
Abstract
Tuberculosis poses one of the greatest infectious disease threats of our time, especially when associated with human immunodeficiency virus (HIV) infection. Very little data is available on the lung microbiome in pulmonary tuberculosis (PTB) in HIV-positive patients. Three patient cohorts were studied: (i) HIV-positive with no respiratory disease (control cohort), (ii) HIV-positive with pneumonia and (iii) HIV-positive with PTB. Sputum specimens were collected in all patients and where possible a paired BALF was collected. DNA extraction was performed using the QIAamp DNA mini kit (QIAGEN, Germany) and extracted DNA specimens were sent to Inqaba Biotechnical Industries (Pty) Ltd for 16S rRNA gene sequence analysis using the Illumina platform (Illumina Inc, USA). Data analysis was performed using QIMME II and R Studio version 3.6.2 (2020). The lung microbiomes of patients with PTB, in the context of HIV co-infection, were dominated by Proteobacteria, Firmicutes, Actinobacteria and Bacteroidetes. Loss of biodiversity and dysbiosis was found in these patients when compared to the HIV-positive control cohort. Microbial community structure was also distinct from the control cohort, with the dominance of genera such as Achromobacter, Mycobacterium, Acinetobacter, Stenotrophomonas and Pseudomonas in those patients with PTB. This is the first study to describe the lung microbiome in patients with HIV and PTB co-infection and to compare findings with an HIV-positive control cohort. The lung microbiomes of patients with HIV and PTB were distinct from the HIV-positive control cohort without PTB, with an associated loss of microbial diversity.
Similar content being viewed by others


Introduction
Tuberculosis (TB) poses a major health threat worldwide, causing an estimated two million deaths annually according to the WHO (2020)1. The morbidity and mortality associated with TB is particularly severe in sub-Saharan Africa, where it is frequently associated with concomitant Human Immunodeficiency Virus (HIV) infection1,2.
The role of microbiota in health and disease has been well-characterised in various body sites3,4,5,6,7, using high-throughput sequencing technologies8. Although initially considered as a ‘sterile’ site, the healthy lung has been found to contain members of the phyla Bacteroides, Firmicutes and Proteobacteria9,10.
The lung microbiome is altered by acute and chronic disease, which alters the local conditions and inflammatory milieu of the lung11. Such alteration has been observed in chronic obstructive airways disease (COPD)12, asthma13, cystic fibrosis (CF)14, pneumonia15 and HIV infection16. When considering the lung microbiome in PTB in populations with a high prevalence of HIV, the effect of HIV itself on the microbiome should be considered.
The lung microbiome in TB has been less well characterized, with a few published studies17,18,19,20,21,22,23, none in the context of HIV. Loss of microbial diversity and the onset of dysbiosis has been illustrated in the lung microbiomes of patients with PTB, when compared with healthy controls22.
This study was the first to describe the lung microbiome of sputum and BALF specimens using 16S rRNA gene sequencing of three patient cohorts consisting of: (i) patients with HIV but no pulmonary disease (control cohort), (ii) patients with HIV and pneumonia, iii) patients with HIV and TB.
Methods
Sample collection
This study was conducted on adult (18 years and older) HIV-infected patients, presenting to Tertiary Academic Hospitals in Pretoria, South Africa, with the diagnosis of pneumonia. Participants for the control cohort were recruited from the Antiretroviral clinic, where patients were referred for initiation of antiretroviral therapy. Patients who had antibiotic exposure in the preceding 6 weeks were excluded from the study. Written informed consent was obtained from all participants. Ethical approval to perform the study was obtained from the Research Ethics Committee, Faculty of Health Sciences, University of Pretoria (REC number 265/2017). All components of the study were performed in alignment with local and international guidelines and regulations, and the research adhered to the principles set out in the Declaration of Helsinki.
Sputum was obtained from all patients enrolled in the study (n = 71), using a lung flute (Medical Acoustics, USA)23. When possible bronchoscopy was performed to collect an additional BALF specimen.
Sample processing and DNA sequencing
Respiratory specimens in those with pneumonia were sent for diagnostic testings: microscopy, culture and antibiotic susceptibility testing, Gene Xpert (Cepheid, South Africa), Pneumocystis PCR (Fast Track diagnostics, Siemens, Germany) at the National Health Laboratory Service (NHLS). The diagnosis of HIV was confirmed in all the participants, by a positive fourth generation ELISA test (Siemens healthcare diagnostics, Germany).
