
Abstract
Ebola virus (EBOV) is a dangerous zoonotic infectious disease. To date, more than 25 EBOV outbreaks have been documented, the majority of which have occurred in Central Africa. The rVSVG-ZEBOV-GP vaccine (ERVEBO), a live attenuated vaccine, has been approved by the US Food and Drug Administration (FDA) to combat EBOV. Because of the several drawbacks of live attenuated vaccines, multi-epitope vaccines probably appear to be safer than live attenuated vaccines. In this work, we employed immunoinformatics tools to design a multi-epitope vaccine against EBOV. We collected sequences of VP35, VP24, VP30, VP40, GP, and NP proteins from the NCBI database. T-cell and linear B-cell epitopes from target proteins were identified and tested for antigenicity, toxicity, allergenicity, and conservancy. The selected epitopes were then linked together in the vaccine's primary structure using appropriate linkers, and the 50S ribosomal L7/L12 (Locus RL7 MYCTU) sequence was added as an adjuvant to the vaccine construct's N-terminal. The physicochemical, antigenicity, and allergenicity parameters of the vaccine were all found to be satisfactory. The 3D model of the vaccine was predicted, refined, and validated. The vaccine construct had a stable and strong interaction with toll-like receptor 4 (TLR4) based on molecular docking and molecular dynamic simulation (MD) analysis. The results of codon optimization and in silico cloning revealed that the proposed vaccine was highly expressed in Escherichia coli (E. coli). The findings of this study are promising; however, experimental validations should be carried out to confirm these findings.
Similar content being viewed by others
Introduction
Ebola virus (EBOV) is a hazardous zoonotic infectious disease that was first identified in 1976 when it spread simultaneously in South Sudan and a village near the Ebola River in Yambuku city of Democratic Republic of Congo1,2. Since then, more than 25 EBOV outbreaks have been reported, the majority of which have occurred in Central Africa1. The 2014–2016 outbreak in West Africa was the largest documented outbreak of EBOV, which infected over 28,000 people and caused over 11,000 deaths3. The African continent's favorable environmental conditions help in promoting EBOV endemicity. At least one human case of one or more Ebolavirus species has been reported in thirteen African countries4. Regrettably, intermittent imported EBOV cases have been reported in the United States, United Kingdom, Thailand, Spain, and Canada5,6.
EBOV is transmitted from wild animals to humans and spreads through human-to-human transmission7. Human-to-human transmission of EBOV occurs through direct contact with infected people's skin, contact with blood and body fluids, and sexual intercourse, handling, secretions, organs, or other bodily fluids of infected people8. The symptoms of EBOV disease, formerly known as EBOV hemorrhagic fever, include fever, headache, fatigue, muscle aches, internal and external bleeding, diarrhea, vomiting, and impaired kidney and liver function5,9,10.
The genus Ebolavirus belongs to the Filoviridae family and includes six species: Bombali ebolavirus, Bundibugyo ebolavirus, Reston ebolavirus, Sudan ebolavirus, Tai Forest ebolavirus, and Zaire ebolavirus11. The EBOV are viruses that contain linear, non-segmented, negative-sense, single-stranded genomic RNA12. Replication promoters are encoded at the 3- and 5-ends of the 19-kb genome, which are known as the 3-leader and 5-trailer, respectively13. The EBOV genome contains seven genes coding for nine mRNAs and proteins, including the nucleoprotein (NP), viral RNA polymerase cofactor (VP35), matrix proteins (VP40), spike glycoprotein (GP) (3 variants), transcriptional activator (VP30), second matrix protein (VP24), and RNA polymerase (L)14. The NP is responsible for genome encapsulation during virus assembly and the protection of viral RNA from degradation15. The VP35 interacts with NP and L during the EBOV RNA replication process, and both VP35-NP and VP35-L interactions are required for viral RNA synthesis16,17. The role of VP40 is vital in the final stage of EBOV replication, when viral particles bud from host cells18,19,20. VP40 attaches the viral nucleocapsid to the inside of the cell membrane and facilitates egress to complete the viral life cycle21. The GP is responsible for EBOV attachment and entry into host cells, as well as inducing immune responses in host cells22,23. The VP30 is a phosphorylation-dependent viral transcription factor that is required to initiate RNA transcription24. The minor viral matrix protein VP24 condenses viral nucleocapsids, which is necessary for efficient nucleocapsid packaging into the virion25,26,27. The L protein is an RNA-dependent polymerase that complexes with VP30 and performs all of the enzymatic activities that are required for virus genome transcription and replication28.
Only a few therapeutics for EBOV disease have been developed and tested to now. The US Food and Drug Administration (FDA) has approved two EBOV-specific monoclonal antibody therapies, REGN-EB3 and mAb114, for the treatment of EBOV disease29. As mentioned, various African countries, the majority of which are low- and middle-income, are more susceptible to this disease. Therefore, more affordable treatments that are easily stored and useful for low- and middle-income countries are required30. Vaccines are critical for managing infectious diseases, particularly among people living in poor health. Vaccines can also have herding effects, resulting in protection even among individuals who have not been vaccinated, which is especially important for the poor who lack access to health care31. The rVSVΔG-ZEBOV-GP vaccine (ERVEBO) was approved on December 19, 2019, by the FDA to prevent EBOV infection in people 18 years of age and older32. The ERVEBO is a live attenuated vaccine that was created by replacing the gene encoding the glycoprotein of the vesicular stomatitis virus strain Indiana with the gene encoding the glycoprotein of the EBOV-Kikwit 1995 strain33. Due to numerous disadvantages of live attenuated vaccines, such as the possibility of reversion to virulent form and instability in various storage conditions, it appears that multi-epitope vaccines appear to be safer than live attenuated vaccines34. The multi-epitope vaccines have received a lot of attention because they have higher immunity, lower allergic reactions, and easier production than conventional vaccines35. The main downside of these vaccines is their low immunogenicity, which is overcome by the use of adjuvants36. The majority of in silico studies on the design of an Ebola vaccine has progressed to the point of predicting epitopes. The vaccine construct has been designed in several studies, although bioinformatics analyses on it have not been extensive. The aim of this study is to in silico design a vaccine construct for the first time that includes the epitopes of VP35, VP24, VP30, VP40, GP, and NP proteins, as well as a 50S ribosomal L7/L12 sequence as an adjuvant. In this regard, target proteins were first selected to predict T-cell and B-cell epitopes. The epitopes were then screened based on their antigenicity, toxicity, allergenicity, conservancy, and potential to produce IFN-gamma and IL-4. The top-ranked epitopes were arranged in a vaccine construct using appropriate linkers. To improve vaccine immunogenicity, the 50S ribosomal L7/L12 (Locus RL7 MYCTU) sequence (as a TLR4 agonist) was added to the vaccine construct's N-terminal as an adjuvant. Through the TLR4, 50S ribosomal L7/L12 can induce immature dendritic cells and naive T cells and elicit an adaptive immune response37. The vaccine construct was evaluated for physicochemical characterisation, solubility, allergenicity, and antigenicity. The secondary structure and 3D structure of the vaccine construct were modeled, and the 3D model was refined. Further, molecular docking of the 3D model with TLR4 was performed, subsequently, the docked complex was subjected to molecular dynamics (MD) simulation to analyze its structural dynamics and stability. Finally, codon optimization and in silico cloning were done to confirm the vaccine's effective expression. Figure 1 depicts the step-by-step workflow employed in this study.

A schematic workflow of constructing a multi-epitope vaccine against EBOV.
Results
Identification and retrieval of viral protein sequences
Based on previous studies38,39,40 and the Protegen server, the VP35, VP24, VP30, VP40, GP, and NP proteins were chosen as target proteins for epitope prediction. The VaxiJen v2.0 server predicted that all target proteins had an antigen score of > 0.4 (threshold value); their accession number and antigenicity scores are given in Table 1.
