improving the binding. Also, a detailed recent study55 shows the importance of the protonation state of the His41 and the keto-warhead, as well as the role of oxyanion hole (comprised of Gly143, Ser144, and Cys145 main chain), in determining the binding affinity of the various inhibitors.
Comparison with other similar ligands
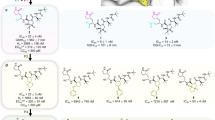
As shown in Fig. 5, we selected three ligands closely related to boceprevir (nirmatrelvir, BBH-2, and L551) to compare their structure and geometry of binding to the Mpro. The main scaffold of these peptidomimetic inhibitors as well as the dimethyl-bicyclo[3.1.0] proline moiety is the same or very similar. The first major difference between boceprevir and Pfizer’s nirmatrelvir (PF-07321332)51 (Fig. 5A) and BBH-255 (Fig. 5B) is the functional group (the warhead) that covalently binds to the Cys145 in the active site. The reactive functional groups of boceprevir and nirmatrelvir are ketoamide and nitrile, respectively. The second major difference is the presence of a γ-lactam 5-membered ring (pyrrolidone-like) as P1 chemical group which significantly increases the binding affinity by making additional hydrogen bonds51,55. The P1 group of boceprevir (a cyclobutyl moiety) cannot form any hydrogen bonds. Nirmatrelvir (PF-07321332) is the active ingredient of the orally bioavailable drug Paxlovid™ by Pfizer which is specifically approved for emergency use authorization (EUA) by the Food and Drug Administration (FDA) for the treatment of COVID-19 patients. The Ki and Kd of nirmatrelvir for main protease are 3 nM51 and 7 nM55, respectively, which are largely driven by the favorable enthalpy55 of the reaction with the main protease. The Kd value of the BBH-2/Mpro complex is 30 nM as reported55 which is comparable to (but less than) the values reported for nirmatrelvir.

Comparison of the binding modes of four closely related ligands to the Mpro. (A) Comparison of boceprevir (PDB: 7K40—this study) and nirmatrelvir51 (PDB: 7RFW) binding modes to the Mpro. The nitrile warhead of the nirmatrelvir can make a 3.0 Å hydrogen bond to the Gly143 (shown in black dotted line) (B) comparison of boceprevir (PDB: 7K40) and BBH-255 (PDB: 7TEH) binding modes to the Mpro. (C) comparison of the designed L551 with the molecular docking binding mode as shown (Fig. S2D—this study) and the binding mode of BBH-2 (PDB: 7TEH) to the Mpro. The chemical structures are shown in their binding form. Structures were drawn using ChemDraw 14 Professional from PerkinElmer (https://perkinelmerinformatics.com/products/research/chemdraw).
Molecular docking suggests that the designed L551 inhibitor (this study) may bind to the main protease with the same geometry as BBH-2 (Fig. 5C). The only difference between L551 and BBH-2 is their reactive warhead group which are ketoamide and nitrile, respectively. As shown in Fig. 1A, boceprevir’s α-ketoamide warhead can make a total of at least three hydrogen bonds as shown: one with His41 (with correct protonation states), one with Thr26 via H2O, and the third one using its amide carbonyl which is within an easy hydrogen bond-making distance with all the N–H groups of the main chain of the oxyanion hole (Gly143-Ser144-Cys145 moieties). In case of the nitrile warhead (nirmatrelvir and BBH-2), it can make only one hydrogen bond to the oxyanion hole55. Since L551 has the same reactive functional group as boceprevir (ketoamide), we anticipate that the binding affinity of the L551 to the Mpro could be higher than, or at least within the same range as, that of the BBH-2/Mpro complex. Also, we anticipate that the affinity of a CF3-substituted derivative of L551 (L546) (Fig. S1J) maybe within the same range as the affinity of the nirmatrelvir to the Mpro, if not better.
The importance of one additional hydrogen bond in increasing the affinity of a ligand to the Mpro is reported for the compound PF-00835231 (Fig. S1K) from Pfizer51, which has an inhibition constant (Ki) of 0.27 nM for the main protease. The reactive warhead group of this compound can make at least one hydrogen bond with the oxyanion hole and one additional hydrogen bond with His41. The same compound with the nitrile warhead (Fig. S1L) can make at least one hydrogen bond only to the oxyanion hole (like nirmatrelvir – Fig. 5A) which reduces its Ki for the Mpro by 100-fold to 28 nM51.
Conclusion
With the SARS-CoV-2 continuously mutating, the need for the design of novel Mpro inhibitors to battle the current and future COVID-19 outbreaks is more than ever. The original HCV protease inhibitors (boceprevir, telaprevir, and narlaprevir) are drugs already approved by FDA and others, and their pharmacokinetics (Table S7) suggest that an inhibitory human blood plasma concentration (Cmax) against Mpro can be reached especially for boceprevir and narlaprevir. Other studies have suggested that these original HCV inhibitors could be used in drug repurposing strategies in combination with other inhibitors38. However, their lower affinity to the Mpro may not justify their use in clinical trials against COVID-19 in terms of economic viability of the treatment options. Therefore, one might consider derivatives51,55 of these HCV inhibitors with a higher affinity to the Mpro for clinical trials as potential COVID-19 therapeutics. Together with broad vaccination campaigns, such treatment options may help significantly to curtail the morbidity and mortality of the current pandemic.
