
Abstract
Clostridioides difficile is one of the most common causes of antibiotic-associated diarrhea in developed countries. As key virulence factors of C. difficile, toxin A (TcdA) and toxin B (TcdB) act by glucosylating and inactivating Rho and Ras family small GTPases in host cells, which leads to actin cytoskeleton disruption, cell rounding, and ultimately cell death. Here we present the co-crystal structure of the glucosyltransferase domain (GTD) of TcdA in complex with its substrate human RhoA at 2.60-angstrom resolution. This structure reveals that TcdA GTD grips RhoA mainly through its switch I and switch II regions, which is complemented by interactions involving RhoA’s pre-switch I region. Comprehensive structural comparisons between the TcdA GTD–RhoA complex and the structures of TcdB GTD in complex with Cdc42 and R-Ras reveal both the conserved and divergent features of these two toxins in terms of substrate recognition. Taken together, these findings establish the structural basis for TcdA recognition of small GTPases and advance our understanding of the substrates selectivity of large clostridial toxins.
Similar content being viewed by others
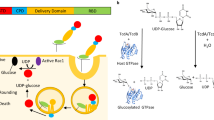


Introduction
Toxin A (TcdA) and toxin B (TcdB) are two exotoxins produced by Clostridioides difficile (C. difficile), which are the main causes of C. difficile infection (CDI) with variable clinical features including life-threatening pseudomembranous colitis1,2,3,4. TcdA and TcdB have modular architectures consisting of four major domains: an N-terminal glucosyltransferase domain (GTD), a cysteine protease domain (CPD), a delivery and receptor binding domain (DRBD), and a combined repetitive oligopeptides (CROPs) domain. TcdA and TcdB recognize and enter host cells via receptor-mediated endocytosis4,5,6,7,8,9. These toxins then transport the GTD and the CPD into the cytosol10,11,12,13, where the CPD cleaves off and release the GTD in the presence of cytosolic inositol hexakisphosphate (InsP6)14,15. Once in the cytosol, the GTD inactivates the Rho/Ras-family of small guanosine triphosphatases (GTPases) via glucosylation, leading to disruption of the actin cytoskeleton in target cells and damage of the barrier function of epithelium in the intestine16,17,18,19,20.
TcdA and TcdB belong to the large clostridial glucosylating toxin (LCGT) family, which also include Paeniclostridium sordellii toxins TcsL and TcsH, Clostridium novyi toxin TcnA, and Clostridium perfringens toxin TpeL4,21. These toxins and many virulence factors from other pathogenic bacteria act by covalently modifying and thus interfering with the physiological functions of small GTPases in host cells, which are essential molecular switches involving in diverse signal transduction pathways22,23. TcdA, TcdB, and other members in the LCGT family glucosylate Rho and/or Ras GTPases at the highly conserved threonine residue (for example T35 in Rac1 and Cdc42, T37 in RhoA) using uridine diphosphate-glucose (UDP-glucose) as the sugar donor18,19,22. Glucosylation prevents these GTPases from binding to their downstream effector proteins and therefore abolish their functions in many crucial signaling pathways related to morphogenesis, polarity, movement, and cell division24.
The sequence identity between the GTD of TcdA and TcdB is only ~ 51%, but both could target the Rho family GTPases (RhoA, Rac1, and Cdc42) with comparable activities, except that GTDTcdA is more efficient at modifying RhoA whereas GTDTcdB is faster at modifying Rac1 based on in vitro time course experiments18,19,25. Besides Rho proteins, GTDTcdA could target other GTPases, such as H/N/K-Ras, as minor substrates26,27,28. In a recent study, we demonstrated the structural mechanism by which GTDTcdB recognizes Rho and R-Ras20. Here we report the co-crystal structure of GTDTcdA from strain VPI10463 in complex with human RhoA in the presence of UDP-glucose, GDP, Mn2+ and Mg2+. Comprehensive structural comparisons between the GTDTcdA–GTPase complex and the GTDTcdB–Cdc42/GTDTcdB–R-Ras complexes reveal both the conserved and divergent features of TcdA and TcdB in terms of their substrate selectivity in host cells. These findings advance our understanding of the glucosyltransferase activities of TcdA and TcdB and pave the structural basis for inhibitor design against the GTD.
