
Abstract
X-linked Alport syndrome is a genetic kidney disease caused by pathogenic COL4A5 variants, but little is known of the consequences of missense variants affecting the NC1 domain of the corresponding collagen IV α5 chain. This study examined these variants in a normal (gnomAD) and other databases (LOVD, Clin Var and 100,000 Genomes Project) to determine their pathogenicity and clinical significance. Males with Cys substitutions in the collagen IV α5 NC1 domain reported in LOVD (n = 25) were examined for typical Alport features, including age at kidney failure. All NC1 variants in LOVD (n = 86) were then assessed for structural damage using an online computational tool, Missense3D. Variants in the ClinVar, gnomAD and 100,000 Genomes Project databases were also examined for structural effects. Predicted damage associated with NC1 substitutions was then correlated with the level of conservation of the affected residues. Cys substitutions in males were associated with the typical features of X-linked Alport syndrome, with a median age at kidney failure of 31 years. NC1 substitutions predicted to cause structural damage were overrepresented in LOVD (p < 0.001), and those affecting Cys residues or ‘buried’ Gly residues were more common than expected (both p < 0.001). Most NC1 substitutions in gnomAD (88%) were predicted to be structurally-neutral. Substitutions affecting conserved residues resulted in more structural damage than those affecting non-conserved residues (p < 0.001). Many pathogenic missense variants affecting the collagen IV α5 NC1 domain have their effect through molecular structural damage and 3D modelling is a useful tool in their assessment.
Similar content being viewed by others


Introduction
Alport syndrome is the commonest genetic kidney disease and results from pathogenic variants in COL4A3, COL4A4 or COL4A51,2. These genes encode the collagen IV α3, α4 and α5 chains that trimerise to form the collagenous network found in the basement membranes of the kidney, ear and eye3.
X-linked Alport syndrome results from pathogenic variants in COL4A5 and is the cause of severe disease. Males typically develop kidney failure before the age of 30, together with hearing loss and ocular abnormalities4,5. Females have a more variable phenotype and are usually less severely affected, with up to 20% developing kidney failure by the age of 606,7. Autosomal recessive Alport syndrome results from pathogenic variants in both copies of COL4A3 or COL4A42,8, resulting in a phenotype similar to X-linked disease in males9. Autosomal dominant Alport syndrome, formerly known as ‘thin basement membrane nephropathy’, results from heterozygous pathogenic variants in COL4A3 or COL4A410. Most affected individuals have only haematuria, some develop late-onset kidney failure, but hearing loss and ocular features are very rare11,12.
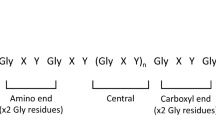
Each collagen IV α-chain comprises three domains: the non-collagenous 7S domain at the amino terminus, the intermediate collagenous (Gly-X–Y) domain, and the non-collagenous NC1 domain at the carboxy terminus3. The mature collagen IV α-chains, unlike the fibril-forming collagens, retain the two non-collagenous termini, which have important structural and functional roles13. The NC1 domains direct chain stoichiometry and trimerisation14,15, and interactions between the termini on neighbouring trimers are responsible for network formation within the basement membranes16. Post-translational modifications and ligand-binding sites have also been identified within the non-collagenous domains3,17.
The six collagen IV NC1 domains are highly conserved. Each comprises two homologous subdomains and is stabilised through six intrachain disulphide bonds16. During trimer formation, the three NC1 domains interact through a domain swapping mechanism where a β-hairpin motif from each chain is exchanged with a docking region (VR3) on the adjacent chain13,14. Following trimerisation, the NC1 domains of two identical trimers interact end-to-end to form a hexamer, which is stabilised by six covalent sulphilimine cross-links and extensive hydrophobic and hydrophilic interactions16,18,19,20. These hexamers have an important structural function within the collagenous network, and are a focal point of bioactivity within the glomerular basement membrane21.
Missense variants affecting the NC1 domains are reported increasingly, but their pathogenicity and clinical significance are often difficult to determine. In the collagen IV α5 chain, many of these variants have been associated with later onset kidney failure in affected males, but the phenotypes and extrarenal features vary22,23,24,25. Cys substitutions are the commonest type, probably because of the structural importance of disulphide bonds for folding of the NC1 domain. The 12 NC1 Cys residues are conserved in all six collagen IV α-chains, and considered ‘critical domains’26. Little is known of other mechanisms underlying disease caused by NC1 missense variants, but conformational changes that interfere with trimer assembly or function are likely to be important15,20.