T-cell epitope prediction
Cytotoxic T lymphocytes (CTLs) are critical elements of adaptive immunity that play a key role in the elimination of infected cells41. CTL epitopes are involved in the development of long-lasting cellular immunity and can remove circulating viruses as well as virus-infected cells42. The NetCTL 1.2 server identified a total of 591 CTL epitopes from six proteins. To determine the best CTL epitopes from a set of CTL epitopes, we first chose CTL epitopes that bind strongly to at least three MHC class I supertypes. These epitopes were then filtered for antigenicity, toxicity, allergenicity, and conservancy. Finally, ten antigenic, non-toxic, and non-allergenic CTL epitopes with more than 80% conservancy were chosen (Tables S1–S6).
Helper T lymphocytes (HTLs) play an important in the elimination of extracellular pathogens using various cytokines and stimulating humoral immune responses43. HTL epitopes help regulate the adaptive immune system by inducing the production of T cell cytokines44. For the six target proteins, 912 HTL epitopes of 15-mer length were predicted by the NetMHCII 2.3 server. Among them, HTL epitopes that could bind strongly to at least 5 human MHC class II alleles were screened for antigenicity, toxicity, allergenicity, and conservancy, as well as the production of cytokines such as IFN-gamma and IL-4. Considering the mentioned characteristics, a total of 17 HTL epitopes were selected (Tables S7–S12).
Linear B-cell epitope prediction
B-cells are the main body of humoral immunity. Antibodies produced by B-cells play a critical role in preventing the spread of viral infections45. Linear B cell epitopes are responsible for producing antigen-specific antibodies46. We predicted 68 linear B-cell epitopes using the IEDB server in this study (Fig. S1). The epitopes with a size of 10–30 mer were tested for antigenicity, toxicity, allergenicity, and conservancy. Finally, seven epitopes were chosen for the VP35, VP24, VP30, VP40, and GP proteins, while no epitopes were chosen for the NP protein (Tables S13–S18).
Multi-epitope vaccine construction
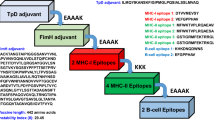
To avoid epitopes repetition in the vaccine construct, epitopes whose sequences were found in other epitopes were eliminated. A total of 7 CTL, 17 HTL, and 6 linear B-cell epitopes were fused using AAY, GPGPG, and KK linkers, respectively (Table 2). In addition, the 50S ribosomal L7/L12 adjuvant (with a length of 130 amino acids) was attached to the N-terminal of the vaccine sequence by an EAAAK linker. The final vaccine structure included 678 amino acids (Fig. 2).

A schematic depiction of the final multi-epitope vaccine construct.
Evaluating physicochemical, solubility, allergenicity, and antigenicity parameters of the designed vaccine
Several important physicochemical features of the multi-epitope vaccine were characterized by the ExPASy ProtParam tool. The theoretical pI and GRAVY of the vaccine construct were estimated to be 9.09 and − 0.264, respectively. The half-life of the vaccine was calculated to be 30 h in mammalian reticulocytes (in vitro), more than 20 h in yeast (in vivo), and more than 10 h in E. coli (in vivo). The molecular weight was computed to be 73.15 kDa. For the vaccine construct, an aliphatic index of 81.47 and an instability index of 26.72 were predicted. The SOLpro server predicted that the multi-epitope vaccine was soluble with a probability of 0.819255. The AllerTOP v. 2.0 server predicted that the vaccine construct was non-allergen. The antigenicity of the vaccine sequence was predicted to be 0.958093 by ANTIGENpro and 0.5179 by the VaxiJen 2.0 server with a virus model and 0.7422 with a bacteria model at a threshold of 0.4.
Secondary structure prediction
The secondary structure of the multi-epitope vaccine was generated by PDBsum. The vaccine construct was composed of 41 helices, 71 helix-helix interactions, 125 beta turns, and 22 gamma turns (Fig. S2).
Tertiary structure prediction, refinement, and validation of the vaccine construct
I-TASSER server generated five 3D structures of the vaccine candidate based on threading templates (PDB Hit: 1rquA, 5lqwQ, 2ftc, 7ey7S, 7eybI, 1dd4, 7f56A, and 4uicA). According to the Z-score values (ranging from 1.26 to 4.74), all of the threading templates were good aligned, the Z-score > 1 indicates that the alignment is good, and vice versa47. Models 1–5 had C-scores of − 0.61, − 2.29, − 2.74, − 2.98, and − 4.36, respectively. For the refining process, the model with the highest C value (− 0.61) was chosen. The GalaxyRefne server was used to refine the selected model. The GalaxyRefne server provided a total of five refined models of the vaccine. Model 3 was considered the best refined model based on its model quality scores (Table 3). Higher GDT-HA values indicate higher quality models48; model 3 has a GDT-HA score of 0.9421, which was higher than all refined models. A lower RMSD value means more stability, and an RMSD score of 0 to 1.2 is normally considered acceptable48. The RMSD score for this model is 0.442. The MolProbity score represents the crystallographic resolution of the protein's 3D model. A lower MolProbity score indicates a less critical error49. Model 3 has a MolProbity score of 2.428, which is significantly lower than the initial model. The Clash score indicates the number of undesirable all-atom spatial overlaps, and the poor rotamers score indicates the number of residues with a lower capacity of conceivable rotation in their side chains50; the lower the score of these parameters, the better the 3D structure of the protein; the Clash and poor rotamers scores of model 3 are 23.9 and 0.9, respectively. The higher the Rama favored value, the higher the model's quality. Model 3 has Rama favored of 89.6. The initial and refined 3D model of the vaccine has been shown in Fig. 3. The quality of the initial and refined models of the multi-epitope vaccine was evaluated using the SWISS-MODEL Structure Assessment and ProSA-web server. Ramachandran plot analysis of the initial model revealed that 72.29% of the residues were in the favoured region, whereas in the refined model 88.76% of the residues were in the favoured region (Fig. S3). The z-scores calculated by the ProSA web server for the initial model and the refined model of the vaccine were − 2.46 and − 3.47, respectively (Fig. S3).

The initial 3D model of the vaccine is shown in blue and the refined 3D model is shown in gold. The UCSF Chimera 1.15rc software was used to visualize them.
Discontinuous B-cell epitope prediction
The ElliPro server predicted 16 discontinuous B-cell epitopes with scores ranging from 0.585 to 0.829 (at a threshold of 0.5). The size of the epitopes varied from 8 to 39 residues (Table 4). Discontinuous B-cell epitopes with a score above 0.8 are shown in (Fig. S4).
Disulfide engineering of the vaccine construct
In the refined model of our vaccine, the DbD2 server predicted 59 residue pairs with the potential to form disulfide bonds. The residue pairs were screened based on χ3 angle (between − 87° and + 97°) and bond energy (less than 2.2 kcal/mol) parameters. Only six residue pairs were chosen for disulfide bond formation after careful consideration, including PHE28-GLY46, THR31-VAL42, PHE32-LYS79, PRO 101-ALA113, PHE284-GLY317, and GLY519-MET549. The original and mutant models of the vaccine candidate are shown in Fig. S5.
Molecular docking analysis
ClusPro 2.0 server conducted molecular docking of the multi-epitope vaccine and TLR4 and generated 30 docked complexes with different cluster members and the lowest energy. Cluster No. 2 had the lowest energy score of − 1279.1 kcal/mol with 38 members, which was the most negative score (strongest interaction) among all docked complexes (Fig. 4). The residues involved in the interaction between TLR4 and vaccine were analyzed by LigPlot software (Fig. 5). Also, the list of residues with hydrogen bonds along with the bond length is given in Tables 5 and 6.

The docked complex of the multi-epitope vaccine and TLR4 was visualized using the UCSF Chimera 1.15rc software. The vaccine construct is shown in the spheres form, while TLR4 is shown in the cartoon form.