Results
Structure determination and overall structure of the TcdA GTD-RhoA complex
Like most other enzymes, the GTD transiently binds to and modifies its substrates, and then releases it before engaging the next substrate. This has posed a great challenge for us to prepare stable GTD–substrate complexes for crystallization. To overcome this obstacle, we have developed a strategy to “freeze” the transient interactions between the GTD and its substrates by protein engineering. As reported in one of our recent studies, we designed a fusion protein where Cdc42 or R-Ras was covalently linked to the N-terminus of the GTD of TcdB via a flexible peptide linker20. The peptide linker does not restrict interactions between GTDTcdB and its substrates, while the covalent linking increases their local concentrations and thus strengthens the protein–protein interactions29,30. At the same time, we mutated the key threonine residue on Rho/Ras (e.g., T35 in Rac1 and Cdc42, T37 in RhoA), the glucosylation target, into an asparagine to prevent the completion of glucosylation in order to stabilize the complex18. In this study, we used the same strategy to design a tandem RhoA (residues 1–181)–GTDTcdA (residues 1–542, strain VPI10463) fusion protein, where the two proteins are linked via an 18-amino acid peptide linker (GGGGSGGGSGTGSGGGGS) (Fig. 1A). RhoA carries the T37N mutation to prevent glucosylation. We also introduced a K190A mutation on GTDTcdA to minimize non-specific degradation at this site during protein expression and purification. This mutation is unlikely to affect the activity of GTDTcdA, because it is located on the surface of GTDTcdA that is far away from the substrate-binding interface and the UDP-glucose-binding pocket.

The overall structure of the GTDTcdA–RhoA complex. (A) A schematic diagram showing the design of the GTDTcdA–RhoA chimera protein. (B/C) Cartoon representations of the GTDTcdA–RhoA complex in two different views. GTDTcdA and RhoA are colored hot pink and cyan, respectively. The W519-loop of GTDTcdA is colored yellow, the lower and upper clamps of GTDTcdA are colored green, the pre-switch I and switch I of RhoA are colored wheat. The α16/17 of GTDTcdA and switch II of RhoA are showed as cylinders in (C). (D/E) Close-up views into the interface between GTDTcdA and RhoA focusing on the pre-switch I (E) and switch I (D/E) from two different viewing angles as indicated by purple and green symbols. The interacting residues are colored using the same scheme as that in (B). (F) Close-up view into the switch II-binding interface, and the interacting residues are colored the same as (C).
The best crystals of the GTDTcdA–RhoA complex were obtained in the presence of Mn2+ and UDP-glucose that bind to the GTD and Mg2+ and GDP that bind to RhoA, and the structure was determined at 2.60 Å resolution (Table S1). The crystals belong to space group C2221, and there is one pair of the GTDTcdA–RhoA complex in an asymmetric unit with a total buried molecular interface of ~ 1434 Å231. The co-factors, Mn2+, Mg2+, GDP, and the UDP moiety of UDP-glucose, have well-defined electron densities, but the glucosyl moiety of UDP-glucose has weaker density that is likely due to partial cleavage of UDP-glucose during crystallization. The flexible 18-amino acid peptide linker has no visible electron density, implying a highly flexible conformation that would not constrain GTD–RhoA interactions.
The crystal structure shows that GTDTcdA mainly recognizes the switch I (residues 33–42) and switch II (residues 61–75) regions of RhoA (Fig. 1B,C, Fig. S1A,B), and the overall architecture of the GTDTcdA–RhoA complex is very similar to the GTDTcdB–Cdc42/R-Ras complex (PDB code: 7S0Y, 7S0Z)20. The overall structure of RhoA-bound GTDTcdA·UDP-glucose is highly similar to the previously reported GTDTcdA·UDP-glucose (PDB code: 3SRZ)25 and GTDTcdA·U2F (a non-hydrolysable UDP-glucose homolog, PDB code: 5UQL)32 complexes, with a root mean square deviation (RMSD) of ~ 0.525/0.504 Å over 408/407 residues, respectively (Fig. 2A). Of note, a loop connecting α20 and α21 helices (residues 514–522, referred to as W519-loop) of GTDTcdA adopts a large conformational change upon UDP-glucose·Mn2+ binding in comparison to the apo state (PDB code: 4DMV) with the Cα of W519 moving ~ 7 Å (Fig. 2B, Fig. S1C). This movement of the W519-loop is triggered by its direct interactions with UDP-glucose and Mn2+25,32,33. Upon RhoA binding, the UDP-glucose·Mn2+-bound conformation of the W519-loop is further stabilized by residues Y34 and T37N on the switch I of RhoA (Fig. 1D), whereas the apo conformation of the W519-loop would clash with RhoA. The homologous W520-loop on GTDTcdB exploits a similar movement to recognize Cdc42 and R-Ras20. These findings suggest that, besides being a glucose donor, UDP-glucose facilitates the GTD of both TcdA and TcdB to engage their substrates.