This study determined the clinical and molecular implications of missense variants affecting the NC1 domain of the collagen IV α5 chain. Firstly, the expected phenotype for Cys substitutions was established and compared with previously-published data for collagenous Gly substitutions in the same molecule27. A structural modelling tool was then used to identify additional variants that resulted in further damaging change. Finally, the importance of conserved residues in the NC1 domain was examined.
Methods
Reference sequences
COL4A5 variants were described at the genomic and protein levels using the following NCBI reference sequences: NM_033380.3, NP_203699.1. Isoform 2 was used, since this is the predominant isoform expressed in the kidney. The NC1 domain was defined as beginning at the first residue immediately following the final Gly-X–Y of the collagenous domain, consistent with the literature and ending at the final residue before the stop codon (Supplemental Table 1). This is different from Uniprot which defines the NC1 domain as beginning at the first secondary structure (beta-strand) following the collagenous domain, meaning that 5 or so residues in each chain are not included in the collagenous or NC1 domain. A further difference is that Uniprot reports positions based on isoform 1.
Variant databases
Variants were primarily examined using the Leiden Open Variation Database (LOVD; https://www.lovd.nl)28. This is an open source database comprising variants, often with associated demographic and clinical data from mainly individuals with X-linked Alport syndrome, which have been extracted from the literature or submitted directly by collaborating laboratories. It now includes more than 2,090 unique COL4A5 variants. Variant pathogenicity was assessed by the submitting laboratory, or where this was not provided, using the American College of Medical Genetics (ACMG) criteria as assessed by VarSome (https://varsome.com).
Three further datasets that included COL4A5 variants were used for comparative analysis with LOVD:
-
Genomics England 100,000 Genomes Project (100kGP)—this database comprises hospital records and genomic data from individuals and families with various diseases, including some with X-linked Alport syndrome or other genetic kidney diseases. Genomic data were extracted from the aggregate multi-sample VCF (aggV2), comprising 78,195 germline genomes from the 100kGP (http://genomicsengland.co.uk; version 10 data release)29.
-
ClinVar—this database contains variants, phenotypes and pathogenicity assessments submitted by testing laboratories, and includes more than 550 Pathogenic or Likely Pathogenic COL4A5 variants (https://www.ncbi.nlm.nih.gov/clinvar; accessed 21 November 2021).
-
Genome Aggregation Database (gnomAD)—this database includes exomic and genomic data from cases and controls recruited for various studies of cardiac, psychiatric and other diseases, which have been combined to represent a cross-section of the general population. Individuals have not been selected for kidney disease, and the database is not expected to be enriched for rare pathogenic COL4A5 variants (https://gnomad.broadinstitute.org; v2.1.1; accessed 15 November 2021)30.
IRB approval
Use of the 100kGP data and publication of this manuscript were approved by the Genomics England Research Ethics Committee. This study did not require Institutional Review Board permission because all other datasets (LOVD, ClinVar, and gnomAD) were de-identified and already in the public domain. All variants in these datasets were from individuals who, at recruitment, had provided written informed consent to testing, the submission of variants to the database, and the use of these variants in research. Testing and submission in each case was performed with Institutional Review Board approval and according to the principles of the Declaration of Helsinki. LOVD also included clinical data associated with variants where this was found in the primary published manuscripts, where Institutional Review Board approval was provided in each case.
Computational tools
Predicted splicing changes
Exonic nucleotide substitutions in COL4A5 close to canonical splice sites sometimes affect normal splicing, especially when the final nucleotide of an exon is affected31,32,33. To ensure that variants with unreported splicing effects were not included unintentionally, all variants within 3 bases of a canonical splice site were assessed using MaxEntScan34. Mutant and wild type sequences were scored using the maximum entropy model, and variants were considered likely to affect splicing where the mutant score was at least 15% lower than the wild type score35. Variants predicted to affect normal splicing were then excluded from the subsequent analyses (Supplemental Table 2).
Predicted structural changes
Structurally-damaging variants were identified using Missense3D (http://missense3d.bc.ic.ac.uk)36,37. This tool uses three-dimensional structural information from experimentally-determined protein models to predict the consequences of amino acid substitutions. Sixteen different structural features were examined (buried charge introduced, buried charge replaced, buried charge switch, etc.), but the ‘cis Pro replaced’ feature was not included in this study because there are no cis Pro residues in the collagen IV α5 NC1 domain. Variants were analysed using the experimentally-determined structure of the collagen IV α5 chain (UniProt ID: P29400, PDB code: 5NAZ).