Interaction of the vaccine with chains B and D of TLR4 was visualized using the LigPlot software. The green dashed lines indicate hydrogen bonds.
Molecular dynamics simulation of the docked complex
The molecular dynamic simulation of the vaccine-TLR4 complex was performed using GROMACS 2019.6 software for 40 ns. The RMSD graph is used to assess the structure's stability during the simulation. The RMSD value of TLR4 increased rapidly at the beginning of the simulation, reaching about 0.3 nm at 3000 ps and remaining stable until the end of the simulation. An initial sudden change occurred in the RMSD graph of the vaccine and after 6000 ps it has reached the value of 1.3 nm and showed slight fluctuations around this value until the end of the simulation (Fig. 6a). The input and output structures of the molecular dynamics simulation are stacked so that any changes can be detected. After simulating molecular dynamics, the vaccine shows a relatively high conformational change, as shown in Fig. 6b. In molecular dynamics studies, RMSF is the most commonly used method for measuring the oscillating motions of macromolecules. As seen in the previous section, the vaccine is attached to chain B of TLR4, and since the chains A and B have the same sequence on the TLR4, to accurately investigate the effect of vaccine binding on chain B flexibility, the RMSF plot for chains A and B is provided. The flexibility of the residues at both ends of the TLR4 sequence (N-terminal and C-terminal) in chain A was greater than in chain B, while the flexibility of the other regions in the two chains was the same and did not show a notable change. The RMSF plot of the vaccine revealed that the majority of the vaccine's residues had slight flexibility, indicating that the vaccine construct has established stable interactions with TLR4 (Fig. 6c).

Molecular dynamics simulation analysis of the vaccine–TLR4 complex. (a) RMSD plot of the vaccine and TLR 4 in the docked complex for a time duration of 40 ns. (b) Conformational changes of the vaccine in the docked complex before (green) and after (red) molecular dynamics simulation was visualized using the UCSF UCSF Chimera 1.15rc software. (c) RMSF plot of the vaccine and TLR 4 in the docked complex.
Codon optimization and in silico cloning
The vaccine construct was back translated using JCat to a cDNA sequence with a length of 2034 nucleotides. The vaccine sequence had a CAI value of 0.9571 and a GC content of 53.09%. The vaccine sequence was cloned into the pET-28a (+) vector by SnapGene software. The length of the cloned vaccine was estimated to be 7.166 kbp (Fig. 7).

In silico cloning of the vaccine construct between XhoI and BglII restriction sites of pET-28a (+) vector using SnapGene sofware free-trial (https://www.snapgene.com/free-trial/). The magenta part represents the vaccine sequence, while the black part represents the backbone of the pET28a (+) vector.
Discussion
EBOV disease is one of the most lethal viral diseases affecting both humans and non-human mammals. The disease's mortality rate is reported to be 90%51. The Ebola outbreak was labeled a public health emergency of international concern by the World Health Organization (WHO) in August 201452. Ebola and Marburg are both members of the Filoviridae family, and they are classified as bioterrorism in category A, alongside diseases such as plague, smallpox, and anthrax5. Because of their high virulence, demonstrated aerosol infectivity in the laboratory, and ability to induce stress and paranoia, the filoviruses have been classified as high priority Category A pathogens by the Centers for Disease Control and Prevention (CDC, USA)53.
Vaccines are a safe and effective way of limiting the spread of lethal infectious diseases and saving millions of lives54. Conventional vaccine development methods are labor-intensive, costly, and time-consuming. Furthermore, the likelihood of failure in subsequent trials is high55. The reverse vaccinology will employ computational approaches and bioinformatics tools for antigen identification. This method can identify new potential antigenic proteins that play an important role in the immunogenicity and the safety of the vaccines56. Recently, reverse vaccinology approaches have been utilized to develop multi-epitope vaccines against dengue virus57, human cytomegalovirus58, SARS-CoV-248,59, Mycobacterium tuberculosis60, Helicobacter pylori61, Leishmania infantum62, Echinococcus granulosus63, and Candida auris64.
In this work, we designed a multi-epitope vaccine against EBOV. The identification of antigenic proteins is an important step in vaccine design. Using computational methods, we conducted an organized and thorough evaluation of the Ebola proteins and selected the VP35, VP24, VP30, VP40, GP, and NP proteins as target proteins for epitope prediction. CTL, HTL, and linear B-cell epitopes were predicted from these proteins. In the first step, epitopes (CTL and HTL epitopes) that interacted with a large number of HLA alleles were chosen for further investigation. To ensure that the selected CTL, HTL, and linear B cell epitopes are sufficiently antigenic and conserved while remaining non-toxic and non-allergenic to vaccine recipients, various immunoinformatics tools were used to screen epitopes for the aforementioned parameters. Aside from the aforementioned parameters, HTL epitopes were tested for their ability to induce IFN-gamma and IL-4 production. IFN-gamma has a crucial function in both adaptive and innate immune activation, in addition to interfering with viral replication65. IL-4 causes allergic reactions. This is due to the fact that IL-4 directs the development of TH2, which results in the production of IgE66. Different linkers were utilized to connect epitopes with various patterns in several primary structures of vaccine construct. After analyzing the physicochemical characteristics, particularly the stability of the generated constructs, it was found that AAY, GPGPG, and KK linkers are appropriate for joining CTL, HTL, and linear B-cell epitopes, respectively. Our vaccine construct's primary structure included 1 adjuvant, 7 CTL epitopes, 17 HTL epitopes, 6 linear B-cell epitopes, 1 EAAAK linker, 6 AAY linkers, 17 GPGPG linkers, and 6 KK linkers. The EAAAK linker is a rigid linker that, due to its helix formation properties, helped improve the immunogenic properties43,67. AAY, GPGPG, and KK linkers are typically made up of flexible and hydrophilic amino acids, which could help to prevent domain disruption68.
The vaccine's physicochemical characteristics were evaluated to facilitate subsequent experimental evaluations of the vaccine and to enable the successful setup of in vitro and in vivo assays. The theoretical pI of the vaccine was determined to be 9.09, suggesting that the vaccine is basic in nature. The vaccine construct's GRAVY was − 0.264; a negative value for this parameter indicates that the vaccine is hydrophilic and also has a high degree of solubility69. Our vaccine construct has a lower GRAVY than the vaccine designed in the study by Kadam et al.38, indicating that our vaccine will interact better with water molecules. The half-life of our vaccine in mammalian, yeast, and E. coli was determined to be 30, 20, and 10 h, respectively, implying that the vaccine is exposed to the immune system for a longer period of time and induces more immune responses70. The predicted half-life of the vaccine designed in the study of Shankar et al.40 was identical to the half-life of our vaccine. The vaccine's molecular weight was determined to be 73.15 kDa; vaccines with molecular weights less than 110 kDa could be a good vaccine candidate since they are easier to clone and express in expression systems than large proteins71. The aliphatic index score was 81.47; an aliphatic index greater than 50 indicates that the vaccine is stable at higher temperatures72. The vaccine instability index was 26.72, indicating its stability. The instability index of the vaccine designed in the study by Kadam et al.38 was 38, indicating that our vaccine is more stable. In general, a protein with an instability index of less than 40 is considered stable73. The Solpro server result showed that our vaccine was soluble. The vaccine construct was identified as non-allergen by the AllerTOP v. 2.0 server. Our vaccine had high antigenicity scores on both the ANTIGENpro and the Vaxijen v2.0 servers. The results of predicting the vaccine's second structure were acceptable because the number of helices in the second structure, which indicates the number of hydrogen bonds and thus the protein's stability, was sufficient74.
