
Abstract
Agroindustrial waste, such as fruit residues, are a renewable, abundant, low-cost, commonly-used carbon source. Biosurfactants are molecules of increasing interest due to their multifunctional properties, biodegradable nature and low toxicity, in comparison to synthetic surfactants. A better understanding of the associated microbial communities will aid prospecting for biosurfactant-producing microorganisms. In this study, six samples of fruit waste, from oranges, mangoes and mixed fruits, were subjected to autochthonous fermentation, so as to promote the growth of their associated microbiota, followed by short-read metagenomic sequencing. Using the DIAMOND+MEGAN analysis pipeline, taxonomic analysis shows that all six samples are dominated by Proteobacteria, in particular, a common core consisting of the genera Klebsiella, Enterobacter, Stenotrophomonas, Acinetobacter and Escherichia. Functional analysis indicates high similarity among samples and a significant number of reads map to genes that are involved in the biosynthesis of lipopeptide-class biosurfactants. Gene-centric analysis reveals Klebsiella as the main assignment for genes related to putisolvins biosynthesis. To simplify the interactive visualization and exploration of the surfactant-related genes in such samples, we have integrated the BiosurfDB classification into MEGAN and make this available. These results indicate that microbiota obtained from autochthonous fermentation have the genetic potential for biosynthesis of biosurfactants, suggesting that fruit wastes may provide a source of biosurfactant-producing microorganisms, with applications in the agricultural, chemical, food and pharmaceutical industries.
Similar content being viewed by others


Introduction
Biosurfactants are surface-active molecules produced by microorganisms that have been highlighted as an environmentally-friendly alternative to their synthetic counterpart, chemical surfactants1,2, which are produced by the petrochemical industry. Microbial surfactants demonstrate higher degradability, lower toxicity, selectivity, antimicrobial and anti-adhesive properties, and applicability in a large range of values of pH, temperature, and salinity3,4,5. They are versatile and have applications in pharmaceutics, food, cosmetics, agriculture, wastewater, bioremediation, enhanced oil recovery, metal removal and other industrial sectors6,7,8.
Biosurfactant-producing microorganisms have been reported and isolated from several sources, including marine habitats, mangroves, freshwater, soil, sludge and fruits9,10,11,12. In particular, fruit residues are important natural microhabitats for microorganisms due to their organic matter content, low pH and high sugar content, making them a source of diverse microorganisms13. Several studies have used microorganisms isolated from fruits in the production of various molecules of biotechnological interest, such as mannitol, hydrogen, organic acids, biofuels and biosurfactants14,15,16,17,18. In addition, fruit waste and residues generated by fruit-processing industries can also be used as a renewable and low-cost carbon source for fermentative processes12,19,20,21. To allow better exploitation of such waste as a source of useful microorganisms, a clearer understanding of the taxonomic and functional diversity of the present microbes is required. Shotgun metagenomic sequencing and subsequent alignment-based analysis allow one to identify specific genes of interest that are related to biosurfactant production.
Various metagenomic approaches combined with bioinformatics tools have been used to study biosurfactants22, such as AntiSMASH, designed for the identification, functional annotation, and analysis of biosynthetic gene clusters23. However, general-purpose functional databases, such as KEGG24, are not ideal for this specific purpose as they tend to focus on other aspects of function. In particular, some genes related to biosurfactant biosynthesis are classified under antibiotics in nonribosomal peptide pathways. Hence, the use of a domain-specific database is of advantage25.
Here we present six short-read metagenomic datasets that we have collected from different fruit autochthonous fermentation batch reactors using short-read Illumina sequencing. We used the DIAMOND+MEGAN26 pipeline to evaluate the microbial diversity present in the samples and to detect and analyze the genes involved in biosurfactant biosynthesis pathways. In more detail, taxonomy analysis was performed by MEGAN based on DIAMOND alignments against the NCBI-nr protein reference database27. For functional analysis, here we present a new extension of MEGAN that allows functional analysis based on the BiosurfDB database, a domain-specific database focused on identifying genes related to biosurfactant production and biodegradation28. To firmly establish the taxonomic identity of the organisms that contain the genes related to biosurfactant production, we applied gene-centric assembly29 to those genes and aligned the resulting gene-length contigs against the NCBI-nt database27 using BLASTN30.
Our analysis indicates that the six samples possess a common core of Gammaproteobacteria, dominated by the genera Klebsiella, Enterobacter, Stenotrophomonas, Escherichia and Acinetobacter. The samples have similar functional profiles and show a potential for biosurfactant biosynthesis, especially lipopeptides. The biosurfactant-related genes are predominately associated with Klebsiella sp. and Enterobacter sp., and, to a lower degree, with Acinetobacter sp. 89 and Stenotrophomonas sp.