Conformational changes on GTDTcdA and RhoA induced by complex formation. (A) Structural superposition of the GTDTcdA–RhoA (hot pink and cyan, respectively) complex, GTDTcdA·U2F (slate, PDB code: 5UQL), and GTDTcdA·UDP-glucose (pale yellow, PDB code: 3SRZ) based on the GTD. (B) The W519-loop of GTDTcdA adopts a similar conformation in the GTDTcdA·UDP-glucose–RhoA complex (hot pink) and GTDTcdA·UDP-glucose (pale yellow), which is drastically different from that of the apo GTDTcdA (slate, PDB code: 4DMV). (C) Comparing the conformations of the switch I of RhoA in the GTDTcdA–RhoA·GDP complex (cyan), RhoA·GTPγs (pale yellow, PDB code: 1A2B), and RhoA·GDP (slate, PDB code: 1FTN). (D) Comparing the conformations of the switch II of RhoA in the GTDTcdA–RhoA·GDP complex (cyan) and RhoA·GDP (slate, PDB code: 1FTN). (E) Superposition of RhoA-bound GTDTcdA·UDP-glucose (hot pink), GTDTcdA·U2F (slate) and GTDTcdA·UDP-glucose (pale yellow) focusing on the α16/17 helixes. (F) GTDTcdA adopts conformational changes to accommodate the pre-switch I and switch I of RhoA when compared to the GTDTcdA·U2F (slate) and GTDTcdA·UDP-glucose (pale yellow) structures.
Structural basis for RhoA recognition by the GTD of TcdA
We next examine the detailed interactions between GTDTcdA and RhoA, which are mainly mediated by the switch I, switch II, and a region right upstream of the switch I of RhoA (residues 27–32, referred to as pre-switch I) (Fig. S1D). A prominent feature of the GTD-bound RhoA is that its switch I adopts a unique conformation that is different from its GTP-bound active form34 or its GDP-bound inactive form35, even though it was crystallized in the presence of GDP and Mg2+ (Fig. 2C). In the GTDTcdA–RhoA complex, the RhoA switch I is stabilized by extensive hydrophobic packing involving residues V33, Y34, V35, P36, V38 and F39 of RhoA and residues M313, A377, L378, V381, I382, I465, P470, A474, L510 and I515 of GTDTcdA, suggesting that this new conformation of the switch I is induced by the GTD (Figs. 1D,E, 2C, Table S2). Despite discontinuity in the primary sequence, these GTDTcdA residues converge in 3D to form a largely hydrophobic groove to accommodate the switch I of RhoA, which is further supported by additional hydrogen bonds involving residues Y34, F39, E40 and N41 of RhoA and K448, T491 and E514 of GTDTcdA (Fig. 1D,E, Table S2).
Triggered by the GTD, such a movement of the switch I positions N37 of RhoA, corresponding to the glucosylation target T37 in the wild type RhoA, into the UDP-glucose binding pocket of the GTD, where N37 interacts with residues R462, S517 and S520 of GTDTcdA and UDP-glucose via hydrogen bonds (Fig. 1D). As the engineered RhoA T37N does not have the hydroxyl group to accept the glucosyl unit from UDP-glucose, the structure captured by our crystal structure probably represents a catalysis intermediate state where T37 on wild-type RhoA is primed to be glucosylated.
The second major interface between GTDTcdA and RhoA is between the switch II region of RhoA and the α16/17 helices of GTDTcdA (Fig. 1C). More specifically, residues H431, F435, A438 on the α16 helix of GTDTcdA and residues S443, L446, T447 and A450 on the α17 helix form a hydrophobic pocket to anchor residues L69 and L72 on the switch II of RhoA (Fig. 1F, Fig. S1E). We also observed that residues G62–L69 of the RhoA switch II exhibited a noticeable reorientation to accommodate GTDTcdA binding. For example, R68 of RhoA forms a salt bridge with D432 of GTDTcdA, while the conformation of the standalone RhoA would clash with GTDTcdA (Fig. 2D). In contrast, the α16/17 helices of GTDTcdA show an almost identical conformation regardless of RhoA binding except for some subtle sidechain reorientation. For example, the side chain of F435 of GTDTcdA exhibited a movement to better interact with RhoA R68 and L72 and avoid potential clash with L69 (Fig. 2E).