The sensitivity and specificity of this tool were examined using the data in ClinVar, where the damage prediction for each missense variant affecting the collagen IV α5 NC1 was compared with the reported pathogenicity. Variants of uncertain significance (VUS) and variants with conflicting interpretations of pathogenicity were excluded. Forty-three unique NC1 missense variants were identified in ClinVar, of which 11 were reported as ‘Pathogenic’ or ‘Likely Pathogenic’, and 8 as ‘Benign’ or ‘Likely Benign’. Missense3D correctly classified 15 of the 19 (79%) variants, with a sensitivity of 0.64 and specificity of 1.00.
Cys substitutions
Individuals with COL4A5 variants resulting in Cys substitutions in the NC1 domain were identified in LOVD. Clinical data were extracted for all affected males, and survival analysis performed to determine the median age at kidney failure. This was compared with the median age at kidney failure in males with non-Cys substitutions in the NC1 domain identified in LOVD, and with previously-published data for males with collagen IV α5 Gly substitutions in the collagenous domain27.
Age at kidney failure was defined as the age at clinical diagnosis of kidney failure, or where this was not available, the age at commencing dialysis, or at the first kidney transplant. Each family was included once only. Where multiple males from the same family developed kidney failure, the mean age (or median age, where only this was available) at kidney failure was used. Where a range of ages was reported, the midpoint was used. Where an affected male had not yet developed kidney failure, his age at the most recent review was included as a censored data point. Males with multiple pathogenic variants in COL4A3-COL4A5, or families with only affected females were not included.
Cys substitutions reported in the ClinVar, gnomAD and 100kGP databases were also examined.
Other structurally-damaging variants
Predicted structural changes were obtained for missense variants affecting the NC1 domain reported in LOVD. The observed proportion of variants with each structural change was then compared with the theoretical expected frequencies to determine whether any changes were overrepresented.
The expected proportions of variants with each structural change were calculated using a neighbour-dependent nucleotide substitution rate model38. This model was chosen since it takes into account the higher rate of substitutions observed for transitions than transversions, as well as the effect of adjacent nucleotides. In brief, all theoretically-possible nucleotide substitutions resulting in missense variants affecting the collagen IV α5 NC1 domain were computed and assessed for structural damage (Supplemental Fig. 1). The relative frequency of each variant was then determined using the relative substitution rates set out by the model, and the total expected proportion of variants causing each structural change calculated.
This analysis was repeated for all collagen IV α5 missense variants affecting the NC1 domain found in 100kGP and gnomAD.
Variants affecting conserved residues
The final part of this study examined whether substitutions affecting conserved residues in the collagen IV α5 NC1 domain were more likely to result in structural damage than those affecting non-conserved residues. This analysis used the cohort of all theoretically possible collagen IV α5 NC1 missense variants described above, but only included unique amino acid substitutions for each residue without regard to the underlying DNA change (Supplemental Fig. 1).
The level of conservation for each residue was determined from multiple sequence alignment of the reference sequences for the six human collagen IV α-chains (Supplemental Table 1) using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). Residues were ‘conserved’ where the same amino acid was present in each of the 6 chains.
Finally, variants affecting conserved residues in LOVD were examined in isolation, and the comparison of observed and expected frequencies of damaging variants performed above was repeated. The expected frequencies were recalculated for this purpose, considering only conserved residues. Variants affecting conserved residues in gnomAD were also examined separately.
Statistical analysis
Categorical data were represented in contingency tables and compared using Fisher’s exact test. Observed and expected frequencies were compared using the exact binomial test.
A p-value less than 0.05 was considered significant, and a Bonferroni correction accounting for 15 tests was applied for comparisons of the individual structural changes. All statistical analysis was performed in R (version 3.6.2) and used the survival and survminer packages39,40,41. Survival curves were produced using the Kaplan–Meier method and compared using the log-rank test.
