
Abstract
Purulent vulvar discharges, primarily caused by genito-urinary tract infections, are an important source of economic loss for swine producers due to sow culling and mortality. However, the agents that compose the vaginal microbiota of sows and their changes during infections are not well understood. The first goal of this study was to characterize and compare the vaginal bacterial content of healthy (HE, n = 40) and purulent vulvar discharge sows (VD, n = 270) by a culture-dependent method and MALDI-TOF MS identification. Secondly, we performed 16S rRNA targeted metagenomic approach (n = 72) to compare the vaginal microbiota between these groups. We found a wide variety of bacteria, with Proteobacteria, Firmicutes, and Bacteroidota being the most abundant phyla in both groups, as well as Escherichia-Shigella, Streptococcus, and Bacteroides at the genus level. Most agents identified in the sequencing method also grew in the culture-dependent method, showing the viability of these bacteria. Alpha diversity did not differ between HE and VD sows, regarding sample richness and diversity, but a beta-diversity index showed a different microbiota composition between these groups in two tested herds. ANCOM analysis revealed that Bacteroides pyogenes were more abundant in VD females and can be a marker for this group. Other agents also require attention, such as the Streptococcus dysgalactiae and Staphylococcus hyicus found in remarkably greater relative abundance in VD sows. Network analysis revealed important positive correlations between some potentially pathogenic genera, such as between Escherichia-Shigella, Trueperella, Streptococcus, Corynebacterium, and Prevotella, which did not occur in healthy sows. We conclude that the alteration of the vaginal microbiota between healthy and purulent vulvar discharge sows, although not extreme, could be due to the increase in the relative abundance of specific agents and to associations between potentially pathogenic bacteria.
Similar content being viewed by others

Introduction
Purulent vulvar discharges caused by bacterial infections are a compelling cause of the decreased reproductive performance of sows1,2,3,4. Metritis and endometritis are the most common cause for these purulent discharges1 and are an important source of sow culling and mortality, which is increasing worldwide2. Infectious agents causing these conditions have been identified by traditional methods that rely on bacterial culture and isolation3,4,5,6, but few studies have characterized the microbiota in the vaginal canal of sows by high-throughput sequencing methods7,8,9,10, and less so compared between healthy sows and those with vulvar discharge11,12.
Previous studies show that the vaginal microbiota can play a significant role as a biological barrier against disease, due to its constitution and secretion of antimicrobial components13,14,15. Thus, a better understanding of the bacterial vaginal composition in healthy and diseased sows has the potential to mitigate urogenital infections with more specific treatments and better prevention strategies, improving sow welfare and reducing economic losses.
Wang et al.11 characterized the vaginal microbiota of sows with and without endometritis and found higher abundances of Escherichia-Shigella, Bacteroides, Fusobacterium, and Clostridium_sensu_stricto_1 genera in diseased than in healthy sows, suggesting that the microbiota of these groups could be different. Zhang et al.12 investigated the effect of intestinal microbiota on vaginal bacterial composition in sows and indicated that some genera such as Escherichia-Shigella and Bacteroides may be linked to the onset of endometritis. However, Wang et al.11 did not perform a differential abundance analysis test and both studies evaluated a small number of females (n = 8) and should be of limited value. Our study hypothesizes that the vaginal microbiota of sows with and without vulvar discharge may present differences in bacterial composition. Therefore, we evaluated 310 vaginal samples by culture and MALDI-TOF (Matrix-Assisted Laser Desorption Ionization Time-of-Flight) mass spectrometry identification, and 72 of these samples were further characterized by targeted metagenomic sequencing to compare the vaginal microbiota between healthy and purulent vulvar discharge sows.
Results
Culture and MALDI-TOF MS identification
Bacterial culture and identification by MALDI-TOF mass spectrometry were able to identify a wide variety of species in sows presenting vulvar discharge. Of the 270 females with this clinical sign, 98.5% (266/270) were positive for bacterial isolation. Eighty-nine percent of the samples generated mixed cultures, with up to eight distinct species isolated from the same animal. For the 40 healthy females, only one did not present bacterial growth and 82% (32/39) of the samples generated mixed cultures.
As there was no statistical difference between the frequencies of the main agents for the different herds in the culture method (Supplementary Table S1), we analyzed the herd data together. The culture method showed that HE sows had a higher frequency of some bacterial species than VD sows (Fig. 1), such as Enterococcus faecalis (33.3% vs. 16.9%, p = 0.02), Streptococcus hyovaginalis (7.7% vs. 1.5%, p = 0.04) and Acinetobacter Iwolffii (5.1% vs. 0.4%, p = 0.04). Although HE sows had a higher frequency of Escherichia coli, we observed no statistically significant difference between groups (p = 0.06). VD sows also showed high frequencies for some species, but also without significant difference, such as Streptococcus dysgalactiae (26.3% vs. 12.8%, p = 0.09) and Staphylococcus hyicus (17.3% vs. 7.7%, p = 0.09). Trueperella pyogenes (5.3% vs. 0%), Bacteroides pyogenes (3.4% vs. 0%) and Corynebacterium diphtheriae (7.9% vs. 0%) were only isolated in VD sows.