The DNA extraction was done with the QIAamp DNA mini kit (QIAGEN, Germany) according to the manufacturer manual for bacteria and extracted DNA samples were sent to Inqaba biotechnical industries (pty) ltd, South Africa, for sequencing. A sequencing library was prepared by random fragmentation of the DNA sample, which was followed by 5’and 3’adapter ligation. The adapter-ligated fragments were PCR amplified using universal primer pairs (341F and 785R, targeting the V3 and V4 regions of the 16S rRNA gene).
Based on the culture results patients were separated into three cohorts: (i) HIV-positive control cohort with no respiratory disease (“HIV” cohort), (ii) HIV-positive with pneumonia but TB cultures negative (“Pneumonia” cohort) and (iii) HIV-positive with pneumonia and TB cultures positive (“TB”cohort). These cohorts are schematically represented in Fig. 1.

Participant cohorts included in the study.
Three specimens that consisted only of 0.9% saline were included as negative controls for quality assurance purposes. These specimens underwent all the processing procedures, including DNA extraction and sequencing.
Statistical analysis
Demographic and laboratory were summarized as a meta-data table. The descriptive data averages were calculated as percentages and mean values between the three cohorts were compared by an Analysis of Variance (ANOVA). Percentages were compared by the Fisher Exact test.
Sequencing data was analysed using the QIIME 225 pipeline. DADA226 was used for quality control and to infer unique sample sequences (amplicon sequence variants, ASVs) from the sequencing data. A quality score threshold of 15 was used to filter out low quality and chimeric reads, with positions 280 and 210 chosen for 3’-terminus trimming of forward and reverse reads, respectively. Clustered ASVs were subsequently assigned to taxonomic groups using the SILVAv138 prokaryotic database27.
Statistical analysis of assigned reads was performed in R Studio version 3.6.2 (2020)28. Before statistical analysis, the ASV table was filtered to remove ASVs that occurred in less than 90% of specimens. In addition, reads present in the negative controls, which were likely to occur due to contamination from the sampling protocol, DNA extraction or sequencing were used to filter out ASVs that are not biologically related to the patients. Specimens were grouped by cohort, using the patient data previously recorded.
Relative abundance values were calculated as fractions of the total sample count for the phyla representing 99% of the reads. The Deseq229 pipeline was used to compare sample communities and calculate taxa that were over-represented in some cohorts when compared to the others.
Alpha diversity was calculated using the Observed, Inverse Simpson (richness) and Shannon (evenness) indices. The Shapiro test was performed to test the normality of distribution of the resulting indexes. Normally distributed data was compared using the ANOVA test30, while non-normal data was compared with the Kruskal–Wallis test31. Pair-wise comparisons were made using the Tukey32 test for normal data and the Wilcox33 test for non-normal data. A p-value of < 0.05 was considered significant.
Beta diversity was calculated using DeSeq229 by the Jaccard34 and Bray Curtis35 indices and plotted in the component analysis (PCoA) -graph.
Ethical approval and consent to participate
Before this study was commenced ethical approval of the research protocol was obtained from the University of Pretoria, Faculty of Health Sciences Research Ethics Committee (ref 265/2017). All patients enrolled in the study first underwent a process of voluntary signed informed consent.
Results
The patient population studied
During the period of February 2018 to January 2019, a total of 71 HIV-positive patients were enrolled in the study: 20 HIV, 31 Pneumonia (TB negative) and 20 with TB. Twenty-eight (20 in the Pneumonia cohort and eight in TB cohort) underwent bronchoscopy for the collection of BALF.
The details of the three cohorts of patients enrolled in the study are presented in Table 1. The patients had a mean age of 39 years and all were of African ethnicity. Fifty-one percent of participants were female and 49% were male, with no significant differences between the three cohorts.
Previous tuberculosis was noted in 15% (10/71) participants, one (5%) in the HIV cohort, 4 (12.9%) in the pneumonia cohort and 5 (25%) in the TB cohort (not statistically significant). There were no statistically significant differences between the HIV, pneumonia and TB cohorts in respect of previous TB infection (p = 0.153), smoking status (p = 0.335) or biomass fuel exposure (p = 0.774).
There was a statistically significant difference between the mean CD4 counts between the three cohorts (p = 0.025). Pair-wise comparisons showed that the mean CD4 counts for the HIV cohort (262 cells/mm3), pneumonia cohort (183 cells/mm3) and TB cohort (127 cells/mm3) differed significantly (t test, p = 0.007). The HIV viral load was only available for 45% (32/71) of the participants. Despite the mean viral load being higher in the TB cohort, no statistically significant difference in HIV viral load between the cohorts was detected (p = 0.188).