The third interface is established between the pre-switch I of RhoA and two discrete regions in the GTD that form a clamp-like motif, whereas the upper and lower clamps in GTDTcdA are composed of residues 307–310 and 378–380, respectively (Fig. 1B, Fig. S1F,G). Most of the interactions with the pre-switch I are mediated by the lower clamp via hydrogen bonds and hydrophobic packing (Fig. 1E). Furthermore, we observed that both the lower clamp and a nearby loop and helix (residues 489–498) of GTDTcdA reorient upon RhoA binding to better recognize the substrate (Fig. 2F). For example, the bulky side chain of K493 of GTDTcdA moves ~ 7.1 Å to avoid conflicting with N41 of RhoA (Fig. 2F, Fig. S1H).
Comparison of the Rho-binding modes between TcdA and TcdB
One of the fascinating features of TcdA and TcdB is their abilities to target different GTPases that are key modulators of diverse signaling pathways. For example, variants of TcdB from diverse C. difficile strains display different selectivity towards Rho or Ras family GTPases, which are linked to two distinct types of cytopathic effects36,37,38,39. In our earlier studies using TcdB GTDs from two different strains VPI10463 and M68 as models, we found that GTDTcdB evolves selective clustering of adaptive mutations in the GTPase-binding sites to adjust their specificities toward Rho or R-Ras, while they share a high sequence identity up to ~ 79% among different variants20. But since the sequence identity between the GTD of TcdA and TcdB from VPI10463 strain is only ~ 51%, how do they manage to recognize the same set of Rho family members such as RhoA, Rac1, and Cdc42? Now that the structures of the GTD–Rho complexes are available for both TcdA and TcdB, they provide a unique opportunity to investigate both the conserved and divergent features of these two toxins in terms of substrate recognition (Fig. 3, Table S2).

Amino acid sequence alignment among the GTDs of TcdA and two TcdB variants. Residue numbers and the secondary structures of the RhoA-bound GTDTcdA are shown on the top. The residues on the GTDs of TcdA-VPI10463, TcdB-VPI10463, and TcdB-M68 that interact with RhoA, Cdc42, and R-Ras are highlighted with red triangles, green stars, and black rhombuses, respectively. The green boxes highlight the upper and lower clamps of the GTD.
We first focused on the interfaces where the GTD recognizes the switch I and the pre-switch I areas. We found that most of the Rho-binding residues in this area are conserved between TcdA and TcdB. For example, residues I382, I465, P470, A474, E514, S517, W519, and S520 of GTDTcdA interact with residues Y34, P36, T37N, V38 and F39 on the switch I of RhoA, and all these interacting residues are conserved on GTDTcdB-VPI10463 and Cdc42, respectively (Fig. 3, Fig. S3, Table S2). However, there are some interactions unique for the GTDTcdA–RhoA complex. For example, K448 of GTDTcdA forms a salt bridge with RhoA E40, which could be applied to interactions with Cdc42 and Rac1 that have a homologous substitution of D38 (Fig. 4A). But GTDTcdB-VPI10463 has E449 at the corresponding position that would weaken this engagement. We also noticed that a hydrophobic surface composed of residues I382, I465 and P470 on GTDTcdA that is also conserved on GTDTcdB (I383, I466 and P471) is expanded by residues A377 and L378 on the lower clamp of GTDTcdA, which enhance interactions with RhoA residues V35, P36 and V38 on the switch I, as well as residues I23 and P31 on the pre-switch I. In contrast, GTDTcdB-VPI10463 has hydrophilic N378 and S379 at the corresponding positions on the lower clamp (Figs. 3, 4B). Moreover, the presence of L378 of GTDTcdA may induce the movement of the bulky side chain of K27 of RhoA on the pre-switch I in order to avoid a clash, and K27 subsequently forms hydrogen bonds with G379 and S380 of GTDTcdA that strengthen interactions (Fig. 4C). This is likely unique to RhoA as Cdc42 and Rac1 have a small Threonine in the place of K27. This finding suggests that the lower clamp of GTDTcdA may match the pre-switch I and switch I of RhoA better than that of GTDTcdB, which might partly explain the observation that GTDTcdA was more efficient than GTDTcdB at modifying RhoA based on a time course in vitro experiment25.