Data availability
All the atomic coordinates and their associated structure factors have been deposited to the Protein Data Bank (PDB) under the accession/entry codes of 7K6D, 7K6E, 7K40, 7K3T, 7JYC, 7MNG, and 7MRR.
References
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Adachi, S. et al. Commentary: origin and evolution of pathogenic coronaviruses. Front. Immunol. 11, 811 (2020).
Gordon, D. E. et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468 (2020).
Huang, Y. et al. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 41, 1141–1149 (2020).
Chai, J. et al. Structural basis for SARS-CoV-2 envelope protein recognition of human cell junction protein PALS1. Nat. Comm. 12, 3433 (2021).
Zheng, Y. et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct. Target. Ther. 5, 299 (2020).
Cubuk, J. et al. The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat. Comm. 12, 1936 (2021).
Hu, X. et al. Non-covalent TMPRSS2 inhibitors identified from viral screening. doi:https://doi.org/10.1101/2020.12.28.424413.
Eastman, R. T. et al. Remdesivir: a review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Cent. Sci. 6, 672–683 (2020).
Bafna, K. et al. Hepatitis C Virus drugs simeprevir and grazoprevir synergize with remdesivir to suppress SARS-CoV-2 replication in cell culture. doi:https://doi.org/10.1101/2020.12.13.422511.
Bafna, K. et al. Structural similarity of SARS-CoV2 Mpro and HCV NS3/4A proteases suggests new approaches for identifying existing drugs useful as COVID-19 therapeutics. https://doi.org/10.26434/chemrxiv.12153615.v1 (2020).
Anson, B. J. et al. Broad-spectrum inhibition of coronavirus main and papain-like proteases by HCV drugs. https://doi.org/10.21203/rs.3.rs-26344/v1 (2020).
Dai, W. et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science https://doi.org/10.1126/science.abb4489 (2020).
Zhang, L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science https://doi.org/10.1126/science.abb3405 (2020).
Banerjee, S. An insight into the interaction between α-ketoamide- based inhibitor and coronavirus main protease: a detailed in silico study. Biophys. Chem. 269, 106510 (2021).
Fu, L. et al. Both boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. https://doi.org/10.1038/s41467-020-18233-x.
Ma, C. et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 30, 678–692 (2020).
Oerlemans, R. et al. Repurposing the HCV NS3-4A protease drug boceprevir as COVID-19 therapeutics. RSC Med. Chem. 12, 370–379 (2021).
Kneller, D. W. et al. Malleability of the SARS-CoV-2 3CL Mpro active-site cavity facilitates binding of clinical antivirals. Structure 28, 1313–1320 (2020).
Ali, E. et al. The temperature-dependent conformational ensemble of SARS-CoV-2 main protease (Mpro). bioRxiv, https://doi.org/10.1101/2021.05.03.437411.
Studier, F. W. Protein Expression Purif., 41, pp. 207–234 (2005).
Kabsch, W. XDS. Acta Cryst., D66, 125–132 (2010).
Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Cryst. D66, 133–144 (2010).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007).
Wojdyr, M. DIMPLE: a pipeline for the rapid generation of difference maps from protein crystals with putatively bound ligands. Acta Cryst. A69, s299 (2013).
Murshudov, G. N. et al. Refmac5 for the refinement of macromolecular crystal structures. Acta Cryst. D67, 355–367 (2011).
Winn, M. D. Overview of CCP4 suite and current developments. Acta Cryst. D67, 235–242 (2011).
Grosse-Kunstleve, R. W. et al. The computational crystallography toolbox: crystallographic algorithms in a reusable software framework. J. Appl. Cryst. 35, 126–136 (2002).
Adams, P. D. et al. PHENIX: building new software for automated crystallographic structure determination. Acta Cryst. D58, 1948–1954 (2002).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Cryst. D75, 861–877 (2019).
Emsley, P. et al. Features and development of coot. Acta Cryst. D66, 486–501 (2010).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 31, 455–461 (2010).
Sterling, T. & Irwin, J. J. ZINC 15: ligand discovery for everyone. J. Chem. Inf. Model. 55, 2324–2337 (2015).
Gilli, G. & Gilli, P. Towards an unified hydrogen-bond theory. J. Mol. Struc. 552, 1–15 (2000).
Jin, Z. et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293 (2020).
Riva, L. et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 586, 113–119 (2020).
Baker, J. D. et al. A drug repurposing screen identifies hepatitis C antivirals as inhibitors of the SARS-CoV2 main protease. PLoS ONE https://doi.org/10.1371/journal.pone.0245962 (2021).