I-TASSER server predicted the vaccine 3D model, which was then refined by the GalaxyRefine server. The Ramachandran plot revealed that 72.29% of the residues in the initial vaccine model were in the favoured region, which increased to 88.76% percent after refining. The initial model had a z-score of − 2.46, whereas the refined model had a z-score of − 3.47. A more negative z-score indicates that the 3D refined model of the vaccine is of higher quality75. In the 3D refined model of the vaccine, 16 discontinuous B-cell epitopes were predicted. Because discontinuous B-cell epitopes play a critical role in humoral immune responses by producing antibodies76, our designed vaccine has the potential to induce large amounts of antibody production. The vaccine's refined structure was subjected to disulfide engineering, and 6 disulfide bonds were introduced in the refined model to increase the vaccine structure's stability77.
Since TLR4 is involved in the activation of proinflammatory mediators after EBOV infection78, the molecular docking analysis of the vaccine construct with TLR4 was carried out. The results of this analysis revealed that the vaccine had a significant affinity for TLR4, implying that it may elicit both an innate and adaptive immune response79. The MD simulation results of the vaccine-TLR4 docked complex revealed that the vaccine reached a stable state in less than 40 ns and that its flexible residues were present in the range of 500–678, because this region had no interaction with TLR4, and can move freely, so its flexibility is higher than other regions of the vaccine construct. JCat server revealed that the vaccine construct had a CAI of 0.9571 and a GC content of 53.09%. CAI greater than 0.89080 and GC content between 30 and 70%76 is ideal for target organism expression. In the present study, we used a set of bioinformatics software to design a multi-epitope vaccine against Ebola, and the results were promising. The only limitation of this study is the need for further in vitro and in vivo studies to demonstrate the vaccine candidate's efficacy and safety.
Conclusion
EBOV is one of the most dangerous viruses among viral hemorrhagic fevers. The purpose of the current study was to design a multi-epitope vaccine against EBOV utilizing immunoinformatics approaches. T-cell and B-cell epitopes of target antigens were predicted, and epitope screening was conducted correctly and sequentially. The vaccine possessed all of the desirable qualities of a vaccine candidate, including good physicochemical properties, solubility, high antigenicity, and non-allergenicity. The vaccine's strong affinity for TLR4 was validated by molecular docking analyses, and the vaccine's stability was ensured by MD simulation. The codon optimization also presented an optimistic CAI value and GC content, confirming the vaccine's expression in a bacterial host. The rational design of the linear structure of the multi-epitope vaccine, which resulted from the proper arrangement of the selected epitopes and adjuvant, made the results of this study more impressive than previous studies. Our findings suggest that the vaccine candidate may elicit appropriate immune responses. As a result, we believe that this vaccine candidate, if further evaluated in vitro and in vivo, could be a promising vaccine against EBOV.
Materials and methods
Identification and retrieval of viral protein sequences
In the first step of the study, antigen proteins of the Zaire Ebola virus were identified using literature38,39,81 and the Protegen server (http://www.violinet.org/protegen/). The Protegen is a web-based central database and processing system for collecting, storing, and analyzing protective antigens82. The reference sequences of the identified proteins were retrieved in FASTA format from the NCBI database (https://www.ncbi.nlm.nih.gov/), and their antigenicity was evaluated using the VaxiJen v2.0 server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html). VaxiJen is the first server for alignment-independent prediction of antigenic peptides or proteins from a variety of organisms such as bacteria, viruses, parasites, fungi, and tumors83,84,85.
T-cell epitope prediction
CTL epitopes were predicted for the selected proteins using the NetCTL 1.2 server (http://www.cbs.dtu.dk/services/NetCTL/). The server predicts CTL epitopes (9-mer) for 12 MHC class I supertypes, including A1, A2, A3, A24, A26, B7, B8, B27, B39, B44, B58, and B62. This server identifies epitopes based on proteasomal C terminal cleavage, MHC class I binding peptide, and TAP transport efficiency scores86. For the present study 12 MHC class I supertypes were selected to predict CTL epitopes, and the cutoff value for epitopes prediction was set at 0.75.
The NetMHCII 2.3 server (http://www.cbs.dtu.dk/services/NetMHCII/) was used to predict HTL epitopes. The NetMHCII 2.3 server identifies the binding of the peptides to the HLA-DR, HLA-DQ, HLA-DP, and mouse MHC class II alleles using artificial neural networks (ANNs)87. The threshold values corresponding to the strong and weak binder were set at 2% and 10%, respectively.
The predicted CTL and HTL epitopes were analyzed for antigenicity, toxicity, allergenicity, and conservancy. The antigenicity of the epitopes was checked with the help of the VaxiJen v2.0 server at a threshold value of 0.4. The toxicity or non-toxicity of the epitopes was determined by the ToxinPred server (https://webs.iiitd.edu.in/raghava/toxinpred/design.php) using the SVM (Swiss-Prot) based method88. The AllerTOP v. 2.0 server (https://www.ddg-pharmfac.net/AllerTOP/method.html) was used to analyze the allergenicity of the epitopes. AllerTOP is an alignment-free server for allergenicity prediction based on protein physicochemical parameters89. The Epitope Conservancy Analysis tool from the Immune Epitope Database (IEDB) (http://tools.iedb.org/conservancy/) was used to determine the degree of the conservancy of predicted epitopes. This tool calculates the degree of conservation of an epitope within a defined protein sequence90. In addition to the above mentioned screenings, HTL epitopes were checked for IFN-gamma and IL-4 production. The IFNepitope server (http://crdd.osdd.net/raghava/ifnepitope/design.php)91 and the IL4pred server (http://crdd.osdd.net/raghava/il4pred/)92 were used to predict the ability of HTL epitopes to induce IFN-gamma and IL-4, respectively.
Linear B-cell epitope prediction
The Antigen Sequence Properties tool from IEDB (http://tools.iedb.org/bcell/) was used to predict linear B-cell epitopes. The epitopes were predicted using the Bepipred Linear Epitope Prediction 2.0 method93. Finally, these predicted epitopes were assessed for antigenicity, toxicity, allergenicity, and conservancy using VaxiJen v2.0, ToxinPred, AllerTOP v. 2.0, and Epitope Conservancy Analysis, respectively.
Multi-epitope vaccine construction
To construct a multi-epitope vaccine, the selected CTL, HTL, and linear B-cell epitopes were incorporated using suitable linkers. AAY (Ala-Ala-Tyr), GPGPG (Gly-Pro-Gly-Pro-Gly), and KK (bi-lysine) linkers were used to connect the CTL, HTL, and linear B-cell epitopes, respectively. These linkers were added to achieve the successful separation of individual epitopes94. Finally, to improve the immunogenicity of the multi-epitope vaccine, 50S ribosomal L7/L12 (Locus RL7_MYCTU) sequence with accession no. P9WHE3 as an adjuvant was added to the N-terminal of the vaccine construct via an EAAAK linker.
Evaluating physicochemical, solubility, allergenicity, and antigenicity parameters of the designed vaccine
The ProtParam tool of the Expasy server (https://web.expasy.org/protparam/) was used to analyze a set of physicochemical parameters of the multi-epitope vaccine, including theoretical pI, grand average of hydropathicity (GRAVY), half-life, molecular weight, aliphatic index, and instability index95. The solubility of the proposed vaccine was predicted by the SOLpro server (http://scratch.proteomics.ics.uci.edu/). The SOLpro uses a two-stage SVM algorithm based on multiple representations of the primary sequence to predict whether a protein would be soluble when overexpressed in Escherichia coli (E. coli)96. AllerTOP v. 2.0 server was utilized to evaluate the allergenicity of the multi-epitope vaccine. The antigenicity of the proposed vaccine was checked by ANTIGENpro and VaxiJen v2.0 servers. ANTIGENpro server (http://scratch.proteomics.ics.uci.edu/) predicts the antigenicity of proteins or peptides using a two-stage architecture of their sequence and five machine learning algorithms97.
Secondary structure prediction
PDBsum (http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=index.html) was employed to predict the secondary structure of the proposed vaccine. PDBsum is a web server that provides information on the protein secondary structure, protein–ligand, protein-DNA interactions, and protein structure quality98.