Significance statement
X-linked Alport syndrome is a genetic kidney disease resulting from pathogenic COL4A5 variants. Missense changes affecting the NC1 domain of the corresponding collagen IV α5 chain are identified increasingly, but their pathogenicity and clinical significance are often uncertain. This study established the expected phenotype for Cys substitutions (the most common NC1 variant), and identified additional likely pathogenic variants using a new 3D structural modelling tool. Predicted structurally-damaging variants were more likely to affect conserved residues and were overrepresented in a pathogenic variant database. Cys substitutions were associated with the typical features of Alport syndrome, and most males with NC1 variants developed later-onset kidney failure. 3D structural modelling represents a useful tool for assessing the pathogenicity of variants in the NC1 domain.
Results
Cys substitutions
Twenty-nine unique Cys substitutions in 49 families were identified in LOVD (Table 1). These variants affected all 12 Cys residues in the collagen IV α5 NC1 domain, with substitutions resulting in all 6 possible amino acids.
Cys substitutions were present in 25 families each with at least one affected male, 7 families with only affected females, and 17 families where the sex of the affected individuals was not reported. In the families with affected males, 13 (of the 18 with clinical data, 72%) had at least one male with kidney failure. The median age at kidney failure was 31 years (n = 13) (Table 2). This was not different from the median age at kidney failure for males with other non-Cys substitutions in the NC1 domain (34.5 years, n = 22, p = 0.23), or males with Gly substitutions in the collagenous domain (26 years, n = 157, p = 0.77) (Table 2, Fig. 1). Eleven of these families (11/15, 73%) had at least one male with a hearing loss, and 3 (3/8, 38%) with ocular changes.

Proportion of cases without kidney failure for males with collagen IV α5 missense variants reported in LOVD. Cys missense variants in the NC1 domain were compared with (a) other missense variants in the NC1 domain (p = 0.23) and (b) Gly missense variants in the Gly-X–Y collagenous domain (p = 0.77). Censored data points are not shown.
Only one Cys substitution (p.Cys1527Ser) was found in gnomAD (Table 1). This was reported in a heterozygous female. The same variant was reported in LOVD in a 40 year old male with proteinuria, stage 3 chronic kidney disease and hearing loss, and also in a female (no age recorded) with haematuria and renal insufficiency.
Twenty-one unique Cys substitutions were found in ClinVar, affecting 10 of the 12 Cys residues (Table 1). Sixteen of these substitutions were also present in LOVD.
No Cys substitution was identified in the 100kGP database.
Other structurally-damaging variants
Eighty-six unique missense variants affecting the NC1 domain were found in LOVD and included in this analysis (Figs. 2, 3). Forty-six (53%) were predicted to cause a structurally-damaging change, which was greater than the expected frequency of 19% (p < 0.001) (Table 3a). Three specific structural changes were found more often than expected. These were ‘disulphide breakage’, ‘clash’ and ‘buried Gly replaced’ (all p < 0.001).

Variant inclusion flowchart for the LOVD study cohort. Counts refer to the number of unique missense variants.

Amino acid sequence of the collagen IV α5 NC1 domain. Substitutions found in LOVD are indicated above the sequence. Substitutions predicted to cause structural damage are in bold, and substitutions where no structural data was available are in italics. Cys residues are indicated with a caret (^), and buried Gly residues with an asterisk (*).
Disulphide breakage from Cys substitutions is a known disease-causing mechanism, and as such, Cys substitutions are reported by testing laboratories as Likely Pathogenic. Interestingly, even after Cys substitutions were excluded to ensure that they did not bias the results, predicted structurally-damaging variants were still found more often than expected in LOVD (p = 0.004) (Table 3b). However, ‘buried Gly replaced’ was the only feature that remained overrepresented in this database (p < 0.001).
Forty unique NC1 missense variants were found in the 100kGP database. Of the 38 variants with corresponding structural data, only 5 (13%) were predicted to be damaging. This was not different from the expected frequency of 19% (p = 0.42). These variants were found in 6 heterozygous females, including one with haematuria, and another with haematuria and proteinuria, but none had evidence of impaired renal function. No male with a predicted structurally-damaging NC1 missense variant was identified in this database.
Seventy-one unique NC1 missense variants were reported in gnomAD. Only 8 of 68 (12%) variants with available structural data were predicted to be structurally-damaging, which was again not different from the expected frequency of 19% (p = 0.16). Most (6/8, 75%) structurally-damaging variants were found once only, and none was found more than 3 times.