A statistically significant difference between the use of antiretrovirals (ARV’s) was seen in the three cohorts (p = 0.002). None of the participants in the HIV cohort were on antiretroviral therapy. The ARV use between the TB positive and TB negative pneumonia cohorts did not differ significantly (p = 0.557). Notably, only 26.7% (19/71) of the study cohort in total were on ARV’s. The difference in severity of pneumonia, as measured by both the CURB-65 and the ACHU scores, was not statistically significant between the pneumonia and TB cohorts. The baseline characteristics of the study participants are detailed in Table 1.
The lung microbiome defined by 16S rRNA sequencing
Due to the nature of the amplicon sequencing methodology used in this study, ASVs could be confidently assigned to genus as the lowest taxonomic rank. Quality and chimeric filtering resulting in a total of 2 089 648 reads were analysed. Thirty-four phyla and 396 genera were identified across all specimens. The dominant microbiota were defined as phyla with a mean relative abundance > 1% across all samples. Five dominant phyla across all specimens were: Proteobacteria, Firmicutes, Actinobacteria, Bacteroides and Patiscibacteria.
Among the HIV control cohort the dominant phyla were: Firmicutes (36%), Actinobacteria (24%), Bacteroidetes (23%), Proteobacteria (16%) and Fusobacteria (4%). In patients with pneumonia who had negative TB cultures, there was a dominance of the phyla: Proteobacteria (28%), Firmicutes (18%), Actinobacteria (10%) and Bacteroides (10%).
The microbiome of patients with PTB was dominated by the following phyla: Proteobacteria (32%), Firmicutes(17%), Actinobacteria(10%) and Bacteroidetes(10%). Proteobacteria and Firmicutes constituting nearly half of the population. Among the phylum Proteobacteria the most common families were: Burkholderiaceae, Enterobacteriaceae and Neisseriaceae. The families Lachnospiraceae, Veilonellaceae, Peptostreptococcaceae, and Staphylococcaceae were the most commonly occurring Firmicutes. Among the phylum Actinobacteria the most common families were: Micrococcaceae, Microbacteriaceae,, Bifidobacteriaceae, Norcardia and Actinomycetaceae. In the phylum Bacteroidetes the prominent families were Prevotellaceae.
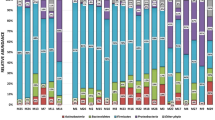
Differences in the abundance of dominant phyla were detected between the study cohorts: the HIV cohort wase dominated by Firmicutes (36%) while communities in patients with pneumonia and TB were dominated by Proteobacteria (28% and 32% respectively). The relative abundances of the dominant phyla in the three cohorts studied is shown in Fig. 2.The relative abundance of Mycobacterium between cohorts is illustrated in Fig. 3. The fact that mycobacteria was over-represented in the cohort with pneumonia who were culture-negative for TB, was further investigated and found to be driven by a patient who had Mycobacterium avium on culture.

Relative abundances of the six dominant phyla between study cohorts, i.e., HIV, Pneumonia and TB across all specimens (sputum (SPU) and BALF).

Relative abundance of mycobacteria in the various cohorts: HIV, Pneumonia and TB in sputum and in BALF.
Impact of different disease states on microbial diversity
A loss in microbial biodiversity (as represented by alpha-diversity indexes used in this study) was detected when in specimens (combined sputum and BALF) of patients with Pneumonia and those with TB when compared to the HIV cohort (Table 2, Fig. 4).

Box plot of the Inverse Simpson alpha diversity between cohorts the HIV (green), Pneumonia (blue) and TB (purple). The negative controls (red) were 0.9% saline samples.
Figure 4 illustrates the Inverse Simpson alpha diversity between the different cohorts.
To investigate differences in community structure between the cohorts the Bray–Curtis and Jaccard beta-diversity indexes of samples in each cohort were calculated and compared. The results from this analysis showed that the TB and pneumonia cohorts shared similar community structures, while being significantly distinct from the HIV cohort. A significant difference in beta diversity was found when comparing the TB cohort with the HIV cohort by abundance, according to the Bray–Curtis index (R2 = 0.118, p = 0.004) and in composition by the Jaccard index (R2 = 0.1, p = 0.005). These results are depicted in Fig. 5.

A PCoA Unirac unweighted analysis of inter- sample variability between the cohorts HIV (green), Pneumonia (blue), TB (purple) and negative controls (red) using the Bray–Curtis dissimilarity index. Principal coordinates 1 and 2 (labelled axis 1 and axis 2) explain 6.7% and 21.7% of the variance in Bray–Curtis dissimilarity, respectively.