Frequency of the main species identified in HE and VD female samples in the culture technique and identification by MALDI-TOF MS. Red: sows with purulent vulvar discharge (VD); Blue: healthy sows (HE). *Fisher Exact Probability Test, 0.05 < p < 0.1. **Indicates significant differences between groups (Fisher Exact Probability Test, p < 0.05).
Figure 2 shows an UpSet plot for the most common bacterial associations that occurred in these groups. VD sows showed more associations between species, with the most frequent one being E. coli and S. dysgalactiae, with 42 animals (15.8%). This was followed by E. coli and Streptococcus suis (12.8%), Escherichia coli and E. faecalis (9.4%), E. coli, and S. hyicus (7.1%). Meanwhile in HE sows, E. coli and E. faecalis (23%), E. coli and S. dysgalactiae (10.2%), E. coli and Trueperella abortisuis (10.2%) were the most frequent associations. Furthermore, the Venn diagram generated for the culture and MALDI-TOF MS identification results (Fig. 3A) shows that of the 115 bacterial species identified, 61% were exclusive to VD sows, 33% shared between VD and HE, and only 12% were exclusive to HE.

UpSet plot for (A) animals with vulvar discharge (VD; n = 266) and (B) UpSet plot for healthy animals (HE, n = 39). The numbers on the bars show the number of animals in which the agents, indicated by the filled dots, were isolated together.

Venn diagram for (A) MALDI-TOF mass spectrometry bacterial identification and (B) OTUs between sows with purulent vulvar discharge (VD) and healthy (HE) sows.
Targeted metagenomic sequencing
We analyzed 72 samples by 16S rRNA gene metagenomic sequencing and a total of 3,760,359 raw reads were obtained. The median number of reads per sample was 39,233, with a maximum of 216,213 and a minimum of 3276. After sequence quality control, denoising, and clustering of amplicons at 97% identity, a total of 1,068 OTUs were obtained from the 72 samples (40 from vulvar discharge sows and 32 from healthy sows).
Alfa and beta diversity analysis
A primary analysis to compare bacterial diversity among herds showed a difference for alpha diversity indices. Herds 1 and 3 had higher richness compared to herds 2 and 4 (p < 0.001, Chao1 index, see Supplementary Fig. S1 online). However, only herd 3 had higher diversity compared to all other herds (p < 0.01, Shannon index). There was also a difference in the beta-diversity analysis comparing herds. In the UniFrac PCoA analysis (see Supplementary Fig. S2 online), only herd 1 and 2 did not show a significant difference from each other, while the other pairwise PERMANOVA results (herd-1 vs. herd-3; herd-1 vs. herd-4; herd-2 vs. herd-3; herd-2 vs. herd-4; herd-3 vs. herd-4) indicated significant differences between bacterial community (p < 0.05 and q < 0.05).
To analyze diversity between groups of sows with and without purulent vulvar discharge, samples were analyzed separately for each herd. There were no differences between these groups for Chao1 and Shannon indices regarding community richness and diversity. The Venn diagram also shows that VD and HE share the most OTUs (69%) (Fig. 3B). For the beta diversity analysis, the PCoA based on Unweighted UniFrac and PERMANOVA analysis showed that for herds 2 and 3 there was a difference between HE sows and VD sows (p < 0.05, q < 0.05). For herd 4 there was a tendency for difference between clusters (p = 0.064, q = 0.064) (Fig. 4).

Unweighted UniFrac Principal Coordinates Analysis (PCoA) showing the clustering between sows with or without purulent vulvar discharge for each herd. Red: sows with purulent vulvar discharge (VD); Blue: healthy sows (HE). * Indicates significant differences between VD and HE (PERMANOVA, p < 0.05; q < 0.05).
Taxonomy composition and differential abundance analysis
Analyzing the herds together at the phylum level, Proteobacteria had the highest relative abundance in healthy and vulvar discharge sows, which accounted for 62.1% and 48.2% of relative abundance, respectively, followed by Firmicutes, which accounted for 29% and 34% of relative abundance, respectively, and Bacteroidota, which accounted for 8.4% and 16.6% of relative abundance, respectively (Fig. 5A). These three phyla represented a bacterial vaginal core for both groups since the other phyla identified represent less than 1% of relative abundance.

Relative abundance of the most influential phyla in bacterial vaginal microbiota of sows for the entire dataset (A) and each herd (B). Red: sows with purulent vulvar discharge (VD); Blue: healthy sows (HE).