Structural comparison of the GTDTcdA–RhoA and the GTDTcdB-VPI10463–Cdc42 complexes. (A) K448 of GTDTcdA (hot pink) but not the equivalent E449 of GTDTcdB (pale yellow, PDB code: 7S0Y) form a salt bridge with E40 (cyan) of RhoA and potentially D38 of Cdc42 (pale yellow). (B) Residues A377 and L378 on the lower clamp of GTDTcdA (hot pink) interact with the switch I of RhoA better than the equivalent N378 and S379 on GTDTcdB-VPI10463 (pale yellow, PDB code: 7S0Y). (C) Examining the interactions between the pre-switch I of RhoA (cyan) and the lower clamp of GTDTcdA (hot pink) in the GTDTcdA–RhoA complex when compared to the GTDTcdB-VPI10463–Cdc42 complex (pale yellow) and RhoA·GDP (slate, PDB code: 1FTN). (D) Comparing the interactions in the switch II area for the GTDTcdA–RhoA (hot pink and cyan, respectively) and the GTDTcdB-VPI10463–Cdc42 (pale yellow) complexes.
We then turned our attention to the Rho switch II binding area. Even though the sequences in the switch II are identical for Rho proteins, RhoA·GDP and Cdc42·GDP exhibited slightly different conformations in this area, suggesting some degree of flexibility35 (Fig. S2A). Interestingly, the switch II is fixed to an identical conformation upon GTD binding in both cases of TcdA and TcdB (Fig. S2B). We found that most of the interactions in this area are similar on GTDTcdA and GTDTcdB-VPI10463. For example, (1) two pairs of salt bridges between K172 of GTDTcdA and RhoA D65, as well as D432 of GTDTcdA and RhoA R68 are identical to that observed in GTDTcdB-VPI10463 and Cdc42 (Fig. 4D); (2) P451 of GTDTcdA forms a hydrophobic packing against RhoA Y66, which is corresponding to a cation-pi interaction between K452 of GTDTcdB-VPI10463 and Cdc42 Y64; (3) GTDTcdA uses residues H431, A450, F435 and L446 to anchor hydrophobic L69 and L72 on RhoA switch II, which are replaced by homologous residues I432, G451, M436 and M447 on GTDTcdB-VPI10463.
Discussion
This work complements our previous studies of GTPase recognition by GTDTcdB20, which together provide a more complete understanding of substrate binding mechanism for TcdA and TcdB. Based on structural and sequence analyses, we found that, despite only ~ 51% overall sequence identity across the whole domain, GTDTcdA and GTDTcdB exploit a similar strategy to target Rho proteins. Moreover, the GTDTcdA-binding residues on RhoA are largely conserved on Rac1 and Cdc42 (Fig. S3, Table S2), which suggest that GTDTcdA may adopt a similar binding pattern to recognize Rac1 and Cdc42. At the same time, structural comparison between the GTDTcdA–RhoA complex and the GTDTcdB-M68–R-Ras complex also provides new insights into how GTDTcdA may target H/N/K-Ras as its minor substrates27,28,40. Based on structural modeling, we were able to map the potential interacting residues on GTDTcdA and H/N/K-Ras, respectively, which reveals interactions that are conserved between GTDTcdA and GTDTcdB in terms of Ras binding, as well as adaptive residue changes on both GTDTcdA and H/N/K-Ras that may establish unique pair-wise interactions (Table S3). This is consistent with our early findings that the GTDs from diverse TcdB natural variants use a common binding mode to target Rho and Ras GTPases by evolving selective amino acid changes at the substrate-binding interface to adjust its substrate specificity20. We expect that the comprehensive structural information reported here and in our earlier work will provide a blueprint to guide future mutagenesis and functional studies, which will reveal a more complete understanding of the glucosyltransferase activities and substrate selectivity of TcdA and TcdB and their contributions to C. difficile pathogenesis.