The analysis indicated that the lung microbial communities from TB and Pneumonia patients clustered together (i.e., microbial communities in in disease-state patients), while HIV microbial communities (cohort without respiratory disease) clustered separately, but with some degree of overlap.
Differential abundance of clinically relevant taxa
To investigate further the identified differences in community structure between cohorts, differential abundance analysis was performed to identify taxa that were specifically enriched in each cohort. Over-representation of ≥ log twofold change was considered significant. The pneumonia cohort showed an over-representation of 6 genera when compared to the HIV cohort: Achromobacter, Acinetobacter, Mycobacterium, Stenotrophomonas, Corynebacterium and Pseudomonas.
Six of these taxa were also found to be over-represented in the TB cohort when compared with the HIV cohort, namely (in descending order of log change): Achromobacter, Mycobacterium, Acinetobacter, Stenotrophomonas and Pseudomonas. By comparison, in the HIV cohort the genera Lautropia, Filifactor, Neisseria and Haemphilus were over-represented when compared to the TB cohort. No differences in genera abundance could be identified between the TB and Pneumonia cohorts, which is consistent with the beta-diversity results showing a big overlap in community structure between these cohorts.
Comparing BALF and sputum samples
Of the 71 patients enrolled in this study only 28 underwent bronchoscopy and BAL. To assess whether the different sampling methods had an impact on the microbial diversity and composition of the specimens the alpha diversity of the sampling strategies was compared.
When only BALF specimens were compared, there was a statistically significant difference with more pronounced dysbiosis in the cohort with TB, when the median values of the Shannon and Inverse Simpson diversity indices were considered (Observed: pneumonia 152, TB 55, p 0.067; Shannon: pneumonia 3.7, TB 3.1 p = 0.017; Inverse Simpson: pneumonia 22.9, TB 14.8. p = 0.014). These results are illustrated in Fig. 6.

Alpha diversity in the BALF and sputum of Pneumonia and TB cohorts by Observed (a), Shannon (b) and Inverse simpson (c) indices.
Looking at the community structure, sampling strategy explained 10% of the dissimilarity between the community of the different specimens. Differential abundance analysis of the specimens obtained with the two sampling strategies identified seven genera that were differently abundant across specimens. In the BALF of the TB specimens, the genus Stenotrophomonas was over-represented when compared with the BALF specimens of the Pneumonia cohort. In the pneumonia cohort Neisseria, Corynebacterium, Dialister and Prevotella were over-represented.
Discussion
This is the first report of the lung microbiome composition in a population of African patients with underlying HIV, with and without pneumonia. The lung microbiome of patients with TB in this study were dominated by Proteobacteria, Firmicutes and Actinobacteria, with a relatively smaller proportion of Bacteroidetes when compared to the HIV cohort. The decrease in frequency of Bacteroidetes when compared to Proteobacteria and Firmicutes has been observed in other pulmonary disease states, such as asthma36, where it is associated with severe inflammation.
As the majority of TB cases reported in South Africa occur in the context of HIV co-infection, the controls selected were HIV positive with no respiratory disease. The rationale for this was the fact that HIV in itself (particularly advanced disease as in the cohort studied) effects a change on the respiratory microbiome. In a previous study HIV infection was associated with increased abundance of Streptococcus whilst there was comparatively less Flavabacterium, curvibacter, Rickettsia and Borellia found37. Advanced HIV infection was associated with a loss of alpha diversity and greater beta diversity37.
In previous studies on the lung microbiome in TB patients, Enterobacteriaceae17, Neisseriaceae22 and Sphingomodaceae18 were also identified as dominant families. The dominance of the Burkholderiaceae family in the cohort with TB is a novel finding in this study. Burkholderia infection in the lung has been described mainly in the context of CF and chronic granulomatous lung disease, where it is associated with poor clinical outcomes36,38. Previous TB infection was reported in 25% (5/20) of those within the cohort with TB and there may have been underlying destructive lung disease (bronchiectasis) in some of the participants—Burkholderia has been described in non-cystic fibrosis bronchiectasis38. The degree of immunosuppression of the patients with TB included in this study (mean CD4 count 127 cells/mm3) could be a further pre-disposing factor to explain the abundance of the genus Burkholderia, but this needs to be further studied.