When herds were analyzed separately, some differences in relative abundance were observed (Fig. 5B). A higher abundance of Proteobacteria was observed for herd-1 (HE: 72%, VD: 57%) and herd-2 (HE: 72.8%, VD: 62.1%) with a lower relative abundance of Bacteroidota (HE: 3%, VD: 4.77%; HE: 2.85%; VD: 3.44%). In contrast, herd-3 had a lower relative abundance of Proteobacteria (HE: 26.5.6%, VD: 28.9%) but a higher relative abundance of Bacteroidota (HE: 23.6%, VD: 38.2%). Herd-3 still had a higher relative abundance of Firmicutes (HE: 49.5%, VD: 32.2%) compared to other herds. Herd-4 had a high relative abundance of Proteobacteria (HE: 71.3%, VD: 48%) and Bacteroidota (HE: 8.38%, VD: 28.6%), but a lower relative abundance of Firmicutes compared to other herds (HE: 18.8%, VD: 20.7%). Except for herd-3, the relative abundance of Proteobacteria was higher in HE females than in VD females in all herds. Conversely, the phylum Bacteroidota was higher in VD females in all herds, especially in herd-3 and herd-4.
At the genus level (Fig. 6A), Escherichia-Shigella was the most abundant in both groups but was higher in healthy than in vulvar discharge sows, accounting for 52.2% and 30.5% of relative abundance when data from all herds were analyzed together. Acinetobacter, Aerococcus, Prevotella, and Pseudomonas also showed a slightly higher relative abundance in healthy sows. Conversely, Streptococcus, Bacteroides, Pasteurella, Porphyromonas, and Staphylococcus were more abundant in sows with purulent vulvar discharge. For Pasteurella, however, all sequences came from a single sample in the VD group.

Relative abundance of the most influential bacterial genera in bacterial vaginal microbiota for the entire dataset (A) and each herd (B). Red: sows with purulent vulvar discharge (VD); Blue: healthy sows (HE).
The graph in Fig. 6B shows the genera with more than 1% of relative abundance for each herd. Escherichia-Shigella, Streptococcus, and Bacteroides remain the main genera, present in all herds. But remarkable differences can be seen for Porphyromonas in herd-3, Bacteroides in herd-3 and herd-4, and Staphylococcus in herd-1 and herd-2.
Since some OTUs were able to be classified at the species level, it is noteworthy that B. pyogenes and S. dysgalactiae were found in remarkable greater relative abundance in females with purulent vulvar discharge than in healthy females (Fig. 7).

Relative abundances between VD and HE sows for (A) B. pyogenes and (B) S. dysgalactiae. Red: sows with purulent vulvar discharge (VD); Blue: healthy sows (HE). *Indicates significant differences between groups by ANCOM test (W = 44).
The differential abundance analysis performed by the ANCOM method between VD and HE females across the entire dataset revealed a significant difference for the species B. pyogenes, with greater relative abundance in VD females (W = 44, Supplementary Table S2). When the test was applied to each farm separately, B. pyogenes were significantly more abundant in VD females only in herd-3 (W = 632, Supplementary Table S3). S. dysgalactiae was also found in greater abundance in VD females (Fig. 7). Interestingly, this difference was not statistically significant by the ANCOM test.
Network analysis
The network analysis shows the correlations between the most influential bacterial genera in vulvar discharge and healthy sows (Fig. 8). For VD females, positive correlations were predominant between potentially pathogenic bacteria such as Escherichia-Shigella, Streptococcus, Trueperella, Corynebacterium, and Prevotella. For healthy sows, however, negative correlations were predominant with Escherichia-Shigella being an important node in these correlations, but with a significant positive correlation with E. faecalis.

Network analysis for the main agents in sows with vulvar discharge (VD) and healthy (HE). Green and red lines indicate positive and negative correlation, respectively. Line thickness is proportional to the strength of the Pearson correlation.
Discussion
Despite the great genetic advances that have made female swine hyper prolific, sow mortality has increased worldwide. Reproductive diseases are responsible for an important part of these deaths2, with vulvar discharge being one of the main clinical signs pointing to infectious problems in the reproductive tract of sows1. Few studies have evaluated the sow vaginal bacterial composition between healthy sows and sows with urogenital infection, with a limited number of animals11,12. To increase knowledge about the vaginal microbiota of healthy and diseased sows, and to fill the gap for this assessment in Brazilian herds, we evaluated the hypothesis that there may be some differences in the bacterial vaginal content of sows with and without purulent vulvar discharge.
The culture-dependent approach revealed a rich population of bacterial species in the vaginal canal of sows, showing that VD and HE females had a predominance of certain taxa such as E. coli, E. faecalis, and Streptococcus and Staphylococcus species, in agreement with previous studies of bacterial isolation from the reproductive tract of sows4,16,17.