Methods
Protein expression and purification
The genes encoding RhoA (residues 1–181, Addgene, plasmid #12959) and TcdA GTD (residues 1–542, strain VPI10463) connected by a peptide linker (GGGGSGGGSGTGSGGGGS) were cloned into the pGEX6p-1 vector via BamH I/Xho I restriction sites. The T37N mutation on RhoA and K190A mutation on TcdA GTD were introduced via QuikChange and verified by DNA sequencing.
The recombinant protein was overexpressed in E. coli strain BL21-star (Invitrogen). Bacteria were cultured at 37 °C in LB medium containing ampicillin. Protein expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) when cell density (OD600) reached ~ 0.8. The temperature was then reduced to 18 °C, and the protein expression continued at 18 °C for 18 h. Cells were harvested by centrifugation and stored at − 80 °C for future use.
For purification, cell pellets were re-suspended in a buffer containing 50 mM HEPEs, pH 7.4, 400 mM NaCl and lysed by sonication, and the fusion protein was purified by Glutathione Sepharose resins (Genesee Scientific). The GST-tag was removed by overnight on-column PreScission Protease cleavage at 4 °C and the flow through was collected, which was exchanged into a buffer containing 20 mM Tris, pH 8.5, 40 mM NaCl, and further purified using Mono-Q ion-exchange chromatography (GE Healthcare). The peak fractions were pooled and exchanged into a buffer containing 20 mM Tris, pH 8.0, 150 mM NaCl, 2 mM MgCl2, 2 mM MnCl2, 0.05 mM GDP, and 2 mM UDP-glucose, which was further concentrated to ~ 10 mg/ml for crystallization.
Crystallization
Initial crystallization screening of the GTDTcdA–RhoA complex was carried out at 18 °C using a Gryphon crystallization robot (Art Robbins Instruments) with sparse matrix screening kits from Hampton Research and Qiagen using the sitting-drop vapor diffusion method (0.2 μl protein + 0.2 μl reservoir equilibrated against 50 μl reservoir). The best crystals were obtained in a condition containing 0.2 M ammonium sulfate, 0.1 M MES, pH 5.9, and 10% (w/v) PEG 8000, after manual optimization and streak-seeding. Crystals were cryoprotected in the mother liquor supplemented with 25% (v/v) ethylene glycerol and snap frozen in liquid nitrogen for data collection.
Data collection and structure determination
The X-ray diffraction data were collected at 100 K at the NE-CAT beamline 24-ID-C, Advanced Photon Source. The data were processed using XDS as implemented in RAPD (https://github.com/RAPD/RAPD)41. The structure was solved by molecular replacement using TcdA GTD (PDB code: 3SRZ)25 and RhoA (PDB code: 1FTN)35 as search models. One GTDTcdA–RhoA complex was positioned in the asymmetric unit using PHENIX.Phaser-MR42. All refinement and model building procedures were carried out with PHENIX.refine43, refmac544, and COOT45. All the refinement progress was monitored with the free R value using a 5% randomly selected test set46. The structure was validated by MolProbity47. Table S1 shows the detailed statistics of data collection and refinement. All the structure figures were prepared by PyMOL (DeLano Scientific).
Data availability
The coordinates and structure factors for the GTDTcdA–RhoA complex has been deposited to the Protein Data Bank under access code 7U2P. All other relevant data are within the manuscript and the Supplemental information.
References
Kuehne, S. A. et al. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467, 711–713. https://doi.org/10.1038/nature09397 (2010).
Leffler, D. A. & Lamont, J. T. Clostridium difficile Infection. N. Engl. J. Med. 372, 1539–1548. https://doi.org/10.1056/NEJMra1403772 (2015).
Sun, X., Savidge, T. & Feng, H. The enterotoxicity of Clostridium difficile toxins. Toxins (Basel) 2, 1848–1880. https://doi.org/10.3390/toxins2071848 (2010).
Aktories, K., Schwan, C. & Jank, T. Clostridium difficile toxin biology. Annu. Rev. Microbiol. 71, 281–307. https://doi.org/10.1146/annurev-micro-090816-093458 (2017).
Papatheodorou, P., Zamboglou, C., Genisyuerek, S., Guttenberg, G. & Aktories, K. Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis. PLoS ONE 5, e10673. https://doi.org/10.1371/journal.pone.0010673 (2010).
Chen, P. et al. Structural basis for recognition of frizzled proteins by Clostridium difficile toxin B. Science 360, 664–669. https://doi.org/10.1126/science.aar1999 (2018).