The Enterobacteriaceae detected in the specimens of patients with TB (Klebsiella, Enterobacter, Serratia and Morganella) are also commonly associated with nosocomial pneumonia39 and community-acquired pneumonia in patients with underlying lung pathology40. Lachnospiraceae was a prominent Firmicute in patients with TB. A recent publication associated the presence of this family with adverse outcomes in the critically ill41. Veilonellaceae and Prevotellaceae are anaerobic families that are found in the oral microbiota42 and when present in the lung microbiome of patients with airways disease, these bacteria are associated with an increased host inflammatory response43. Veilonella was lso a dominant family in a previous study of HIV-negative patients with TB22. Enrichment of the lung microbiome with Veilonella and Prevotella is associated with increased concentrations of arachidonic acid and interleukin-17, constituting a pro-inflammatory environment43. In HIV patients with pneumonia, increases in the relative abundance of Prevotella are associated with a unique metabolomic fingerprint (enriched with amino acid metabolites and monoacylglycerols), a pro-inflammatory milieu with increased IL-17A and an increased risk for mortality44.
Mycobacteria was not found on 16S rRNA sequencing of all the specimens that were culture positive for M. tuberculosis. The gold standard for diagnosis of M. tuberculosis is culturing, and 16S rRNA sequencing is not routinely used for diagnosis of PTB. Cheung and colleagues (Cheung et al., 2013) found that M. tuberculosis represented a very small relative abundance in patients where it was considered a causative microorganism18. The cell walls of mycobacteria, by virtue of the richness in fatty acids and waxes, are resistant to some DNA extraction processes45. As the aim of the study was to describe the microbiome associated with active pulmonary TB, rather than to diagnose M. tuberculosis itself, DNA extraction did not include a process of mycobacterial cell wall lysis and during sequencing no primers that are specific for the genus Mycobacterium were used.
Previous studies have observed a loss of alpha diversity in lung samples from patients with advanced HIV infection46, which is why the control cohort selected for this study was HIV-positive. The CD4 count of the HIV-positive control cohort was higher than that of the TB cohort, which if used as a surrogate for may indicate more advanced disease in the latter cohort (the stage of HIV has previously been found to be associated with a loss of alpha diversity in the lung microbiome)11. Active pulmonary tuberculosis in itself may cause a modest decrease in CD4 cell count47, so the difference may not be a pure reflection of the stage of HIV.
Beta diversity was different between the HIV cohort and the cohorts with respiratory disease, be it Pneumonia or TB. The Jaccard analysis considers the number of species present, while the Bray–Curtis considers abundance of species. There was over-representation of the Lautropia, Filifactor, Neisseria and Hemophilus genera in the HIV cohort. In the TB cohort, the dissimilarity was driven by the over-representation of the following genera: Achromobacter, Mycobacterium, Acinetobacter, Stenotrophomonas and Pseudomonas, all of which are typically considered pathogenic bacteria. Lautropia has previously been isolated in the lung microbiome of children with HIV48, where it was not associated with clinical disease. Achromobacter spp. (predominant genus with the TB cohort) are frequent pathogens in healthcare-associated infections and has been associated with CF49. The study was performed in a referral hospital and many patients have frequent exposure to healthcare facilities before presentation to an Academic centre, potentially exposing them to nosocomial pathogens.
The over-representation of mycobacteria in the Pneumonia cohort, when compared to the HIV cohort, is partially explained by the presence of M. avium intracellularae cultured in one patient. Three other patients were found to have Mycobacterium spp. but the species subtypes were not classified in this study. The mycobacteria identified by 16S rRNA sequencing in those who were culture negative for TB may, therefore, either represent mycobacteria other than M. tuberculosis, or alternatively represent patients with M. tuberculosis who were missed by conventional culture50. Culture-negative pulmonary TB can occur in the early stages of the disease51.
When the TB cohort was compared with the Pneumonia cohort, no bacteria were found to be over-represented in either cohort. Lung microbiota have been correlated with specific metabolic signatures in the BAL of HIV-positive patients52. Metabolomic analysis may be useful to delineate possible functional differences in the lung microbiome in TB when compared with pneumonia in future studies.