Tertiary structure prediction, refinement, and validation of the vaccine construct
The tertiary structure of the multi-epitope vaccine construct was modeled using the I-TASSER server (https://zhanggroup.org/I-TASSER/). I-TASSER server builds 3D structures from the amino acid sequence by reconfiguring the excised sections from the threading templates and computes the C-score to determine the correctness of the predicted models47,99,100. The GalaxyRefne server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) then refined the selected 3D model of the vaccine. The GalaxyRefine server employs a refining approach that was validated in CASP10. After reconstructing and repacking the side chains, this approach uses molecular dynamics simulation to achieve a general relaxation of the three-dimensional structure. Based on the CASP10 evaluation, this method performed the best in terms of improving the quality of the local structure101,102. The SWISS-MODEL Structure Assessment (https://swissmodel.expasy.org/assess) and ProSA-web server (https://prosa.services.came.sbg.ac.at/prosa.php) were used to compare the quality of the initial 3D structure and the refined structure of the vaccine. The Ramachandran plot is obtained using SWISS-MODEL Structure Assessment103. The Ramachandran plot illustrates the allowed and disallowed dihedral angles psi (ψ) and phi (ϕ) of amino acid, which are calculated based on the van der Waals radius of the side chain104. ProSA web server estimates the overall quality of the model as a z-score. If this z-score falls outside of the normal range for native proteins, the structure most likely contains errors105,106.
Discontinuous B-cell epitope prediction
The ElliPro server (http://tools.iedb.org/ellipro/) was utilized to identify discontinuous B-cell epitopes in the refined 3D model of the vaccine. ElliPro assigns a score to each predicted epitope, which is characterized as a Protrusion Index (PI) value averaged over epitope residues107. The minimum score in this analysis was set at 0.5, while the maximum distance was set at 6 Å.
Disulfide engineering of the vaccine construct
Disulfide engineering is a new strategy of designing new disulfide bonds in the target protein through cysteine mutation of protein structure residues, resulting in increased protein structure stability108. Therefore, the Disulfide by Design 2 (DbD2) server (http://cptweb.cpt.wayne.edu/DbD2/) was used to identify residue pairs that had the potential for mutation and could be used in disulfide engineering109.
Molecular docking analysis
Molecular docking is a computational approach that predicts the preferred orientation of the ligand to the receptor when they are joined together to form a stable complex110. We used the ClusPro 2.0 server (https://cluspro.org/login.php) to perform molecular docking111,112,113,114. Initially, the 3D structure of TLR4 in PDB format (PDB ID: 4G8A) was obtained from the protein data bank (RCSB). The ligands attached to the retrieved TLR4 structure were then removed using Chimera 1.15rc software. The refined 3D model of the multi-epitope vaccine and TLR4 was submitted in the ClusPro 2.0 server as ligand and receptor, respectively. The LigPlot software was also used to analyze the bonds established between the ligand and receptor residues in the docked complex115.
Molecular dynamics simulation of the docked complex
MD simulation is an effective method for studying ligand and receptor stability in a docked complex at the microscopic level116. In this study, MD simulation was run for 40 ns by GROMACS 2019.6 software117. Firstly, the input structure was prepared using the ff99SB force field. Following that, Na+ and Cl− ions were introduced into the system to neutralize the net charge of the system. The complex was then solvated into a 10 Å layer of TIP3P using the gmx solvate software. The energy minimization of the solvated system was performed using the steepest descent approach to eliminate steric clashes. The system was gradually heated to 300 K for 20 ps and then the system was equilibrated at constant pressure, and the SHAKE algorithm was used to keep the hydrogen bond restrained. Finally, the root mean square deviation (RMSD) and root mean square fluctuation (RMSF) were generated using the gmx rms and gmx rmsf modules, respectively.
Codon optimization and in silico cloning
The Java Codon Adaptation Tool (JCat) (http://www.jcat.de/) was utilized for reverse translation, codon optimization, and calculating the codon adaptation index (CAI) value and GC content of the vaccine construct in E. coli (strain K12)118. Three options were chosen here: avoid rho-independent transcription terminators, avoid prokaryotic ribosome binding sites, and avoid cleavage sites of restriction enzyme. The XhoI and BglII restriction sites were tagged at the 5′ and 3′ ends of the vaccine's DNA sequence, respectively. Finally, using SnapGene software (https://www.snapgene.com/free-trial/), the obtained sequence was inserted into the pET28a (+) expression vector between the XhoI and BglII restriction sites.
References
Tomori, O. & Kolawole, M. O. Ebola virus disease: Current vaccine solutions. Curr. Opin. Immunol. 71, 27–33. https://doi.org/10.1016/j.coi.2021.03.008 (2021).
Rajak, H., Jain, D. K., Singh, A., Sharma, A. K. & Dixit, A. Ebola virus disease: Past, present and future. Asian Pac. J. Trop. Biomed. 5, 337–343 (2015).
Schreiber-Stainthorp, W. et al. Longitudinal in vivo imaging of acute neuropathology in a monkey model of Ebola virus infection. Nat. Commun. 12, 2855. https://doi.org/10.1038/s41467-021-23088-x (2021).
Walldorf, J. A., Cloessner, E. A., Hyde, T. B. & MacNeil, A. Considerations for use of Ebola vaccine during an emergency response. Vaccine 37, 7190–7200. https://doi.org/10.1016/j.vaccine.2017.08.058 (2019).
Feldmann, H. & Geisbert, T. W. Ebola haemorrhagic fever. Lancet 377, 849–862. https://doi.org/10.1016/s0140-6736(10)60667-8 (2011).
Maganga, G. D. et al. Ebola virus disease in the Democratic Republic of Congo. N. Engl. J. Med. 371, 2083–2091. https://doi.org/10.1056/NEJMoa1411099 (2014).
Nnaji, N. D. et al. The deuce-ace of Lassa Fever, Ebola virus disease and COVID-19 simultaneous infections and epidemics in West Africa: Clinical and public health implications. Trop. Med. Health 49, 102. https://doi.org/10.1186/s41182-021-00390-4 (2021).
Osterholm, M. T. et al. Transmission of Ebola viruses: What we know and what we do not know. MBio 6, e00137. https://doi.org/10.1128/mBio.00137-15 (2015).
Etard, J.-F. et al. Multidisciplinary assessment of post-Ebola sequelae in Guinea (Postebogui): An observational cohort study. Lancet Infect. Dis. 17, 545–552. https://doi.org/10.1016/S1473-3099(16)30516-3 (2017).
Brasel, T. et al. Mucosal challenge ferret models of Ebola virus disease. Pathogens 10, 292. https://doi.org/10.3390/pathogens10030292 (2021).
O’Donnell, K. L. & Marzi, A. Immunotherapeutics for Ebola virus disease: Hope on the horizon. Biologics 15, 79–86. https://doi.org/10.2147/btt.S259069 (2021).
Ruigrok, R. W. H., Crépin, T. & Kolakofsky, D. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr. Opin. Microbiol. 14, 504–510. https://doi.org/10.1016/j.mib.2011.07.011 (2011).
Bach, S., Demper, J. C., Biedenkopf, N., Becker, S. & Hartmann, R. K. RNA secondary structure at the transcription start site influences EBOV transcription initiation and replication in a length- and stability-dependent manner. RNA Biol. 18, 523–536. https://doi.org/10.1080/15476286.2020.1818459 (2021).
Farman, A., Badshah, S. L. , Khan, K., Ahmad, N., & Naeem, A. Ebola, the Negative Stranded RNA Virus. In Some RNA Viruses (eds Shah, Y. & Abuelzein, E.) https://doi.org/10.5772/intechopen.91776 (IntechOpen, 2020).