Chen, P. et al. Structural basis for CSPG4 as a receptor for TcdB and a therapeutic target in Clostridioides difficile infection. Nat. Commun. 12, 3748. https://doi.org/10.1038/s41467-021-23878-3 (2021).
Tao, L. et al. Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature 538, 350–355. https://doi.org/10.1038/nature19799 (2016).
Tao, L. et al. Sulfated glycosaminoglycans and low-density lipoprotein receptor contribute to Clostridium difficile toxin A entry into cells. Nat. Microbiol. 4, 1760–1769. https://doi.org/10.1038/s41564-019-0464-z (2019).
Qa’Dan, M., Spyres, L. M. & Ballard, J. D. pH-induced conformational changes in Clostridium difficile toxin B. Infect Immun. 68, 2470–2474 (2000).
Genisyuerek, S. et al. Structural determinants for membrane insertion, pore formation and translocation of Clostridium difficile toxin B. Mol. Microbiol. https://doi.org/10.1111/j.1365-2958.2011.07549.x (2011).
Zhang, Z. et al. Translocation domain mutations affecting cellular toxicity identify the Clostridium difficile toxin B pore. Proc. Natl. Acad. Sci. USA 111, 3721–3726. https://doi.org/10.1073/pnas.1400680111 (2014).
Barth, H. et al. Low pH-induced formation of ion channels by Clostridium difficile toxin B in target cells. J. Biol. Chem. 276, 10670–10676. https://doi.org/10.1074/jbc.M009445200 (2001).
Reineke, J. et al. Autocatalytic cleavage of Clostridium difficile toxin B. Nature 446, 415–419 (2007).
Egerer, M., Giesemann, T., Jank, T., Satchell, K. J. & Aktories, K. Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J. Biol. Chem. 282, 25314–25321 (2007).
Kordus, S. L., Thomas, A. K. & Lacy, D. B. Clostridioides difficile toxins: Mechanisms of action and antitoxin therapeutics. Nat. Rev. Microbiol. https://doi.org/10.1038/s41579-021-00660-2 (2021).
Chen, S., Sun, C., Wang, H. & Wang, J. The role of rho GTPases in toxicity of Clostridium difficile toxins. Toxins 7, 5254–5267. https://doi.org/10.3390/toxins7124874 (2015).
Just, I. et al. Glucosylation of rho proteins by Clostridium difficile toxin B. Nature 375, 500–503. https://doi.org/10.1038/375500a0 (1995).
Just, I. et al. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J. Biol. Chem. 270, 13932–13936. https://doi.org/10.1074/jbc.270.23.13932 (1995).
Liu, Z. et al. Structural basis for selective modification of Rho and Ras GTPases by Clostridioides difficile toxin B. Sci. Adv. 7, eabi582. https://doi.org/10.1126/sciadv.abi4582 (2021).
Orrell, K. E. & Melnyk, R. A. Large clostridial toxins: Mechanisms and roles in disease. Microbiol. Mol. Biol. Rev. 85, e0006421. https://doi.org/10.1128/mmbr.00064-21 (2021).
Aktories, K. Bacterial protein toxins that modify host regulatory GTPases. Nat. Rev. Microbiol. 9, 487–498. https://doi.org/10.1038/nrmicro2592 (2011).
Lemichez, E. & Aktories, K. Hijacking of Rho GTPases during bacterial infection. Exp. Cell Res. 319, 2329–2336. https://doi.org/10.1016/j.yexcr.2013.04.021 (2013).
Jaffe, A. B. & Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269. https://doi.org/10.1146/annurev.cellbio.21.020604.150721 (2005).
Pruitt, R. N. et al. Structural determinants of Clostridium difficile toxin A glucosyltransferase activity. J. Biol. Chem. 287, 8013–8020. https://doi.org/10.1074/jbc.M111.298414 (2012).
Genth, H. et al. Haemorrhagic toxin and lethal toxin from Clostridium sordellii strain vpi9048: molecular characterization and comparative analysis of substrate specificity of the large clostridial glucosylating toxins. Cell Microbiol. 16, 1706–1721. https://doi.org/10.1111/cmi.12321 (2014).
Zeiser, J., Gerhard, R., Just, I. & Pich, A. Substrate specificity of clostridial glucosylating toxins and their function on colonocytes analyzed by proteomics techniques. J. Proteome Res. 12, 1604–1618. https://doi.org/10.1021/pr300973q (2013).