A statistically significant difference in alpha diversity was found when BALF samples were compared between the two disease states (eight BALF specimens in the TB cohort and 20 in the Pneumonia cohort), with a relatively lower alpha diversity in patients with M. tuberculosis. This held true for both the Shannon and Inverse Simpson indices. Although the dominant phyla (Proteobacteria, Firmicutes and Actinobacteria) were similar in both cohorts of patients, a loss of diversity was shown in those with TB when BALF was considered. This is a significant finding, as the primary target for TB is the lower airway53, which typically has low microbial biomass. BALF is superior to sputum for the detection of TB in patients with HIV and low CD4 counts53 and BALF are likely to better represent the microbiome in M. tuberculosis. The over-expression of Stenotrophomonas in the BALF of patients with TB, when compared with the BALF of those with pneumonia is an interesting finding. Stenotrophomonas maltophillia is a pathogen that is well-recognized in hospital-associated infections54, but it is also emerging as a community-acquired pathogen55 which is associated with significant morbidity and mortality in immunocompromised patients56. Cui and colleagues (2012) described Stenotrophamonas as a dominant constituent in the sputum microbiome of patients with PTB57. In the analysis of specimens from an explanted lung in COPD, Stenotrophomonas was found in the more distal part of the lung, whilst P. aeruginosa dominated the microbial composition more proximally12. The over-expression of Stenotrophomonas in the BAL of patients with active pulmonary TB and the potential networks between the two microbial species needs to be further investigated. A limitation of this study was the small sample size, which limits the generalizability.
This study considered the lung microbiome of patients with TB in the context of HIV-co-infection, which is particularly applicable to Sub-Saharan Africa. It was limited by the number of BALF specimens available for comparison with sputum, and the signal for differences in diversity may have been stronger if more BALF specimens were available. Further research is needed to prove the significance of Bcc in the lung microbiome of patients with HIV and TB. The metabolomic signatures of the lung microbiome, the importance of the virome and mycobiome, as well as functional relationships between microorganism in the lung microbiome warrant further research.
Conclusion
The lung microbiome of patients with active PTB showed a dominance of the phyla: Proteobacteria, Firmicutes and Actinobacteria. When compared to the control population with HIV only, there was a decreased abundance observed in the phyla Bacteroidetes; a finding previously associated with pulmonary disease such as asthma and indicative of a pro-inflammatory milieu It is likely therefore, that in patients with TB a similar inflammatory state exists.
The abundance of Burhkolderia in this cohort of patients with PTB is a novel finding that should be further investigated and may be related to underlying destructive lung disease from previous infection with M. tuberculosis. In this study, there is clear evidence of dysbiosis in the lung microbiome of HIV-positive patients with active PTB. No distinct relationships of M. tuberculosis with other bacteria could be elucidated and metabolomic analysis may provide insight into functional relationships that may exist beyond mere species identification. Based on the results of this study the BALF is likely to be more representative of the lung microbiome in M. tuberculosis infection, as it retrieves bacteria from the site of disease. Furthermore, the dominance of Stenotrophomonas in the BAL of patients with PTB may be indicative of a functional relationship between these two bacteria and should be further studied by network analyses, growth rates and transcriptomics.
Data availability
The datasets generated and analysed during the current study are available in the University of Pretoria data repository and can be accessed at https://doi.org/10.25403/UPresearchdata.19491317.v1.
Abbreviations
- BAL:
-
Broncho-alveolar lavage
- BALF:
-
Broncho-alveolar lavage fluid
- CD:
-
Cluster of differentiation
- CF:
-
Cystic fibrosis
- COPD:
-
Chronic obstructive pulmonary disease
- DNA:
-
Deoxyribonucleic acid
- HAART:
-
Highly active antiretroviral therapy
- HIV:
-
Human immunodeficiency virus
- M. tuberculosis :
-
Mycobacterium tuberculosis
- NGS:
-
Next generation sequencing
- PTB:
-
Pulmonary tuberculosis
- rRNA:
-
Ribosomal ribonucleic acid
- TB:
-
Tuberculosis
References
Harding, E. WHO global progress report on tuberculosis elimination. Lancet Respir. Med. 8(1), 19 (2020).
Byrne, A. L., Marais, B. J., Mitnick, C. D., Lecca, L. & Marks, G. B. Tuberculosis and chronic respiratory disease: A systematic review. Int. J. Infect. Dis. 32, 138–146 (2015).
The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012).
Lazarevic, V. et al. Metagenomic study of the oral microbiota by Illumina high-throughput sequencing. J. Microbiol. Methods 79(3), 266–271 (2009).
Zaura, E., Keijser, B. J., Huse, S. M. & Crielaard, W. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 9, 259 (2009).
Ling, Z. et al. Molecular analysis of the diversity of vaginal microbiota associated with bacterial vaginosis. BMC Genom. 11, 488 (2010).
Charlson, E. S. et al. Assessing bacterial populations in the lung by replicate analysis of specimens from the upper and lower respiratory tracts. PLoS ONE 7(9), e42786 (2012).