Baseler, L., Chertow, D. S., Johnson, K. M., Feldmann, H. & Morens, D. M. The pathogenesis of Ebola virus disease. Annu. Rev. Pathol. 12, 387–418. https://doi.org/10.1146/annurev-pathol-052016-100506 (2017).
Leung, D. W. et al. An intrinsically disordered peptide from Ebola virus VP35 controls viral RNA synthesis by modulating nucleoprotein–RNA interactions. Cell Rep. 11, 376–389. https://doi.org/10.1016/j.celrep.2015.03.034 (2015).
Prins, K. C. et al. Basic residues within the ebolavirus VP35 protein are required for its viral polymerase cofactor function. J. Virol. 84, 10581–10591. https://doi.org/10.1128/jvi.00925-10 (2010).
Harty, R. N. No exit: Targeting the budding process to inhibit filovirus replication. Antiviral Res. 81, 189–197. https://doi.org/10.1016/j.antiviral.2008.12.003 (2009).
Hoenen, T. et al. VP40 octamers are essential for Ebola virus replication. J. Virol. 79, 1898–1905. https://doi.org/10.1128/jvi.79.3.1898-1905.2005 (2005).
Timmins, J., Scianimanico, S., Schoehn, G. & Weissenhorn, W. Vesicular release of Ebola virus matrix protein VP40. Virology 283, 1–6. https://doi.org/10.1006/viro.2001.0860 (2001).
Noda, T., Watanabe, S., Sagara, H. & Kawaoka, Y. Mapping of the VP40-binding regions of the nucleoprotein of Ebola virus. J. Virol. 81, 3554–3562. https://doi.org/10.1128/jvi.02183-06 (2007).
Dolnik, O., Kolesnikova, L. & Becker, S. Filoviruses: Interactions with the host cell. Cell. Mol. Life Sci. 65, 756. https://doi.org/10.1007/s00018-007-7406-2 (2007).
Lee, J. E. et al. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 454, 177–182. https://doi.org/10.1038/nature07082 (2008).
Biedenkopf, N., Lier, C. & Becker, S. Dynamic phosphorylation of VP30 is essential for ebola virus life cycle. J. Virol. 90, 4914–4925. https://doi.org/10.1128/jvi.03257-15 (2016).
Mateo, M. et al. Knockdown of Ebola virus VP24 impairs viral nucleocapsid assembly and prevents virus replication. J. Infect. Dis. 204(Suppl 3), S892-896. https://doi.org/10.1093/infdis/jir311 (2011).
Noda, T., Halfmann, P., Sagara, H. & Kawaoka, Y. Regions in Ebola virus VP24 that are important for nucleocapsid formation. J. Infect. Dis. 196(Suppl 2), S247–S250. https://doi.org/10.1086/520596 (2007).
Banadyga, L. et al. Ebola virus VP24 interacts with NP to facilitate nucleocapsid assembly and genome packaging. Sci. Rep. 7, 7698. https://doi.org/10.1038/s41598-017-08167-8 (2017).
Morales-Tenorio, M. et al. Potential pharmacological strategies targeting the Niemann–Pick C1 receptor and Ebola virus glycoprotein interaction. Eur. J. Med. Chem. 223, 113654. https://doi.org/10.1016/j.ejmech.2021.113654 (2021).
Hargreaves, A. et al. Filovirus neutralising antibodies: Mechanisms of action and therapeutic application. Pathogens 10, 1201. https://doi.org/10.3390/pathogens10091201 (2021).
Chakraborty, C. Therapeutics development for Ebola virus disease: A recent scenario. Curr. Opin. Pharmacol. 60, 208–215. https://doi.org/10.1016/j.coph.2021.07.020 (2021).
Ho, P. L. et al. Economical value of vaccines for the developing countries—The case of Instituto Butantan, a public institution in Brazil. PLoS Negl. Trop. Dis. 5, e1300. https://doi.org/10.1371/journal.pntd.0001300 (2011).
Choi, M. J. et al. Use of Ebola Vaccine: Recommendations of the Advisory Committee on Immunization Practices, United States, 2020. MMWR Recomm. Rep. 70, 1 (2021).
Mahon, B. E. et al. Baseline asymptomatic malaria infection and immunogenicity of recombinant vesicular stomatitis virus-Zaire Ebola virus envelope glycoprotein: vaccine: The Sierra Leone Trial to Introduce a Vaccine Against Ebola (STRIVE). J. Infect. Dis. https://doi.org/10.1093/infdis/jiab243 (2021).
Pastor, M., Pedraz, J. L. & Esquisabel, A. The state-of-the-art of approved and under-development cholera vaccines. Vaccine 31, 4069–4078. https://doi.org/10.1016/j.vaccine.2013.06.096 (2013).
Negahdaripour, M. et al. Harnessing self-assembled peptide nanoparticles in epitope vaccine design. Biotechnol. Adv. 35, 575–596. https://doi.org/10.1016/j.biotechadv.2017.05.002 (2017).
Nezafat, N., Eslami, M., Negahdaripour, M., Rahbar, M. R. & Ghasemi, Y. Designing an efficient multi-epitope oral vaccine against Helicobacter pylori using immunoinformatics and structural vaccinology approaches. Mol. Biosyst. 13, 699–713. https://doi.org/10.1039/c6mb00772d (2017).
Rahmani, A. et al. Development of a conserved chimeric vaccine based on helper T-cell and CTL epitopes for induction of strong immune response against Schistosoma mansoni using immunoinformatics approaches. Int. J. Biol. Macromol. 141, 125–136. https://doi.org/10.1016/j.ijbiomac.2019.08.259 (2019).
Kadam, A., Sasidharan, S. & Saudagar, P. Computational design of a potential multi-epitope subunit vaccine using immunoinformatics to fight Ebola virus. Infect. Genet. Evol. 85, 104464. https://doi.org/10.1016/j.meegid.2020.104464 (2020).
Ullah, M. A., Sarkar, B. & Islam, S. S. Exploiting the reverse vaccinology approach to design novel subunit vaccines against Ebola virus. Immunobiology 225, 151949. https://doi.org/10.1016/j.imbio.2020.151949 (2020).
Shankar, U. et al. Mining of Ebola virus genome for the construction of multi-epitope vaccine to combat its infection. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2021.1874529 (2021).
Kumar, B. V., Connors, T. J. & Farber, D. L. Human T cell development, localization, and function throughout life. Immunity 48, 202–213. https://doi.org/10.1016/j.immuni.2018.01.007 (2018).
Doherty, P. C., Allan, W., Eichelberger, M. & Carding, S. R. Roles of alpha beta and gamma delta T cell subsets in viral immunity. Annu. Rev. Immunol. 10, 123–151. https://doi.org/10.1146/annurev.iy.10.040192.001011 (1992).
Pandey, R. K., Sundar, S. & Prajapati, V. K. Differential expression of miRNA regulates T cell differentiation and plasticity during visceral leishmaniasis infection. Front. Microbiol. 7, 206. https://doi.org/10.3389/fmicb.2016.00206 (2016).
Kalita, P., Lyngdoh, D. L., Padhi, A. K., Shukla, H. & Tripathi, T. Development of multi-epitope driven subunit vaccine against Fasciola gigantica using immunoinformatics approach. Int. J. Biol. Macromol. 138, 224–233. https://doi.org/10.1016/j.ijbiomac.2019.07.024 (2019).
Kurosaki, T., Kometani, K. & Ise, W. Memory B cells. Nat. Rev. Immunol. 15, 149–159. https://doi.org/10.1038/nri3802 (2015).
Alexander, J. et al. The optimization of helper T lymphocyte (HTL) function in vaccine development. Immunol. Res. 18, 79–92. https://doi.org/10.1007/bf02788751 (1998).
Roy, A., Kucukural, A. & Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738. https://doi.org/10.1038/nprot.2010.5 (2010).
Yang, Z., Bogdan, P. & Nazarian, S. An in silico deep learning approach to multi-epitope vaccine design: A SARS-CoV-2 case study. Sci. Rep. 11, 3238. https://doi.org/10.1038/s41598-021-81749-9 (2021).