Buried Gly variants
The collagen IV α5 NC1 domain includes 4 buried Gly residues, each of which is conserved in all six human collagen IV α-chains (Table 4). Buried Gly residues were identified using Missense3D. The α5 chain also includes 13 non-buried Gly residues, but none of the variants affecting these residues in LOVD or ClinVar was reported as pathogenic.
Seven unique missense variants affecting buried Gly residues were found in 21 families in LOVD, and all involved Gly1492 or Gly1602 (Fig. 4). They included 10 families with at least one affected male, 6 families with only affected females, and 4 families where the sex of the affected individual was not reported. One additional male demonstrated a mosaic pattern of expression for a buried Gly variant and was excluded.

(a,b) Space-filling models showing the location of the two commonly affected buried Gly residues (shown in green) within the collagen IV α5 NC1 domain. Models were produced using UniProt (https://www.uniprot.org/). (c,d) Ribbon diagrams showing the effect of two substitutions affecting these buried Gly residues (shown in blue). In both cases the affected residue is located within a β-strand, where the larger side-chain of the substituting residue (shown in red) encroaches upon the neighbouring secondary structure. Diagrams were produced using JSmol, accessed through Missense3D (http://missense3d.bc.ic.ac.uk).
Clinical data were rarely reported for these variants. Of the few males with clinical data, only 1 of 6 (17%) had kidney failure. This was less often than described above for Cys substitutions (p = 0.05). Four of 5 (80%) males also had a hearing loss, but none of three had ocular abnormalities.
Many of the same buried Gly variants found in LOVD were also reported in ClinVar (Table 4). Interestingly, ClinVar also included a pathogenic variant affecting the Gly1510 residue (p.Gly1510Arg), which was likely to affect splicing (Supplemental Table 2).
Only one buried Gly variant (p.Gly1624Cys) was found in the 100kGP database. This was in a heterozygous female with kidney stones and unexplained hearing loss, but no evidence of impaired kidney function. A different variant affecting the same residue (p.Gly1624Ala) was found in gnomAD, and also in a heterozygous female. This was the only buried Gly variant found in gnomAD. Neither variant was present in LOVD or ClinVar.
Variants affecting conserved residues
The collagen IV α5 NC1 domain comprises 231 residues, 105 (45%) of which are fully conserved in all six human collagen IV α-chains (Supplemental Fig. 2). Considering all possible exonic single-nucleotide substitutions, 1,345 unique missense variants are possible, of which structural data were available for 1,316 (Supplemental Fig. 1). Variants affecting conserved residues were more likely to result in a structurally-damaging change compared with variants affecting non-conserved residues (p < 0.001) (Supplemental Table 3a). This association persisted even after excluding all Cys variants (p < 0.001) (Supplemental Table 3b).
Sixty-three (63/86, 73%) unique collagen IV α5 NC1 missense variants in LOVD affected a conserved residue. To determine whether the conservation status of a residue could be used as a proxy for whether a variant affecting that residue were structurally-damaging, these 63 variants were examined in isolation. The expected frequencies of structurally-damaging variants were also recalculated using only conserved residues. Interestingly, even when considering only conserved residues, predicted structurally-damaging variants were still found more often in LOVD (44/63, 70%) than the new expected frequency of 32% (p < 0.001). This increase persisted even after excluding all Cys variants (p = 0.006). In contrast, of the 22 unique collagen IV α5 NC1 missense variants affecting a conserved residue in gnomAD, only 4 (18%) were predicted to cause structural damage. This was a lower proportion than found in LOVD (p < 0.001). These results suggest that structural modelling provides useful information about a variant’s likely pathogenicity that the level of conservation alone does not fully capture.
Discussion
In X-linked Alport syndrome, missense variants affecting the NC1 domain of the collagen IV α5 chain are recognised increasingly but little has been known of their pathogenicity or clinical consequences. This study used a new computational tool to predict the structurally-damaging changes associated with missense variants reported in four variant databases.
Cys residues in the NC1 domain of the collagen IV α5 chain are highly conserved and have been recognised recently as ‘critical domains’ in assessing pathogenicity26. Variants resulting in Cys substitutions were the most common pathogenic change in the NC1 domain and rare in the general population. In the collagen IV α5 chain, substitutions have been reported for all 12 NC1 Cys residues and were associated with all the typical clinical features of Alport syndrome. The median age at kidney failure for males was more than 30 years, but not different from that for other NC1 substitutions, nor for collagenous domain Gly substitutions27. However, the median age at kidney failure for both NC1 groups suggest that variants in this region cause later-onset disease, and the lack of a difference with the Gly substitutions may have been due to the small cohort.