We found potentially harmful bacteria in a higher frequency in VD sows. Winter et al.18 reproduced the vulvar discharge syndrome through uterine experimental inoculation with S. hyicus in swine females. This species was isolated with high frequency in our study in VD sows. C. diphtheriae, already described in genitourinary infection of sows and public health importance for its role in human infections19,20,21, as well as T. pyogenes, B. pyogenes, and S. dysgalactiae, also reported in reproductive disorders in cows and sows9,22,23,24,25,26,27,28,29, were also isolated more frequently in VD sows. But there was no statistical difference in the frequency of these agents between HE and VD sows. These agents were also associated with a high frequency in VD sows, suggesting that these associations may be important for causing disease.
However, since the culture-dependent method used does not quantify specific bacteria and may also be biased since most agents do not grow easily in culture media30, we performed a 16S rRNA metagenomic sequencing approach in 72 of these vaginal samples to characterize the bacterial population between VD and HE sows.
Our study showed that sows’ vaginal bacterial communities differed between herds. This can be due to several uncontrolled factors, such as nutritional, environmental, and genetic differences, hygiene level of the facilities, and microclimate. Although there are some fluctuations in relative abundance at the phylum and genus level between herds, which explains the difference in the beta-diversity index, they share most of the agents, with the phyla Proteobacteria, Firmicutes, and Bacteroidota and the genera Escherichia-Shigella, Streptococcus, Enterococcus, and Bacteroides were among the most relatively abundant in all herds.
Beta diversity analysis revealed a significant difference between HE and VD sows by the unweighted UniFrac PCoA analysis for two of the four herds, which indicates that the difference between bacterial communities could be in specific features31, such as B. pyogenes, S. dysgalactiae and S. hyicus, found in greater relative abundance in VD sows.
Proteobacteria, Firmicutes, and Bacteroidetes were the predominant phyla for both HE and VD groups. Remarkably comparable results were found by other studies in both healthy and diseased animals7,9,10,11,12, indicating that the vaginal microbiota of sows is dominated by these phyla. At the genus level, there was a high agreement with the culture-dependent method results, since Escherichia, Streptococcus, Enterococcus, and Staphylococcus are among the highest relative abundances. Interestingly, the phylum Bacteroidetes and the genus Bacteroides were found in high relative abundances only in the metagenomic approach, in contrast to the culture-dependent method. This may be due to the difficulty of traditional culture methods to cover fastidious bacteria, such as the obligate anaerobes Bacteroides members32. This reinforces that the metagenomic approach may be more effective in characterizing the microbiota due to its higher resolution to identify low-abundance or fastidious microbes, while culture-dependent methods can confirm the viability of the identified agents, making them complementary techniques.
The species Acinetobacter lwoffii was significantly more frequent in HE sows in the culture method. Luque et al.7 also found these genera in vaginal samples of healthy sows. As these authors point out, despite not being described as causing urogenital infection in sows, these genera have been described as carriers and spreaders of resistance genes for antimicrobials used in veterinary practice and human medicine33,34 and their presence requires attention.
Bacteroides, Staphylococcus, and Porphyromonas, found in higher relative abundance in VD than in HE sows, were also detected in higher relative abundance in sows with endometritis in recent studies11,12. Even though the sequencing did not differentiate Staphylococcus species, we suggest that part of this high relative abundance in VD sows may be represented by S. hyicus, as shown by culture and identification by MALDI-TOF MS. S. hyicus has been described as capable of causing uterine infection in an experimental infection. Also, S. dysgalactiae had a notably higher relative abundance also in VD sows. Kiefer et al.9 found that S. dysgalactiae was significantly more abundant in sows with a high score for pelvic organ prolapse, which reinforces the importance of this species.
We used the ANCOM test to perform a differential abundant analysis between specific taxa to compare VD and HE sows. Previous researchers showed that statistical methods commonly used in differential abundance analysis, such as ANOVA or Kruskal–Wallis, are not appropriate for compositional data because they inflate false discovery rates (FDR) and should be avoided35,36,37. Recent studies showed that the ANCOM test was superior in controlling FDR while retaining great statistical power36,37. In our study, the ANCOM test revealed that B. pyogenes relative abundance in VD sows is higher than in HE sows, being a marker for vulvar discharge and suggesting its importance in vulvar discharges syndromes since this agent has been identified in uterine infections in cows and sows12,38,39. Indeed, Zhang et al.12 suggest a link between the increase of some bacteria in the gut microbiota and the onset of endometritis in sows, including the Bacteroides genus. The pathogenesis of B. pyogenes infections is still unclear, especially in the swine species. But recent studies described this agent in important infections in humans, such as purulent abscesses, infected wounds, and urinary tract infections32. Only herd-3 showed a significant difference in the ANCOM method for the higher relative abundance of B. pyogenes in VD females, which could explain the difference in beta-diversity observed between the HE and VD groups for this herd. Moreover, we found that the relative abundance of B. pyogenes was also significantly higher in VD females for the ANCOM method when samples from all herds were analyzed together. In addtion, the graphs show that for all herds the relative abundance of B. pyogenes was greater in VD females. Although this difference was most notable for herd-3 and herd-4, this is an important finding across the entire dataset.