Results
Taxonomic analysis
Each of the six metagenomic datasets was sequenced to over 20 million reads per sample, using Illumina sequencing in a 2 \(\times\) 150 bp layout. Of 140, 183, 232 total input sequencing reads, 121, 875, 304 obtained at least one alignment against the NCBI-nr database, and of these, 121, 561, 304 could be assigned to a taxon in the NCBI taxonomy, leading to an assignment rate of \(87\%\).
In all samples, reads were predominantly assigned to bacteria by an NCBI-nr run of the DIAMOND+MEGAN pipeline. The three main phyla are Proteobacteria followed by Firmicutes and Actinobacteria (Table 1). Rarefaction analysis (see Supplementary Figure S1) showed that the number of genera detected reached a plateau for each of the samples, indicating that the amount of sequencing performed was sufficient to obtain a stable representation of the taxonomic content of the samples. Rarefaction analysis suggests that the Mango and Mix samples have slightly higher diversity than the Orange samples, which is also reflected in the Shannon–Weaver diversity indices (\(\alpha\)-diversity measure) calculated from the genus-level assignments, which are 2.0, 1.6, 2.0, 1.9, 0.5 and 1.0, for the Mango-1, Mango-2, Mix-1, Mix-2, Orange-1 and Orange-2 samples, respectively. Further, a \(\beta\)-diversity analysis at genus-level (see Supplementary Figure S2) shows that all pairs of samples cluster with each other.
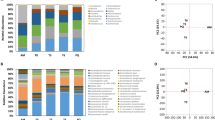
Most of the taxonomic content of the six samples falls into 19 genera (Fig. 1). Klebsiella dominates all six samples, accounting for 91.8 % and 83.6 % of genus-level assignments in the Orange-1 and Orange-2 samples respectively, and with assignment rates of 32.2 %, 68.9 %, 44.3 % and 49.5 %, for Mango-1, Mango-2, Mix-1 and Mix-2, respectively. Differences in the proportion of reads assigned to specific genera were observed between all six samples, which is to be expected even between the duplicates, as they are biological replicates (Table 2).

For each of the six fruit samples, we show the number of reads assigned to each of 19 occurring genera, based on an NCBI-nr run of the DIAMOND+MEGAN pipeline. Note that “taxonomic projection” was performed to map all read assignments to the genus level.
The NCBI-nr runs of MEGAN indicate that the six fruit samples studied here are dominated by Gammaproteobacteria, with most reads assigned to the genera Klebsiella (\(32.2{-}91.8\) %, average 61.7 %), Enterobacter (\(6.3{-}31.8\) %, average 18.3 %), Stenotrophomonas (\(0.0{-}31.3\) %, average 7.0 %), Escherichia (\(0.5{-}21.5\) %, average 6.8 %) and Acinetobacter (\(0.0{-}6.0\) %, average 2.4 %). The first, second and fourth genera are members of the Enterobacteriaceae family, whereas the third and last belong to the family of Lysobacteriaceae and Moraxellaceae, respectively.
Functional analysis
The results of the BioSurfDB runs of the DIAMOND+MEGAN pipeline were loaded into MEGAN to obtain an overview of the functional potential present in the samples and to allow an assessment of the relative abundances of genes related to biosynthesis of biosurfactants (Fig. 2). Strikingly, more than 60% of all reads that map to a Surfactant class were assigned to a biosynthesis-of-lipopeptide biosurfactant subclass. In addition, a fair number of reads were aligned to genes crucial for synthesizing non-ribosomal lipopeptide structures, namely surfactin, mycosubtilin, iturin A, lichenysin D, bacillomycin D and fengycin (Fig. 3).

For each of the six fruit samples, we report the number of reads that are assigned to a surfactant class, based on a BioSurfDB run of the DIAMOND+MEGAN pipeline.

For each of the six fruit-based samples, we indicate the number of reads assigned to the different genes involved in non-ribosomal synthesis. The illustration is based on KEGG pathway ko01054 “Nonribosomal peptide structures”24. Each gene is represented by a rectangle and inside each rectangle we use a bar chart to indicate the number of reads assigned from each of the six samples, namely Mango-1 and Mango-2 (yellow), Mix-1 and Mix2 (green) and Orange-1 and Orange-2 (gray).

Comparison of functional profiles of fruit samples. Here we only show those biosurfactant types whose mean proportions of assigned reads showed significant differences between samples (p < 0.05, Tukey’s adjustment), computed using STAMP31.