Carney, S. M. et al. Methods in lung microbiome research. Am. J. Respir. Cell Mol. Biol. 62(3), 283–299 (2020).
Hui, A. W., Lau, H., Chan, T. H. & Tsui, S. K. The Human Microbiota: A new direction in the investigation of thoracic diseases. J. Thorac. Dis. 5(Suppl 2), S127–S131 (2013).
Charlson, E. S. et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am. J. Respir. Crit. Care Med. 184(8), 957–963 (2011).
Dickson, R. P. & Huffinagle, G. B. The lung microbiome: New principles for respiratory bacteriology in health and disease. PLoS Pathog. 11(7), e1004923 (2015).
Erb-Downward, J. R. et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS ONE 6(2), e16384 (2011).
Huang, Y. J. et al. National heart, lung, and blood institute’s asthma clinical research network: Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J. Allergy Clin. Immunol. 127(2), 372–381 (2011).
Cox, M. J. et al. Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS ONE 5(6), e11044 (2010).
Wu, B. G. & Segal, L. N. The lung microbiome and its role in pneumonia. Clin Chest Med. 39(4), 677–689 (2018).
Iwai, S. et al. Oral and airway microbiota in HIV-infected pneumonia patients. J Clin Microbiol. 50(9), 2995–3002 (2012).
Cui, Z. et al. Complex sputum microbial composition in patients with pulmonary tuberculosis. BMC Microbiol. 12, 276 (2012).
Cheung, M. K. et al. Sputum microbiota in tuberculosis as revealed by 16S rRNA pyrosequencing. PLoS ONE 8(1), e54574 (2013).
Wu, J. et al. Sputum microbiota associated with new, recurrent and treatment failure tuberculosis. PLoS ONE 8(12), e83445 (2013).
Botero, L. E. et al. Respiratory tract clinical sample selection for microbiota analysis in patients with pulmonary tuberculosis. Microbiome. 2, 29 (2014).
Zhou, Y. et al. Correlation between either Cupriavidus or Porphyromonas and primary pulmonary tuberculosis found by analysing the microbiota in patients’ bronchoalveolar lavage fluid. PLoS ONE 10(5), e0124194 (2015).
Krishna, P., Jain, A. & Bisen, P. S. Microbiome diversity in the sputum of patients with pulmonary tuberculosis. Eur. J. Clin. Microbiol. Infect. Dis. 35(7), 1205–1210 (2016).
Medical Acoustics. Product Overview. Lung Flute operation. Accessed December 2018. Accessed on www.medicalacoustics.com
Fujita, A., Murata, K. & Takamori, M. Novel method for sputum induction using the lung flute in patients with suspected pulmonary tuberculosis. Respirology 14(6), 899–902 (2009).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotech. 37, 852–857 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Yilmaz, P. et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucl. Acids Res. 42, 643–648 (2014).
RStudio Team (2020). RStudio: Integrated Development for R. RStudio, PBC, Boston, MA URL http://www.rstudio.com/.
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Girden ER. ANOVA: Repeated measures. Sage; 1992.
Kruskal, W. H. & Wallisc, W. A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 147, 583–621 (1952).
Haynes, W. Tukey’s Test. In: Dubitzky, W., Wolkenhauer, O., Cho, K. H., Yokota, H. (Eds.) Encyclopedia of Systems Biology. Springer, New York, NY (2013).
Rey, D., Neuhäuser, M. Wilcoxon-Signed-Rank Test. In: Lovric M. (eds) International Encyclopedia of Statistical Science. Springer, Berlin (2011)
Chung, N. C., Miasojedow, B., Startek, M. & Gambin, A. Jaccard/Tanimoto similarity test and estimation methods for biological presence-absence data. BMC Bioinf. 20(Suppl 15), 644 (2019).
Beals, E. W. Bray-curtis ordination: An effective strategy for analysis of multivariate ecological data. Adv. Ecol. Res. 14, 1–55 (1984).
Haung, Y. J. et al. Airway microbiota and bronchial hyperresponsiveness in patients with sub-optimally controlled asthma. J. Allergy Clin. Immunol. 127(2), 372–381 (2011).
Stressmann, F. A. et al. Analysis of the bacterial communities present in lungs of patients with cystic fibrosis from American and British centers. J. Clin. Microbiol. 49(1), 281–291 (2011).
Regan, K. H. & Bhatt, J. Eradication therapy for Burkholderia cepacia complex in people with cystic fibrosis. Cochr. Database Syst. Rev. 18(4), CD009876 (2019).