Network analysis correlations between the genera identified by metagenomic sequencing show that VD sows had more positive correlations between agents recognized as pathogenic for the reproductive tract, such as Escherichia-Shigella and Trueperella, and Escherichia-Shigella and Streptococcus18,25,26,27. Significantly, the association between these species also occurred in the culture-dependent method. A positive correlation between Corynebacterium and Prevotella may also be important since these agents have already been described in reproductive disorders of sows9,20 and women40. Such correlations did not occur for HE females, in which Escherichia-Shigella and Enterococcus are dominant nodes, with negative correlations, showing dominance for these agents. This is also corroborated by the MALDI-TOF MS and relative abundance graphs since Escherichia and Enterococcus present higher frequency and relative abundance in HE females in all herds. The Enterococcus genus also has a strong positive correlation with the Veillonella genus commonly found in considerable abundance in the vaginal microbiota of sows, between 2 and 6%, but without a relationship with disease9,41.
This study provides new evidence on the vaginal bacterial composition of sows with and without purulent vulvar discharge. Most bacteria obtained in culture were also present in the metagenomic sequencing, showing the viability of the detected agents. The vaginal microbiota of these groups shares most of the features identified by 16S rRNA sequencing, but punctual changes in the abundance of certain agents differentiated VD and HE females, especially B. pyogenes, which can be proposed as a marker for VD sows. Higher frequencies or relative abundances of specific agents in VD sows, and the association between them, also require attention. Evidence suggests that dysbiosis in the vaginal canal of female swine that presents purulent vulvar discharge occurs by the association and increase of some potentially pathogenic bacteria such as B. pyogenes, S. dysgalactiae, S. hyicus, C. diphtheriae, and T. pyogenes.
Methods
Animals and sample collection
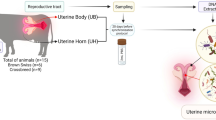
Three hundred ten vaginal samples were collected from commercial crossbred sows housed in four commercial farms in different Brazilian states: São Paulo, Minas Gerais, Paraná, and Mato Grosso. Two hundred seventy samples were collected from sows presenting purulent vulvar discharge (VD) and 40 were collected from healthy sows (HE).
Animals were inspected for purulent vulvar discharge with the use of a sterile and disposable vaginal speculum during pregnancy or in the lactation period, at least three days after the farrowing process, combined with the observation of the presence of purulent material on the floor and in the bars below the female. The introduction of the speculum was always performed by two veterinarians, one dealing with the vulva and exposing the vaginal canal and the other introducing the speculum to avoid external contamination Samples were only collected if purulent secretion was present in the deep region of the vaginal canal, after the passage of the speculum, which also excludes the possibility that this purulent material originates from vulvar lesions or vaginitis. For the collection, two sterile swabs (one for culture and one for metagenomic approaches) were collected, placed in transport media, and immediately refrigerated until arrival at the laboratory for further processing. Secretions considered to be normal-appearing and lochia were not collected. Only secretions very suggestive of infectious processes, intensely purulent and with lumps were collected and were considered in further analyses. Sows that had just urinated were not collected. If antimicrobials were used in any of the animals in the previous 30 days before collection, these females were not included in further analysis. Healthy females were collected if they did not present vulvar discharge, hyperthermia, low body condition score, had been medicated, or had any other disease condition.
Ethics approval
This study was evaluated and approved by the Animal Use Ethics Committee (CEUA) of the Faculty of Veterinary Medicine and Animal Science of the University of São Paulo, Brazil (CEUA Process Number 1875170317). This Committee follow all international guidelines and regulations to evaluate studies and experiments on live vertebrates. The authors declare that the methods are in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Bacterial culture
Swabs were plated on MacConkey agar, Chromagar Orientation®, and Columbia agar with 5% sheep blood (Difco-BBL, Detroit, MI /USA), and the plates were incubated at 37ºC for 24 to 48 h under aerobic conditions. For anaerobic growth, swabs were seeded on Brucella agar with 5% sheep blood supplemented with hemin and menadione (Difco-BBL, Detroit, MI, USA) and incubated at 37ºC for 24 to 48 h. After growth, the isolated bacterial colonies were transferred to 3.0 ml of BHI broth (Brain Heart Infusion – Difco), and from this culture, aliquots were separated for storage at -80º C and for protein extraction for identification by Matrix Associated Laser Desorption Ionization Time-of-Flight mass spectrometry (MALDI-TOF MS).
Identification of bacterial strains by MALDI-TOF MS
For bacterial identification by MALDI-TOF MS, the colonies were subjected to ribosomal protein extraction using the protocol described by Hijazin et al.42. The Microflex™ mass spectrophotometer (Bruker Daltonik) from the São Paulo State Environmental Company (CETESB) was used. To capture the protein spectra, 1.0 µl of protein suspension was transferred to a 96-well stainless-steel plate and, after drying at room temperature, 1.0 µl of the polymer matrix (α-cyano-4-hydroxycinnamic acid) was added. Each strain was distributed in three wells and for each plate, two readings were performed with the FlexControl™ software (Bruker Daltonik) using the MTB_autoX method. Finally, the BioTyper™ 3.0 software (Bruker Daltonik) compared the captured spectra for each strain with the manufacturer's library for bacterial identification. By comparing the presence/absence of specific peaks, a log (score) value was obtained. The criteria for interpreting the standards used in this study were those of the manufacturer Bruker Daltonik: scores ≥ 2.0 were accepted for species attribution, and scores ≥ 1.7 and < 2.0 were used only for genus identification.