In more detail, the most abundant surfactant-related proteins present in the samples were associated with the biosynthesis of pultisolvins followed by trehalolipids, mycosubtilin and iturin A. Trehalolipids are classified as glycolipids while putisolvins, mycosubtilin and iturin A are considered lipopeptides. Significant differences were observed only in the lipopeptide biosynthesis category: iturin A biosynthesis (Fig. 4a) was more abundant in Orange samples than Mix and Mango samples and amphisin biosynthesis (Fig. 4b) was more abundant in Orange samples than in Mix samples.
To address the question of which taxa contain the detected biosurfactant genes, we applied gene-centric assembly29 to reads assigned to a number of such genes and then determined the taxonomic assignment of the resulting contigs. For a cluster of genes involved in putisolvins production, the resulting contigs were mainly assigned to genera in the above described common core of genera found in the fruit samples. The class Gammaproteobacteria, the family Enterobacteriaceae, and their representatives Klebsiella sp. and Enterobacter sp. (Fig. 5) were the most assigned to. At a lower abundance, Acinetobacter sp. and Stenotrophomonas sp. were also assigned to.

For six different genes involved in putisolvins biosynthesis, under (A–F), we report the percentage of reads assigned to the gene, and we also show a heatmap indicating how many contigs obtained by gene-centric assembly were assigned to certain taxa, based on their alignment against the NCBI-nt database.
The taxonomic assignments of contigs obtained from genes involved in the synthesis of mycosubtilin, iturin A and lichenysin (Fig. 3) were mainly to classes and families associated with the mentioned common core of genera. The genera Brevibacillus, Klebsiella and Enterobacter received the highest assignments (Figure S2).
These results suggest that all six fruit-waste samples have a similar functional profile related to biosurfactant biosynthesis and all samples appear to have a significant potential for the production of biosurfactants.
Discussion
Our analysis suggests that the microbiota of the six investigated fruit samples share a common core of Gammaproteobacteria and that Enterobacteriaceae is the most abundant family. This is compatible with previous studies that indicate that Enterobacteriaceae are predominant in plant-associated microbiomes32,33. Enterobacter and Klebsiella are endophytic bacteria that are involved in plant survival, production of toxins, antimicrobial compounds, and contribute to plant-growth promotion34. Fruit microbiota are diverse and their composition are influenced by management practices, pesticide use, external factors, growing season, etc.35. Several genera of Proteobacteria are considered efficient oil-degrading and biosurfactant-producing microorganisms, including Pseudomonas, Streptomyces, Enterobacter, Acinetobacter, Escherichia, Klebsiella, and Stenotrophomonas3,36,37,38,39,40. Some of these appear as members of the microbial common core presented here.
The results summarized in Figs. 2 and 5 suggest that the functional profiles of the microbiota present in the six fruit samples are quite similar to each other and have the potential for biosurfactant biosynthesis, especially of lipopeptides. Lipopeptides biosurfactants are considered potent antimicrobials that are capable of disrupting the cell membrane41. In addition, they also exhibit antitumor, immunomodulatory, emulsifying activities and their production is also closely related to motility and biofilm formation or inhibition22,42,43. These biosurfactants are synthesized by non-ribosomal peptide synthases (NRPSs). They have diverse structures, and nutritional parameters influence their composition44,45. The high occurrence of genes related to the biosynthesis of lipopeptide biosurfactants may be due to the fact that some endophytic bacteria produce such antimicrobial molecules to help protect plants against pathogens45,46.
Previous studies have reported that putisolvins biosurfactants are associated with motility, the dispersion of naphthalene and phenanthrene crystals and also with breaking or inhibiting biofilms47. Their production depends on several regulatory genes such as the heat shock system DnaK and the two component system GacA/GacS48. Previously, their production has been attributed to the Pseudomonas putida species47,48, whereas our study now links them to Klebsiella, Enterobacter and Acinetobacter, as well. This suggests the presence of certain regulatory genes for secondary metabolites that are common to gram-negative bacteria48. The fact that these bacteria possess genes involved in the biosynthesis of putisolvins may direct studies on the production of these molecules and their applicability in biofilm removal.
Another biosurfactant present is iturin A1,46. The genera Brevibacillus, Klebsiella and Enterobacter have the highest number of assignments to genes related to the biosynthesis of members of the iturins family. In addition, all samples have reads that align to genes involved in the synthesis of molecules that are related to surfactin, lichenysin, mycosubitilin, iturin A and bacillomycin, respectively. This suggests that fruit-autochthonous microbiota are a promising source for genes for producing these molecules.