Twigg, H. et al. Effect of advanced HIV infection on the respiratory microbiome. Am. J. Resp. Crit. Care Med. 179, 97–107 (2017).
Green, H. & Jones, A. M. The microbiome and emerging pathogens in cystic fibrosis and non-cystic fibrosis bronchiectasis. Semin. Respir. Crit. Care Med. 36(2), 225–235 (2015).
Peleg, A. Y. & Hooper, D. C. Hospital-acquired infections due to Gram-negative bacteria. N. Engl. J. Med. 362(19), 1804–1813 (2010).
Von Baum, H., Welte, T., Marre, R., Suttorp, N. & Ewigl, S. Community-acquired pneumonia through Enterobacteriaceae and Pseudomonas aeruginosa: diagnosis, incidence and predictors. Eur. Respir. J. 35, 598–660 (2010).
Dickson, R. et al. Lung microbiota predict clinical outcomes in critically ill patients. Am. J. Resp. Crit. Care Med. 201(5), 555–563 (2020).
Caverly, L., Haung, M. & Sze, M. Past, present and future research on the Lung Microbiome in inflammatory airway diseases. Chest 156(2), 376–383 (2019).
Segal, L. N. et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat. Microbiol. 1, 16031 (2016).
Shenoy, M. K. et al. Immune response and mortality risk relate to distinct lung microbiomes in HIV-pneumonia patients. Am. J. Respir. Crit. Care Med. 195(1), 104–114 (2017).
Sulaiman, I. et al. Evaluation of the airway microbiome in non-tuberculous mycobacteria. Eur. Respir. J. 52, 1800810 (2018).
Saxena, D. et al. Human microbiome and HIV/AIDS. Curr HIV/AIDS Rep. 9(1), 44–51 (2012).
Wejse, C. et al. Impact of tuberculosis treatment on CD4 cell count, HIV RNA, and p24 antigen in patients with HIV and tuberculosis. Int J Infect. Dis. 17(10), e907–e912 (2013).
Rossmann, S. et al. Isolation of Lautropia mirabilis from oral cavities of Human-immunodeficiency virus-infected children. J. Clin. Micro 36(6), 1756–1760 (1998).
Swenson, C. & Sadikot, R. Achromobacter respiratory infections. Ann. Am. Thorac. Soc. 12, 252–259 (2015).
Hu, Y. et al. Metagenomic analysis of the lung microbiome in pulmonary tuberculosis – a pilot study. Emerg. Microb. Infect. 9(1), 1444–1452 (2020).
Fan, L. et al. Parallel tests using culture, Xpert M tuberculosis/RIF, and SAT-TB in sputum plus bronchial alveolar lavage fluid significantly increase diagnostic performance of smear-negative pulmonary tuberculosis. Front. Microbiol. 15(9), 1107 (2018).
Cribbs, S. et al. Correlation of the lung microbiota with metabolic profiles in bronchoalveolar lavage fluid in HIV infection. Microbiome. 4, 3 (2016).
Santoso, P. et al. Improving diagnosis of pulmonary Tuberculosis in HIV patients by bronchoscopy: A cross sectional study. Acta Med. Indones. 49(4), 330–335 (2017).
Chawla, K., Vishwanath, S. & Gupta, A. Stenotrophomonas maltophilia in lower respiratory tract infections. J. Clin. Diagn. Res. 8(12), 20–22 (2014).
An, S. & Berg, G. Stenotrophomonas maltophilia. Trends Microbiol. 26(7), 637–638 (2018).
Brooke, J. S. Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin. Microbiol. Rev. 25(1), 2–41 (2012).
Cui, Z. et al. Complex sputum microbial composition in patients with pulmonary tuberculosis. BMC Microbiol. 12, 276 (2012).
Funding
Funding for this project was from the Infectious diseases Research Fund.
Author information
Authors and Affiliations
Contributions
V.U. was the primary investigator and author of this manuscript. M.E. was the senior author for the manuscript and edited the manuscript. J.G. assisted with DNA extraction at Ampath Laboratories. E.H. provided assistance with data interpretation. P.L. and D.C. assisted with data analysis and editing of the manuscript. L.J. assisted with lung-flute sputum induction for participants.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ueckermann, V., Lebre, P., Geldenhuys, J. et al. The lung microbiome in HIV-positive patients with active pulmonary tuberculosis. Sci Rep 12, 8975 (2022). https://doi.org/10.1038/s41598-022-12970-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-12970-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