Missense variants predicted to cause structural damage to the NC1 domain were overrepresented in LOVD, suggesting that this was a major cause of pathogenicity. Three types of damage were more common than expected: disulphide breakages, clashes, and substitutions of buried Gly residues. Interestingly, many Cys substitutions flagged the ‘clash’ feature in addition to the expected ‘disulphide breakage’, possibly because disulphide bonds necessarily bring two segments of the chain into close proximity. Thus clashes were not more common after Cys variants had been excluded. In contrast, only a small number of variants in gnomAD were predicted to cause structural damage, consistent with structurally-neutral variants being more common in the general population. Interestingly, about 12% of the NC1 variants in gnomAD were predicted to be structurally-damaging, which was similar to the frequency of structurally-damaging variants reported for non-disease associated variants in the original Missense3D study (11%)36.
Missense variants affecting a buried Gly residue were common in LOVD, and all involved Gly1492 or Gly1602. These two residues are located in analogous positions in the two homologous NC1 subdomains, so that they may have an important structural role. However, only one of the affected males had kidney failure, suggesting that they may be associated with milder disease. Gly substitutions affecting the Gly-X–Y collagenous domain are the most common missense variants reported in Alport syndrome26. These Gly residues are located within the core of the triple helix, where their small size is essential for close packing of the chains42. Substitution with any other amino acid destabilises heterotrimer formation, resulting in disease43. Buried Gly variants in the NC1 domain may have their effect through a similar mechanism, where substitutions with larger amino acids destabilise the molecule or impair folding of the NC1 domain.
Missense variants affecting conserved residues were more likely to result in structural damage than those affecting non-conserved residues. This suggests an important structural function for conserved residues in the NC1 domain of the α5 chain, and considering the homology between α-chains similar findings are to be expected for the α3 and α4 chains. Interestingly, a greater proportion of variants affecting conserved residues in LOVD were predicted to be structurally-damaging than those affecting conserved residues in gnomAD. This highlights the importance of using structural data in addition to conservation status when assessing a variant’s likely pathogenicity. Residues that are fully conserved in all six α-chains are likely to be critical for the NC1 domain structure. However, many chain-specific residues may also be important for functions such as chain recognition during trimerization13,14.
Many in silico pathogenicity prediction tools return only a binary outcome for variants such as ‘benign' or ‘deleterious'. While this is useful for identifying likely pathogenicity it provides little explanation of the mechanism by which the amino acid substitution affects protein structure. Missense3D addresses this issue by examining sixteen specific structural changes that have previously been associated with disease-causing variants36. Using this tool we have identified buried Gly residues in the NC1 domain that represent novel mutational ‘hotspots’ in Alport syndrome. Missense3D also allows for the analysis of both experimentally-determined protein structures as well as predicted structures. This means that Missense3D represents a valuable addition to the tools available for variant assessment.
This study examined the molecular and clinical implications of missense variants affecting the NC1 domain of the collagen IV α5 chain. It characterised the expected phenotype of Cys substitutions, evaluated a new computational tool for assessing pathogenicity, and identified additional likely pathogenic variants. However, variant classification in the NC1 domain remains challenging, and other molecular factors such as the effect on ligand binding sites may also be important17. Further studies will confirm whether the present study's findings are also applicable to NC1 variants in the collagen IV α3 and α4 chains, and the implications for autosomal dominant and recessive Alport syndrome. Structural modelling is one of many tools now available for assessing variants in at least X-linked Alport syndrome.
Data availability
The public datasets analysed in the present study are freely available in the LOVD (https://databases.lovd.nl/shared/genes/COL4A5), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and gnomAD (https://gnomad.broadinstitute.org/) repositories. Data from the 100,000 Genomes Project is available to authorised users for approved projects through the Genomics England Research Environment (http://genomicsengland.co.uk).
References
Barker, D. F. et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248, 1224–1227 (1990).
Mochizuki, T. et al. Identification of mutations in the α3(IV) and α4(IV) collagen genes in autosomal recessive Alport syndrome. Nat. Genet. 8, 77–82 (1994).
Khoshnoodi, J., Pedchenko, V. & Hudson, B. G. Mammalian collagen IV. Microsc. Res. Tech. 71, 357–370 (2008).