Tina Elie, B. et al. Identification and pre-clinical testing of a reversible cathepsin protease inhibitor reveals anti-tumor efficacy in a pancreatic cancer model. Biochimie 92, 1618–1624 (2010).
Simmons, G. et al. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. PNAS 102, 11876–11881 (2005).
Chung, M. K. et al. COVID-19 and cardiovascular disease. Circ. Res. 128, 1214–1236 (2021).
Wu, C. et al., Furin: A potential therapeutic target for COVID-19. iScience 23, 101642 (2020).
Aoyagi, T., et al. Biological activities of leupeptins. J. Antibiotics, 558–568 (1969).
Günther, S. et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 372, 642–646 (2021).
Korber, B. et al. Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182, 812–827 (2020).
Kemp, S. A. et al. Neutralising antibodies drive spike mediated SARS-CoV-2 evasion. https://doi.org/10.1101/2020.12.05.20241927.
Iketani, S. et al. Lead compounds for the development of SARS-CoV-2 3CL protease inhibitors. Nat. Commun. 12, 2016 (2021).
Wang, Y. et al. Structural basis of SARS-CoV-2 main protease inhibition by a broad-spectrum anti-coronaviral drug. Am. J. Cancer Res. 10(8), 2535–2545 (2020).
Daina, A. et al. SwissADME: a free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Halford, B. Pfizer unveils its oral SARS-CoV-2 inhibitor. c&en 99 (13), (April 7, 2021). https://cen.acs.org/acs-news/acs-meeting-news/Pfizer-unveils-oral-SARS-CoV/99/i13 (2021).
Owen, D. R. et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science https://doi.org/10.1126/science.abl4784 (2021).
Zhao, Y. et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell https://doi.org/10.1007/s13238-021-00883-2 (2021).
Laskowski, R. A. & Swindells, M. B. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 (2011).
Vuong, W. et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 11(4282), 1–8 (2020).
Kneller, D. W. et al. Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat. Commun. 13, 2268 (2022).
Acknowledgements
We would like to thank Herbert Bernstein, Dean Hidas, and Hubertus Van Dam, for their help with molecular docking studies. J.K. and J.S. were supported by BES grant (DOE 456 KC0304000). This research was supported by the DOE Office of Science through the National Virtual Biotechnology Laboratory (NVBL) with funding from the Coronavirus CARES Act and additional funding was provided by Brookhaven National Laboratory (BNL) for research on COVID-19 (LDRD 20-042). We received beamline-support donation from Pfizer & Co. This research used 17-ID-1 and 17-ID-2 beamlines of the National Synchrotron Light Source II; a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The Center for BioMolecular Structure (CBMS) is primarily supported by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS) through a Center Core P30 Grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1605010).
Author information
Authors and Affiliations
Contributions
B.A., D.F.K., and J.J. performed molecular docking studies. D.K. expressed and purified the Mpro. B.A., D.K., D.F.K., E.O.L., and A.S.S. crystallized the Mpro. B.A., D.F.K., E.O.L., and A.S.S. prepared the drug libraries. B.A., D.K., D.F.K., A.S.S., J.J., W.S. and M.R.F. prepared the beamlines, collected, and processed the data. B.A., D.K., D.F.K., A.S.S., W.S., and J.J. determined, refined, and deposited the structures. J.K., and J.S. performed binding assay measurements. B.A. designed new inhibitors with M.S.H., J.G.S. and S.M. as collaborators. J.S., P.D.A., M.S.H., and S.M. oversighted all the associated reduction to practices and provided critical comments and editing. R.M.S., J.S., P.D.A., M.S.H., and S.M. secured funding through grants, awards, and donations. B.A., D.K., and S.M. conceived the experiment. B.A., D.K., and J.K. wrote the initial draft of the manuscript with input from all the other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Andi, B., Kumaran, D., Kreitler, D.F. et al. Hepatitis C virus NS3/4A inhibitors and other drug-like compounds as covalent binders of SARS-CoV-2 main protease. Sci Rep 12, 12197 (2022). https://doi.org/10.1038/s41598-022-15930-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-15930-z
This article is cited by
-
Electrochemical synthesis of peptide aldehydes via C‒N bond cleavage of cyclic amines
Nature Communications (2024)
-
Machine learning combines atomistic simulations to predict SARS-CoV-2 Mpro inhibitors from natural compounds
Molecular Diversity (2024)
-
In-silico study: docking simulation and molecular dynamics of peptidomimetic fullerene-based derivatives against SARS-CoV-2 Mpro
3 Biotech (2023)
-
Exploring potential SARS-CoV-2 Mpro non-covalent inhibitors through docking, pharmacophore profile matching, molecular dynamic simulation, and MM-GBSA
Journal of Molecular Modeling (2023)
-
Structural and functional characterization of NEMO cleavage by SARS-CoV-2 3CLpro
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