Biosurfactants mainly arise in response to environmental conditions imposed on the microorganism, acting in physiological functions such as motility, protection from toxins, adhesion to substrates and cell interactions49. The microbiota of hydrocarbon-contaminated sites have been widely studied, isolated and used in the production of biosurfactants, due to the ability of these microorganisms to utilize hydrophobic carbon sources, and thus play a role in bioremediation. Our work provides a new perspective for prospecting for biosurfactant-producing microorganisms, as it suggests that fruit-waste samples are a promising source of bacteria capable of producing biosurfactants, mainly lipopeptides, which may have application in several industrial sectors. Further, isolation of culture-dependent strains from these fruit residues might be usable in fermentative processes and we intend to explore this further.
Our analysis determined similar profiles directed towards biosurfactant biosynthesis. The samples show high reads counts for biosurfactant production, mainly lipopeptides, a potential source of novel antibiotic and antifungal molecules. Furthermore, in line with the result of taxonomic analysis, the results of the gene-centric analysis showed that the common core found in the samples is directly related to the genes of interest.
The BiosurfDB domain-specific database is an essential resource for detecting the presence of genes associated biosurfactant biosynthesis. Integration of the BiosurfDB classification into our interactive metagenome analysis tool allows the user to interactively explore and compare the reads assigned to biosurfactant genes. While the putisolvins genes detected in our samples are represented in BioSurfDB by reference genes obtained from Pseudomonas genomes, gene-centric assembly and DNA alignment to the NCBI-nt database clearly show that the organisms carrying these genes in the six samples are not Pseudomonas, but rather belong to the genera Klebsiella, Enterobacter and Acinetobacter.
Methods
Sampling and autochthonous fermentation
Fruit residues were collected from open fairs in the city of Sorocaba, São Paulo, Brazil. The fruit residues were separated into three distinct samples: orange bagasse, mango residue and mixed fruit residue (using a mixture of fruits that includes papaya, pear, avocado, grapes, guava and banana residues). Each fruit sample was crushed and blended before use. Subsequently, the samples were subjected to autochthonous fermentation in order to promote the growth of the associated microbiota of the residues.
For the autochthonous fermentation, 12.5 g of each fruit sample was added separately into reagent bottles containing 500 mL of Luria-Bertami (LB) medium for enrichment. The autochthonous fermentation occurred for 5 days at 32\(^\circ\)C and 150 rpm, in duplicates, totaling six batch reactors. Later, the composition of microbiota obtained was analyzed through metagenome sequencing.
DNA extraction and metagenome sequencing
DNA from each autochthonous fermentation was extracted and purified using the PowerSoil DNA kit (MoBio Laboratories, Inc., Carlsbad, CA, USA), following the manufacturer’s instructions. DNA concentration and quality were estimated using a ND-2000 spectrophotometer (Nanodrop Inc, Wilmington, DE), using a ratio of 260/280 nm > 1.8. For each extraction, the DNA was sequenced in a \(2 \times 150\) bp format using a Illumina HiSeq at the Animal Biotechnology Laboratory, Department of Animal Science (ESALQ/USP, Piracicaba, São Paulo, Brazil) according to the manufacturer’s guidelines. This resulted in six datasets, which we will refer to as Orange-1, Orange-2, Mango-1, Mango-2, Mix-1 and Mix-2. These contain very similar numbers of reads, namely \(23\,407\,430\), \(22\,989\,500\), \(24\,136\,468\), \(24\,072\,864\), \(21\,872\,692\), and 23 704 278, respectively.
All sequencing reads were submitted to the European Nucleotide Archive under project accession PRJEB47062 and sample accessions ERS7265231 (Mango-1), ERS7265232 (Mango-2), ERS7265233 (Mix-1), ERS7265234 (Mix-2), ERS7265235 (Orange-1) and ERS7265236 (Orange-2).
BioSurfDB representation in MEGAN
The MEGAN software allows incorporation of additional classifications. Here we describe how to add a new functional classification to MEGAN that represents data provided by the BioSurfDB28 database, which is focused on biosurfactants and biodegradation. Using the URLs https://www.biosurfdb.org/api/get/pathway, https://www.biosurfdb.org/api/get/protein and https://www.biosurfdb.org/api/get/organism_pathway, we downloaded three files in JSON format from BioSurfDB, which we called pathway.json, protein.json and organism_pathway.json, respectively.
The first file, pathway.json, contains information about pathways. We parsed it to create the basic hierarchical tree representation of entities in the BioSurfDB classification. This representation uses integer identifiers for all nodes in the tree and a separate mapping of those identifiers to names. We provide the tree representation and the mapping in two files called biosurfdb.tre and biosurfdb.map, respectively.