Jais, J. P. et al. X-linked Alport syndrome: Natural history in 195 families and genotype-phenotype correlations in males. J. Am. Soc. Nephrol. 11, 649–657 (2000).
Bekheirnia, M. R. et al. Genotype–phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 21, 876–883 (2010).
Jais, J. P. et al. X-linked Alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: A “European Community Alport Syndrome Concerted Action” study. J. Am. Soc. Nephrol. 14, 2603–2610 (2003).
Rheault, M. N. Women and Alport syndrome. Pediatr. Nephrol. 27, 41–46 (2012).
Lemmink, H. H. et al. Mutations in the type IV collagen α3 (COL4A3) gene in autosomal recessive Alport syndrome. Hum. Mol. Genet. 3, 1269–1273 (1994).
Storey, H., Savige, J., Sivakumar, V., Abbs, S. & Flinter, F. A. COL4A3/COL4A4 mutations and features in individuals with autosomal recessive Alport syndrome. J. Am. Soc. Nephrol. 24, 1945–1954 (2013).
Savige, J. et al. Thin basement membrane nephropathy. Kidney Int. 64, 1169–1178 (2003).
Matthaiou, A., Poulli, T. & Deltas, C. Prevalence of clinical, pathological and molecular features of glomerular basement membrane nephropathy caused by COL4A3 or COL4A4 mutations: A systematic review. Clin. Kidney J. 13, 1025–1036 (2020).
Furlano, M. et al. Clinical and genetic features of autosomal dominant Alport syndrome: A cohort study. Am. J. Kidney Dis. 78, 560–570 (2021).
Khoshnoodi, J., Cartailler, J.-P., Alvares, K., Veis, A. & Hudson, B. G. Molecular recognition in the assembly of collagens: Terminal noncollagenous domains are key recognition modules in the formation of triple helical protomers. J. Biol. Chem. 281, 38117–38121 (2006).
Khoshnoodi, J. et al. Mechanism of chain selection in the assembly of collagen IV: A prominent role for the α2 chain. J. Biol. Chem. 281, 6058–6069 (2006).
Pedchenko, V. et al. Collagen IVα345 dysfunction in glomerular basement membrane diseases. III. A functional framework for α345 hexamer assembly. J. Biol. Chem. 296, 100592 (2021).
Sundaramoorthy, M., Meiyappan, M., Todd, P. & Hudson, B. G. Crystal structure of NC1 domains: Structural basis for type IV collagen assembly in basement membranes. J. Biol. Chem. 277, 31142–31153 (2002).
Parkin, J. D. et al. Mapping structural landmarks, ligand binding sites, and missense mutations to the collagen IV heterotrimers predicts major functional domains, novel interactions, and variation in phenotypes in inherited diseases affecting basement membranes. Hum. Mutat. 32, 127–143 (2011).
Vanacore, R. M. et al. A role for collagen IV cross-links in conferring immune privilege to the Goodpasture autoantigen: Structural basis for the crypticity of B cell epitopes. J. Biol. Chem. 283, 22737–22748 (2008).
Vanacore, R. et al. A sulfilimine bond identified in collagen IV. Science 325, 1230–1234 (2009).
Boudko, S. P. et al. Collagen IVα345 dysfunction in glomerular basement membrane diseases. II. Crystal structure of the α345 hexamer. J. Biol. Chem. 296, 100591 (2021).
Pokidysheva, E. N. et al. Collagen IVα345 dysfunction in glomerular basement membrane diseases. I. Discovery of a COL4A3 variant in familial Goodpasture’s and Alport diseases. J. Biol. Chem. 296, 100590 (2021).
Pont-Kingdon, G. et al. Molecular testing for adult type Alport syndrome. BMC Nephrol. 10, 1–6 (2009).
Barker, D. F. et al. A mutation causing Alport syndrome with tardive hearing loss is common in the western United States. Am. J. Hum. Genet. 58, 1157–1165 (1996).
Barker, D. F., Denison, J. C., Atkin, C. L. & Gregory, M. C. Common ancestry of three Ashkenazi-American families with Alport syndrome and COL4A5 R1677Q. Hum. Genet. 99, 681–684 (1997).
Wilson, J. C., Yoon, H.-S., Walker, R. J. & Eccles, M. R. A novel Cys1638Tyr NC1 domain substitution in α5(IV) collagen causes Alport syndrome with late onset renal failure without hearing loss or eye abnormalities. Nephrol. Dial. Transplant. 22, 1338–1346 (2007).