Junemann, J. et al. Quantification of small GTPase glucosylation by clostridial glucosylating toxins using multiplexed MRM analysis. Proteomics https://doi.org/10.1002/pmic.201700016 (2017).
Yao, G. et al. N-linked glycosylation of SV2 is required for binding and uptake of botulinum neurotoxin A. Nat. Struct. Mol. Biol. 23, 656–662. https://doi.org/10.1038/nsmb.3245 (2016).
Jin, R., Rummel, A., Binz, T. & Brunger, A. T. Botulinum neurotoxin B recognizes its protein receptor with high affinity and specificity. Nature 444, 1092–1095 (2006).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797. https://doi.org/10.1016/j.jmb.2007.05.022 (2007).
Alvin, J. W. & Lacy, D. B. Clostridium difficile toxin glucosyltransferase domains in complex with a non-hydrolyzable UDP-glucose analogue. J. Struct. Biol. 198, 203–209. https://doi.org/10.1016/j.jsb.2017.04.006 (2017).
Ziegler, M. O., Jank, T., Aktories, K. & Schulz, G. E. Conformational changes and reaction of clostridial glycosylating toxins. J. Mol. Biol. 377, 1346–1356. https://doi.org/10.1016/j.jmb.2007.12.065 (2008).
Ihara, K. et al. Crystal structure of human RhoA in a dominantly active form complexed with a GTP analogue. J. Biol. Chem. 273, 9656–9666. https://doi.org/10.1074/jbc.273.16.9656 (1998).
Wei, Y. et al. Crystal structure of RhoA-GDP and its functional implications. Nat. Struct. Biol. 4, 699–703. https://doi.org/10.1038/nsb0997-699 (1997).
Chaves-Olarte, E. et al. R-Ras glucosylation and transient RhoA activation determine the cytopathic effect produced by toxin B variants from toxin A-negative strains of Clostridium difficile. J. Biol. Chem. 278, 7956–7963. https://doi.org/10.1074/jbc.M209244200 (2003).
Mansfield, M. J. et al. Phylogenomics of 8839 Clostridioides difficile genomes reveals recombination-driven evolution and diversification of toxin A and B. PLoS Pathog. 16, e1009181. https://doi.org/10.1371/journal.ppat.1009181 (2020).
Li, Z. et al. Ribotype classification of Clostridioides difficile isolates is not predictive of the amino acid sequence diversity of the toxin virulence factors TcdA and TcdB. Front. Microbiol. 11, 1310. https://doi.org/10.3389/fmicb.2020.01310 (2020).
Shen, E. et al. Subtyping analysis reveals new variants and accelerated evolution of Clostridioides difficile toxin B. Commun. Biol. 3, 347. https://doi.org/10.1038/s42003-020-1078-y (2020).
Genth, H. et al. Difference in mono-O-glucosylation of ras subtype GTPases between toxin A and toxin B from Clostridioides difficile strain 10463 and lethal toxin from Clostridium sordellii strain 6018. Front. Microbiol. 9, 3078. https://doi.org/10.3389/fmicb.2018.03078 (2018).
Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132. https://doi.org/10.1107/S0907444909047337 (2010).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007).
Adams, P. D. et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221. https://doi.org/10.1107/S0907444909052925 (2010).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D 67, 355–367. https://doi.org/10.1107/S0907444911001314 (2011).
Emsley, P. & Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132. https://doi.org/10.1107/S0907444904019158 (2004).
Brunger, A. T. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 (1992).
Chen, V. B. et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21. https://doi.org/10.1107/S0907444909042073 (2010).
Acknowledgements
This work was partly supported by National Institute of Allergy and Infectious Diseases (NIAID) Grants R01AI125704, R01AI139087, R01AI158503, and R21AI156092 to R.J. NE-CAT at the Advanced Photon Source (APS) is supported by a Grant from the National Institute of General Medical Sciences (P30 GM124165). Use of the APS, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.
Author information
Authors and Affiliations
Contributions
Conceptualization: B.C. and R.J.; Investigation: B.C., Z.L., and K.P.; Supervision and funding acquisition: R.J.; Writing: B.C. and R.J.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, B., Liu, Z., Perry, K. et al. Structure of the glucosyltransferase domain of TcdA in complex with RhoA provides insights into substrate recognition. Sci Rep 12, 9028 (2022). https://doi.org/10.1038/s41598-022-12909-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-12909-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