Majidiani, H., Dalimi, A., Ghaffarifar, F. & Pirestani, M. Multi-epitope vaccine expressed in Leishmania tarentolae confers protective immunity to Toxoplasma gondii in BALB/c mice. Microb. Pathog. 155, 104925. https://doi.org/10.1016/j.micpath.2021.104925 (2021).
Narula, A., Pandey, R. K., Khatoon, N., Mishra, A. & Prajapati, V. K. Excavating chikungunya genome to design B and T cell multi-epitope subunit vaccine using comprehensive immunoinformatics approach to control chikungunya infection. Infect. Genet. Evol. 61, 4–15. https://doi.org/10.1016/j.meegid.2018.03.007 (2018).
Taylor, D. J., Leach, R. W. & Bruenn, J. Filoviruses are ancient and integrated into mammalian genomes. BMC Evol. Biol. 10, 193. https://doi.org/10.1186/1471-2148-10-193 (2010).
Nyarko, Y., Goldfrank, L., Ogedegbe, G., Soghoian, S. & de-Graft Aikins, A. Preparing for Ebola virus disease in West African countries not yet affected: Perspectives from Ghanaian health professionals. Global Health 11, 7. https://doi.org/10.1186/s12992-015-0094-z (2015).
Gupta, S., Gupta, N., Yadav, P. & Patil, D. Ebola virus outbreak preparedness plan for developing Nations: Lessons learnt from affected countries. J. Infect. Public Health 14, 293–305. https://doi.org/10.1016/j.jiph.2020.12.030 (2021).
Alom, M. W., Shehab, M. N., Sujon, K. M. & Akter, F. Exploring E, NS3, and NS5 proteins to design a novel multi-epitope vaccine candidate against West Nile Virus: An in-silico approach. Informatics in Medicine Unlocked, 100644 (2021).
Nain, Z., Karim, M. M., Sen, M. K. & Adhikari, U. K. Structural basis and designing of peptide vaccine using PE-PGRS family protein of Mycobacterium ulcerans—An integrated vaccinomics approach. Mol. Immunol. 120, 146–163. https://doi.org/10.1016/j.molimm.2020.02.009 (2020).
Davies, M. N. & Flower, D. R. Harnessing bioinformatics to discover new vaccines. Drug Discov. Today 12, 389–395. https://doi.org/10.1016/j.drudis.2007.03.010 (2007).
Sunil Krishnan, G., Joshi, A. & Kaushik, V. T cell epitope designing for dengue peptide vaccine using docking and molecular simulation studies. Mol. Simul. 46, 787–795. https://doi.org/10.1080/08927022.2020.1772970 (2020).
Akhtar, N., Joshi, A., Singh, J. & Kaushik, V. Design of a novel and potent multivalent epitope based human cytomegalovirus peptide vaccine: An immunoinformatics approach. J. Mol. Liq. 335, 116586. https://doi.org/10.1016/j.molliq.2021.116586 (2021).
Kar, T. et al. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 10, 10895. https://doi.org/10.1038/s41598-020-67749-1 (2020).
Bibi, S. et al. In silico analysis of epitope-based vaccine candidate against tuberculosis using reverse vaccinology. Sci. Rep. 11, 1249. https://doi.org/10.1038/s41598-020-80899-6 (2021).
Meza, B., Ascencio, F., Sierra-Beltrán, A. P., Torres, J. & Angulo, C. A novel design of a multi-antigenic, multistage and multi-epitope vaccine against Helicobacter pylori: An in silico approach. Infect. Genet. Evol. 49, 309–317. https://doi.org/10.1016/j.meegid.2017.02.007 (2017).
Vakili, B. et al. Immunoinformatics-aided design of a potential multi-epitope peptide vaccine against Leishmania infantum. Int. J. Biol. Macromol. 120, 1127–1139. https://doi.org/10.1016/j.ijbiomac.2018.08.125 (2018).
Zhao, X. et al. Bioinformatics analysis of EgA31 and EgG1Y162 proteins for designing a multi-epitope vaccine against Echinococcus granulosus. Infect. Genet. Evol. 73, 98–108. https://doi.org/10.1016/j.meegid.2019.04.017 (2019).
Akhtar, N., Joshi, A., Kaushik, V., Kumar, M. & Mannan, M.A.-U. In-silico design of a multivalent epitope-based vaccine against Candida auris. Microb. Pathog. 155, 104879. https://doi.org/10.1016/j.micpath.2021.104879 (2021).
Kang, S., Brown, H. M. & Hwang, S. Direct antiviral mechanisms of interferon-gamma. Immune Netw. 18, e33. https://doi.org/10.4110/in.2018.18.e33 (2018).
Ryan, J. J. Interleukin-4 and its receptor: Essential mediators of the allergic response. J. Allergy Clin. Immunol. 99, 1–5. https://doi.org/10.1016/S0091-6749(97)81037-8 (1997).
Hajighahramani, N. et al. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infect. Genet. Evol. 48, 83–94. https://doi.org/10.1016/j.meegid.2016.12.010 (2017).
Aslam, S. et al. Designing a multi-epitope vaccine against Chlamydia trachomatis by employing integrated core proteomics, immuno-informatics and in silico approaches. Biology (Basel) 10, 997. https://doi.org/10.3390/biology10100997 (2021).
Mahmud, S. et al. Designing a multi-epitope vaccine candidate to combat MERS-CoV by employing an immunoinformatics approach. Sci. Rep. 11, 15431. https://doi.org/10.1038/s41598-021-92176-1 (2021).
Nikolich-Žugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 19, 10–19. https://doi.org/10.1038/s41590-017-0006-x (2018).
Tahir Ul Qamar, M. et al. Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput. Biol. Med. 132, 104389. https://doi.org/10.1016/j.compbiomed.2021.104389 (2021).
Solanki, V. & Tiwari, V. Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacter baumannii. Sci. Rep. 8, 9044. https://doi.org/10.1038/s41598-018-26689-7 (2018).
Gasteiger, E. et al. Protein identification and analysis tools on the ExPASy server. The Proteomics Protocols Handbook, 571–607 (2005).
Alihodžić, S. et al. Current trends in macrocyclic drug discovery and beyond-Ro5. Prog. Med. Chem. 57, 113–233. https://doi.org/10.1016/bs.pmch.2018.01.002 (2018).
Yakubu, A., De Donato, M. & Imumorin, I. Modelling functional and structural impact of non-synonymous single nucleotide polymorphisms of the DQA1 gene of three Nigerian goat breeds. South Afr. J. Anim. Sci. 47, 146–156 (2017).
Ali, M. et al. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci. Rep. 7, 9232. https://doi.org/10.1038/s41598-017-09199-w (2017).
Donnarumma, D., Faleri, A., Costantino, P., Rappuoli, R. & Norais, N. The role of structural proteomics in vaccine development: Recent advances and future prospects. Expert Rev. Proteomics 13, 55–68. https://doi.org/10.1586/14789450.2016.1121113 (2016).
Olejnik, J. et al. Ebolaviruses associated with differential pathogenicity induce distinct host responses in human macrophages. J. Virol. 91, e00179-17. https://doi.org/10.1128/jvi.00179-17 (2017).
Saghazadeh, A. & Rezaei, N. Implications of Toll-like receptors in Ebola infection. Expert Opin. Ther. Targets 21, 415–425. https://doi.org/10.1080/14728222.2017.1299128 (2017).
Morla, S., Makhija, A. & Kumar, S. Synonymous codon usage pattern in glycoprotein gene of rabies virus. Gene 584, 1–6. https://doi.org/10.1016/j.gene.2016.02.047 (2016).