DNA extraction and 16S rRNA gene sequencing
For the metagenomic approach, 72 swabs (40 VD and 32 HE) had their bacterial DNA extracted using the DNeasy® Blood & Tissue Kit (QIAGEN), following the manufacturer's recommendations. DNA concentration and integrity were verified in the NanoDrop® 2000 equipment and by agarose gel electrophoresis before being sent to the sequencing facility. Briefly, 16S rRNA amplicons were produced using specific primers for the 16S-V4 region (515F-806R) along with barcodes. All PCR reactions were conducted with Phusion® High-Fidelity PCR Master Mix (New England Biolabs). The mixed PCR products were purified with Qiagen Gel Extraction Kit (Qiagen, Germany) and the libraries generated with NEBNext® UltraTM DNA Library Prep Kit for Illumina and quantified via Qubit and qPCR. Amplicons were sequenced on the Illumina MiSeq platform to generate 250 bp paired-end reads.
Bioinformatics analysis
Downstream analyses were performed using the QIIME 2 platform43. Paired-end reads were assigned to samples based on their unique barcodes and truncated by cutting off the barcode and primer sequences with the cutadapt tool44 and the union of reads was performed with the Vsearch plugin45. A quality filter was applied with the quality-filter plugin by the q-score-joined method46 and a denoising step was performed using the deblur plugin with the denoise-16S method47. Sequences with ≥ 97% or more similarity were assigned to the same OTU (Operating Taxonomic Unit) with the Vsearch plugin45 and were taxonomically classified using a pre-trained classifier trained on the SILVA database48,49.
Statistical analyses
Fisher's exact test was used to compare the frequencies of the main agents isolated in the culture method and identified by MALDI-TOF MS, between herds and between HE and VD sows, under the significance level of 5%. For statistical analysis of alpha diversity comparing the herds and groups of healthy females and those with purulent vulvar discharge, the Kruskal–Wallis test was performed, with a significance level of 5%. For the comparison of beta diversity, the permutational multivariate analysis of variance (PERMANOVA) was performed with 999 permutations. A significance level of 5% for false discovery rate adjusted p-value (q-value) was considered. Differential abundance analysis was performed using the ANCOM37 test on the QIIME 2 platform43. Figures and graphics were performed with the qiime2R package50 and ggplot2 package51 in the R-Studio software52.
For network analysis, the absolute abundance data were submitted to non-paranormal transformation according to Liu et al.53 to build a correlation matrix based on Pearson correlation. Plots were generated using the qgraph package54.
Data availability
The targeted metagenomic dataset was deposited in the NCBI’s Sequence Read Archive (SRA) database under the accession code PRJNA773131.
References
Kirkwood, R. N., Althouse, G. C., Yaeger, M. J., Carr, J. & Almond, G. W. Diseases of Reproductive System. in Diseases of Swine (eds. Zimmerman, J. J., Karriker, L. A., Ramirez, A., Schwartz, K. J. & Stevenson, G. W.) 329–347 (Wiley-Blackwell, Amsterdam, 2012). doi:https://doi.org/10.1017/CBO9781107415324.004.
China Supakorn, C. S. A review of aetiology and risk factors affecting sow mortality. CAB Rev. Perspect. Agric. Vet. Sci. Nutr. Nat. Resour. 14 (2019).
Carabin, H., Martineau, G.-P., Vaillancourt, D., Higgins, R. & Bigras-Poulin, M. Detection of cervical bacterial contamination in swine by two methods of swabbing in relation to artificial insemination. Can. J. Vet. Res. 60, 40–44 (1996).
Bara, M. R. et al. A study of the microbial flora of the anterior vagina of normal sows during different stages of the reproductive cycle. Aust. Vet. J. 70, 256–259 (1993).
de Winter, P. J. J., Verdonck, M., de Kruif, A., Devriese, L. A. A. & Haesebrouck, F. Endometritis and vaginal discharge in the sow. Anim. Reprod. Sci. 28, 51–58 (1992).
Wanyoike, S. K. & Bilkei, C. Concurrent pathological and bacteriological findings in the urogenital organs and mammary glands of sows culled because of chronic vulvovaginal discharge and swine urogenital disease (SUGD): a case study. Tijdschr. Diergeneeskd. 131, 686–691 (2006).