Savige, J. et al. Consensus Statement on Standards and Guidelines for the Molecular Diagnostics of Alport Syndrome: Refining the ACMG Criteria
Gibson, J. T. et al. Genotype-phenotype correlations for COL4A3-COL4A5 variants that result in Gly substitutions in Alport syndrome. Sci. Rep. 12, 1–3 (2022).
Fokkema, I. F. A. C. et al. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 32, 557–563 (2011).
Genomics England: The National Genomics Research and Healthcare Knowledgebase v5. (2019).
Karczewski, K. J. et al. Genome Aggregation Database Consortium, Neale, BM, Daly, MJ, MacArthur, DG: The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Nozu, K. et al. X-linked Alport syndrome caused by splicing mutations in COL4A5. Clin. J. Am. Soc. Nephrol. 9, 1958–1964 (2014).
Horinouchi, T. et al. Detection of splicing abnormalities and genotype-phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 29, 2244–2254 (2018).
Aoto, Y. et al. Last nucleotide substitutions of COL4A5 exons cause aberrant splicing. Kidney Int. Rep. 7, 108–116 (2021).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394 (2004).
Houdayer, C. et al. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum. Mutat. 33, 1228–1238 (2012).
Ittisoponpisan, S. et al. Can predicted protein 3D structures provide reliable insights into whether missense variants are disease associated?. J. Mol. Biol. 431, 2197–2212 (2019).
Khanna, T., Hanna, G., Sternberg, M. J. E. & David, A. Missense 3D-DB web catalogue: An atom-based analysis and repository of 4M human protein-coding genetic variants. Hum. Genet. 140, 805–812 (2021).
Hess, S. T., Blake, J. D. & Blake, R. D. Wide variations in neighbor-dependent substitution rates. J. Mol. Biol. 236, 1022–1033 (1994).
Therneau, T. M. & Grambsch, P. M. Modeling survival data: Extending the Cox model (Springer, 2000).
Therneau, T. A. Package for survival analysis in R. R package version 3.2–11. https://CRAN.R-project.org/package=survival (2021).
Kassambara, A., Kosinski, M. & Biecek, P. Survminer: Drawing survival curves using 'ggplot2'. R package version 0.4.9. https://CRAN.R-project.org/package=survminer (2021).
Bella, J., Eaton, M., Brodsky, B. & Berman, H. M. Crystal and molecular structure of a collagen-like peptide at 1.9 Å resolution. Science 266, 75–81 (1994).
Persikov, A. V. et al. Stability related bias in residues replacing glycines within the collagen triple helix (Gly-Xaa-Yaa) in inherited connective tissue disorders. Hum. Mutat. 24, 330–337 (2004).
King, K., Flinter, F. A. & Green, P. M. A two-tier approach to mutation detection in the COL4A5 gene for Alport syndrome. Hum. Mutat. 27, 1061–1061 (2006).
Acknowledgements
The authors would like to thank the Leiden Open Variation Database (LOVD), the Genome Aggregation Database (gnomAD), ClinVar, VarSome, and the many groups that contribute to and maintain these resources. We particularly acknowledge the Imperial College London Structural Bioinformatics Group, whose in-house pipeline, Missense3D, formed the basis of this study. This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project is funded by the National Institute for Health Research and National Health Service England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the NHS as part of their care and support. Prof Daniel P Gale is the contact person for the work that involved the 100,000 Genomes Project data at d.gale@ucl.ac.uk. Authors of this manuscript who were also Consortium members were Omid Sadeghi-Alavijeh, Daniel P Gale and Judy Savige. Other authors are indicated in the Supplementary file.
Author information
Authors and Affiliations
Consortia
Contributions
J.T.G.: Study design, methodology, data acquisition, analysis, visualisation, writing—original draft. O.S.-A.: Methodology, data acquisition, writing—review & editing. D.P.G.: Writing—review & editing. H.R.: Data acquisition, writing—review & editing. J.S.: Study design, methodology, data acquisition, writing—original draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gibson, J.T., Sadeghi-Alavijeh, O., Gale, D.P. et al. Pathogenicity of missense variants affecting the collagen IV α5 carboxy non-collagenous domain in X-linked Alport syndrome. Sci Rep 12, 11257 (2022). https://doi.org/10.1038/s41598-022-14928-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-14928-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