Ahmad, B., Ashfaq, U. A., Rahman, M. U., Masoud, M. S. & Yousaf, M. Z. Conserved B and T cell epitopes prediction of ebola virus glycoprotein for vaccine development: An immuno-informatics approach. Microb. Pathog. 132, 243–253. https://doi.org/10.1016/j.micpath.2019.05.010 (2019).
Yang, B., Sayers, S., Xiang, Z. & He, Y. Protegen: A web-based protective antigen database and analysis system. Nucleic Acids Res. 39, D1073-1078. https://doi.org/10.1093/nar/gkq944 (2011).
Doytchinova, I. A. & Flower, D. R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 8, 4. https://doi.org/10.1186/1471-2105-8-4 (2007).
Doytchinova, I. A. & Flower, D. R. Identifying candidate subunit vaccines using an alignment-independent method based on principal amino acid properties. Vaccine 25, 856–866. https://doi.org/10.1016/j.vaccine.2006.09.032 (2007).
Doytchinova, I. A. & Flower, D. R. Bioinformatic approach for identifying parasite and fungal candidate subunit vaccines. Open Vaccine J. 1, 4 (2008).
Larsen, M. V. et al. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 8, 424. https://doi.org/10.1186/1471-2105-8-424 (2007).
Jensen, K. K. et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 154, 394–406. https://doi.org/10.1111/imm.12889 (2018).
Gupta, S. et al. In silico approach for predicting toxicity of peptides and proteins. PLoS One 8, e73957. https://doi.org/10.1371/journal.pone.0073957 (2013).
Dimitrov, I., Flower, D. R. & Doytchinova, I. AllerTOP—A server for in silico prediction of allergens. BMC Bioinform. 14(Suppl 6), S4. https://doi.org/10.1186/1471-2105-14-s6-s4 (2013).
Bui, H. H., Sidney, J., Li, W., Fusseder, N. & Sette, A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinform. 8, 361. https://doi.org/10.1186/1471-2105-8-361 (2007).
Dhanda, S. K., Vir, P. & Raghava, G. P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 8, 30. https://doi.org/10.1186/1745-6150-8-30 (2013).
Dhanda, S. K., Gupta, S., Vir, P. & Raghava, G. P. Prediction of IL4 inducing peptides. Clin. Dev. Immunol. 2013, 263952. https://doi.org/10.1155/2013/263952 (2013).
Jespersen, M. C., Peters, B., Nielsen, M. & Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 45, W24–W29. https://doi.org/10.1093/nar/gkx346 (2017).
Pandey, R. K., Ojha, R., Aathmanathan, V. S., Krishnan, M. & Prajapati, V. K. Immunoinformatics approaches to design a novel multi-epitope subunit vaccine against HIV infection. Vaccine 36, 2262–2272. https://doi.org/10.1016/j.vaccine.2018.03.042 (2018).
Walker, J. M. The Proteomics Protocols Handbook (Springer, 2005).
Magnan, C. N., Randall, A. & Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 25, 2200–2207. https://doi.org/10.1093/bioinformatics/btp386 (2009).
Magnan, C. N. et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 26, 2936–2943. https://doi.org/10.1093/bioinformatics/btq551 (2010).
Laskowski, R. A., Jabłońska, J., Pravda, L., Vařeková, R. S. & Thornton, J. M. PDBsum: Structural summaries of PDB entries. Protein Sci. 27, 129–134. https://doi.org/10.1002/pro.3289 (2018).
Yang, J. et al. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 12, 7–8. https://doi.org/10.1038/nmeth.3213 (2015).
Yang, J. & Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181. https://doi.org/10.1093/nar/gkv342 (2015).
Heo, L., Park, H. & Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 41, W384–W388. https://doi.org/10.1093/nar/gkt458 (2013).
Lee, G. R., Heo, L. & Seok, C. Effective protein model structure refinement by loop modeling and overall relaxation. Proteins 84(Suppl 1), 293–301. https://doi.org/10.1002/prot.24858 (2016).
Mariani, V., Biasini, M., Barbato, A. & Schwede, T. lDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 29, 2722–2728. https://doi.org/10.1093/bioinformatics/btt473 (2013).
Shey, R. A. et al. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci. Rep. 9, 4409. https://doi.org/10.1038/s41598-019-40833-x (2019).
Wiederstein, M. & Sippl, M. J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–W410. https://doi.org/10.1093/nar/gkm290 (2007).
Sippl, M. J. Recognition of errors in three-dimensional structures of proteins. Proteins 17, 355–362. https://doi.org/10.1002/prot.340170404 (1993).
Ponomarenko, J. et al. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 9, 514. https://doi.org/10.1186/1471-2105-9-514 (2008).
Mugunthan, S. P. & Harish, M. C. Multi-epitope-based vaccine designed by targeting cytoadherence proteins of Mycoplasma gallisepticum. ACS Omega 6, 13742–13755. https://doi.org/10.1021/acsomega.1c01032 (2021).
Craig, D. B. & Dombkowski, A. A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 14, 346. https://doi.org/10.1186/1471-2105-14-346 (2013).
Tabassum, R., Abbas, G. & Azam, S. S. Immunoinformatics based designing and simulation of multi-epitope vaccine against multi-drug resistant Stenotrophomonas maltophilia. J. Mol. Liq. 340, 116899. https://doi.org/10.1016/j.molliq.2021.116899 (2021).
Desta, I. T., Porter, K. A., Xia, B., Kozakov, D. & Vajda, S. Performance and its limits in rigid body protein-protein docking. Structure 28, 1071-1081.e1073. https://doi.org/10.1016/j.str.2020.06.006 (2020).
Kozakov, D. et al. The ClusPro web server for protein–protein docking. Nat. Protoc. 12, 255–278. https://doi.org/10.1038/nprot.2016.169 (2017).
Vajda, S. et al. New additions to the ClusPro server motivated by CAPRI. Proteins 85, 435–444. https://doi.org/10.1002/prot.25219 (2017).
Kozakov, D. et al. How good is automated protein docking?. Proteins 81, 2159–2166. https://doi.org/10.1002/prot.24403 (2013).
Wallace, A. C., Laskowski, R. A. & Thornton, J. M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 8, 127–134. https://doi.org/10.1093/protein/8.2.127 (1995).
Nain, Z. et al. Proteome-wide screening for designing a multi-epitope vaccine against emerging pathogen Elizabethkingia anophelis using immunoinformatic approaches. J. Biomol. Struct. Dyn. 38, 4850–4867. https://doi.org/10.1080/07391102.2019.1692072 (2020).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. https://doi.org/10.1016/j.softx.2015.06.001 (2015).
Grote, A. et al. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33, W526–W531. https://doi.org/10.1093/nar/gki376 (2005).
Acknowledgements
The authors would like to acknowledge the Medical Plants Research Center of Shahrekord University of Medical Sciences for providing facilities for this study.
Author information
Authors and Affiliations
Contributions
M.A., S.S., and N.B.: conceptualization, methodology, software, writing- original draft preparation. H.A.K.: drawing Figs. 1 and 2. S.T., M.G.S, and K.G.S.: reviewing, editing. Y.E., H.A.K and S.R.: software, validation. M.S. and M.G.D., F.A.D.: writing, reviewing. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alizadeh, M., Amini-Khoei, H., Tahmasebian, S. et al. Designing a novel multi‑epitope vaccine against Ebola virus using reverse vaccinology approach. Sci Rep 12, 7757 (2022). https://doi.org/10.1038/s41598-022-11851-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-11851-z
This article is cited by
-
Proteomics-based vaccine targets annotation and design of multi-epitope vaccine against antibiotic-resistant Streptococcus gallolyticus
Scientific Reports (2024)
-
Immunoinformatic-guided designing and evaluating protein and mRNA-based vaccines against Cryptococcus neoformans for immunocompromised patients
Journal of Genetic Engineering and Biotechnology (2023)
-
Design of a multi-epitope protein as a subunit vaccine against lumpy skin disease using an immunoinformatics approach
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