Luque, A. T. et al. Vaginal bacterial diversity from healthy gilts and pregnant sows subjected to natural mating or artificial insemination. Res. Vet. Sci. 140, 26–37. https://doi.org/10.1016/j.rvs.c.2021.07.023 (2021).
Sanglard, L. P. et al. Vaginal microbiota diverges in sows with low and high reproductive performance after porcine reproductive and respiratory syndrome vaccination. Sci. Rep. 10, 3046 (2020).
Kiefer, Z. E. et al. Vaginal microbiome and serum metabolite differences in late gestation commercial sows at risk for pelvic organ prolapse. Sci. Rep. 11, 1–11 (2021).
Xu, S. et al. Responses of vaginal microbiota to dietary supplementation with lysozyme and its relationship with rectal microbiota and sow performance from late gestation to early lactation. Animals 11, 1–16 (2021).
Wang, J. et al. Characterization of vaginal microbiota of endometritis and healthy sows using high-throughput pyrosequencing of 16S rRNA gene. Microb. Pathog. 111, 325–330 (2017).
Zhang, L. et al. Effect of Sow Intestinal Flora on the Formation of Endometritis. Front. Vet. Sci. 8, 1–8 (2021).
Doerflinger, S. Y., Throop, A. L. & Herbst-Kralovetz, M. M. Bacteria in the vaginal microbiome alter the innate immune response and barrier properties of the human vaginal epithelia in a species-specific manner. J. Infect. Dis. 209, 1989–1999 (2014).
Oakley, B. B., Fiedler, T. L., Marrazzo, J. M. & Fredricks, D. N. Diversity of human vaginal bacterial communities and associations with clinically defined bacterial vaginosis. Appl. Environ. Microbiol. 74, 4898–4909 (2008).
Lamont, R. et al. The vaginal microbiome: new information about genital tract flora using molecular based techniques. BJOG An Int. J. Obstet. Gynaecol. 118, 533–549 (2011).
Maes, D., Verdonck, M. & Kruif, A. D. E. Vaginal microecology and vulvar discharge in swine. Old Herborn Univ. Semin. Monogr. 50, 39–50 (1999).
Scofield, A. M., Clegg, F. G. & Lamming, G. E. Embryonic mortality and uterine infection in pig. J. Reprod. Fertil. 36, 353–361 (1974).
de Winter, P. J. J., Verdonck, M., de Kruif, A., Devriese, L. A. & Haesebrouck, F. Bacterial endometritis and vaginal discharge in the sow: prevalence of different bacterial species and experimental reproduction of the syndrome. Anim. Reprod. Sci. 37, 325–335 (1995).
Scheifer, C. et al. Re-emergence of Corynebacterium diphtheriae. Méd. Mal. Infect. 49, 463–466 (2019).
Poor, A. P. et al. Characterization of Corynebacterium diphtheriae, C. confusum and C. amycolatum isolated from sows with genitourinary infection. Vet. Microbiol. 207, 149–152 (2017).
Zasada, A. A., Baczewska-Rej, M. & Wardak, S. An increase in non-toxigenic Corynebacterium diphtheriae infections in Poland: molecular epidemiology and antimicrobial susceptibility of strains isolated from past outbreaks and those currently circulating in Poland. Int. J. Infect. Dis. 14, e907–e912 (2010).
Cunha, F. et al. Quantifying known and emerging uterine pathogens, and evaluating their association with metritis and fever in dairy cows. Theriogenology 114, 25–33 (2018).
Yang, L. et al. Using PacBio sequencing to investigate the effects of treatment with lactic acid bacteria or antibiotics on cow endometritis. Electron. J. Biotechnol. 51, 67–78 (2021).
Wagener, K., Grunert, T., Prunner, I., Ehling-Schulz, M. & Drillich, M. Dynamics of uterine infections with Escherichia coli, Streptococcus uberis and Trueperella pyogenes in post-partum dairy cows and their association with clinical endometritis. Vet. J. 202, 527–532 (2014).
Brodzki, P., Bochniarz, M., Brodzki, A., Wrona, Z. & Wawron, W. Trueperella pyogenes and Escherichia coli as an etiological factor of endometritis in cows and the susceptibility of these bacteria to selected antibiotics. Pol. J. Vet. Sci. 17, 657–664 (2014).
Sens, A. & Heuwieser, W. Presence of Escherichia coli, Trueperella pyogenes, α-hemolytic streptococci, and coagulase-negative staphylococci and prevalence of subclinical endometritis. J. Dairy Sci. 96, 6347–6354 (2013).
Carneiro, L. C., Cronin, J. G. & Sheldon, I. M. Mechanisms linking bacterial infections of the bovine endometrium to disease and infertility. Reprod. Biol. 16, 1–7 (2016).
Wang, M. L. et al. Uterine microbiota of dairy cows with clinical and subclinical endometritis. Front. Microbiol. 9, 1–11 (2018).
Jarosz, S., Grądzki, Z. & Kalinowski, M. Trueperella pyogenes infections in swine: clinical course and pathology. Pol. J. Vet. Sci. 17, 395–404 (2014).
Hugenholtz, P. & Tyson, G. W. Metagenomics. Nature 455, 481–483 (2008).
Lozupone, C. A., Hamady, M., Kelley, S. T. & Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585 (2007).
Majewska, A., Kierzkowska, M. & Kawecki, D. What we actually know about the pathogenicity of Bacteroides pyogenes. Med. Microbiol. Immunol. 210, 157–163 (2021).
Gao, F.-Z. et al. Swine farming elevated the proliferation of Acinetobacter with the prevalence of antibiotic resistance genes in the groundwater. Environ. Int. 136, 105484 (2020).
Lupo, A., Haenni, M. & Madec, J.-Y. Antimicrobial resistance in Acinetobacter spp. and Pseudomonas spp. Microbiol. Spectr. (2018). doi: https://doi.org/10.1128/microbiolspec.ARBA-0007-2017
Gloor, G. B., Macklaim, J. M., Pawlowsky-Glahn, V. & Egozcue, J. J. Microbiome datasets are compositional: and this is not optional. Front. Microbiol. 8, 1–6 (2017).
Lin, H. & Peddada, S. D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 11, 1–11 (2020).
Mandal, S. et al. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Heal. Dis. 26, 27663. https://doi.org/10.3402/mehd.v26.27663 (2015).
Jeon, S. J. et al. Uterine microbiota and immune parameters associated with fever in dairy cows with metritis. PLoS ONE 11, e0165740. https://doi.org/10.1371/journal.pone.0165740 (2016).
Galvão, K. N., Bicalho, R. C. & Jeon, S. J. Symposium review: The uterine microbiome associated with the development of uterine disease in dairy cows. J. Dairy Sci. 102, 11786–11797 (2019).
Si, J., You, H. J., Yu, J., Sung, J. & Ko, G. Prevotella as a hub for vaginal microbiota under the influence of host genetics and their association with obesity. Cell Host Microbe 21, 97–105 (2017).
Sanglard, L. P. et al. Investigating the relationship between vaginal microbiota and host genetics and their impact on immune response and farrowing traits in commercial gilts. J. Anim. Breed. Genet. 137, 84–102 (2020).
Hijazin, M. et al. Identification of trueperella (Arcanobacterium) bernardiae by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry analysis and by species-specific PCR. J. Med. Microbiol. 61, 457–459 (2012).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10 (2011). doi: https://doi.org/10.14806/ej.17.1.200
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584. https://doi.org/10.7717/peerj.2584 (2016).
Bokulich, N. A. et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59 (2013).
Amir, A. et al. Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2, 1–7 (2017).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90. https://doi.org/10.1186/s40168-018-0470-z (2018).
Yilmaz, P. et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648 (2014).
Jordan & Bisanz. qiime2R: Importing QIIME2 artifacts and associated data into R sessions. unpublished https://github.com/jbisanz/qiime2R (2018).
Wickham, H. ggplot2. (Springer New York, 2009). doi:https://doi.org/10.1007/978-0-387-98141-3.
RStudio Team. RStudio: integrated development environment for R. (2020).
Liu, H., Lafferty, J. & Wasserman, L. The nonparanormal: semiparametric estimation of high dimensional undirected graphs. J. Mach. Learn. Res. 10, 2295–2328 (2009).
Epskamp, S., Cramer, A. O. J., Waldorp, L. J., Schmittmann, V. D. & Borsboom, D. qgraph : network visualizations of relationships in psychometric data. J. Stat. Softw. https://doi.org/10.18637/jss.v048.i04 (2012).
Acknowledgements
This study was supported by the Coordination of Improvement of Higher Education Personnel-Brazil (CAPES)—Finance Code 001, and the National Council for Scientific and Technological Development (CNPq)—Universal Grant 436370/2018-2. A.P.P., L.Z.M., C.E.C.M., and M.S.M. were recipients of São Paulo Research Foundation (FAPESP) fellowships (Grants 2019/01192-7, 2016/25745-7, 2015/26159-1, 2019/17683-0). V.T.M.G was the recipient of the CNPq fellowship (Grant 400267/2017-9). A.M.M. is a CNPq fellow (Grant 310736/2018-8).
Author information
Authors and Affiliations
Contributions
A.P.P. conducted the experiment, analyzed the results, and wrote the main manuscript text; L.Z.M. conceived the experiment and analyzed the results; M.S.M., C.E.C.M., M.C.D., and D.F.L. contributed to the acquisition and analysis of material and data; V.T.M.G., A.P.S.S., M.R.F.B., M.I.Z.S. contributed samples analysis; A.M.M. conceived the experiment and analyzed the results. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Poor, A.P., Moreno, L.Z., Monteiro, M.S. et al. Vaginal microbiota signatures in healthy and purulent vulvar discharge sows. Sci Rep 12, 9106 (2022). https://doi.org/10.1038/s41598-022-13090-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-13090-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