The second downloaded file, protein.json, provides all protein accessions and sequences that occur in the classification. We provide these in a fasta file called biosurfdb.fasta. Finally, the third downloaded file, organism_pathway.json, was used to place the proteins into the tree representation. As the number of proteins is small (\(\approx\) 6,000), their accessions were also added to the tree as leaves. The downloaded file was also used to create a mapping of protein accessions to the integer node identifiers. We provide this mapping as the file acc2biosurfdb.map.
As mentioned, MEGAN allows the user to incorporate new classifications into the software. This requires that one prepares two files that describe the hierarchical structure of the classification, such as the two files biosurfdb.tre and biosurfdb.map described above. Use the Edit-Preferences-Add Classification... menu item to select the tree file so as to add the new classification to MEGAN. The program will provide a viewer for the classification and will integrate it into all menus, toolbars and dialogs.
To enable MEGAN to assign reads to a new classification, during the meganization or import of an alignment file, the user must specify an appropriate mapping file that maps reference sequence accessions to entities in the classification. In the case of alignment against the BioSurfDB sequences in biosurfdb.fasta, the appropriate mapping file is acc2biosurfdb.map.
DIAMOND+MEGAN analysis
All six samples were first aligned against the NCBI-nr database27 (downloaded from ftp://ftp.ncbi.nih.gov/blast/db/FASTA/nr.gz in January 2021) using DIAMOND50 (version 2.0.0, default alignment options). The resulting DAA files were “meganized”, i.e. subjected to taxonomic and functional binning, using MEGAN26 (version 6.21.2, default options). We will refer to this DIAMOND+MEGAN analysis as an NCBI-nr run.
We also aligned all six datasets against the BioSurfDB reference file using DIAMOND (default alignment options), The resulting DAA files were “meganized” with the help of the BioSurfDB-specific mapping file acc2biosurf.map. We will refer to this DIAMOND+MEGAN analysis as a BioSurfDB run.
Taxonomic and functional profiles were exported from MEGAN and statistical analysis was performed using STAMP31. In more detail, analysis of variance (ANOVA) were conducted to compare these three groups of functional profiles and post hoc tests by Tukey’s adjustment were examined to determine the significant differences between samples. Unclassified reads were removed from the analysis and results with \(p< 0.05\) (corrected p-value) were considered significant.
To determine the genus-level taxonomic content of the samples, we employed taxonomic projection as implemented in MEGAN. In this calculation, all reads assigned at a taxonomic rank that is more specific than genus are “projected up” to the appropriate genus, whereas all reads that are assigned at a higher taxonomic rank are “projected down” onto the subsequent genera, in proportion to the number of reads assigned to each such genus.
Gene-centric assembly
MEGAN provides an algorithm for assembling all reads that align to a specific class of reference sequences, using protein-alignment-guided assembly, as described in29. For each of the six samples, we applied this algorithm (default parameters) to genes associated with Surfactant classes in the BioSurfDB classification.
For the Putisolvins Biosynthesis class we assembled the genes for heat-shock protein, membrane fusion protein, molecular chaperone, outer membrane protein, putisolvin related integral membrane protein, and putisolvin synthetase (see Table 3). For the classes Iturin A Biosynthesis, Lichenysin Biosynthesis and Mycosubtilin Biosynthesis, we assembled the genes for Iturin A synthetase C, lichenysin synthetase A, and both mycosubtilin synthase subunit A and Mycosubtilin synthase subunit B, respectively.
The resulting contigs were aligned against the NCBI-nt database (downloaded October, 2021) using BLASTN30 (default parameters) and the results were then imported into MEGAN so as to perform taxonomic analysis (default parameters, using the megan-nucl-Jan2021.db mapping file). The heatmaps in Fig. 5 (and in supplementary Figure S3) are based on the number of contigs assigned to each taxon, for a given gene and for each sample.
Data availability
The six sequences are available from the European Nucleotide Archive under project accession PRJEB47062 and sample accessions ERS7265231 (Mango-1), ERS7265232 (Mango-2), ERS7265233 (Mix-1), ERS7265234 (Mix-2), ERS7265235 (Orange-1) and ERS7265236 (Orange-2). The BioSurfDB extension for MEGAN is available here: https://software-ab.informatik.uni-tuebingen.de/download/megan6/biosurfdb.zip. The MEGAN files for all six samples, and the results of gene-centric assembly, are available here: https://software-ab.informatik.uni-tuebingen.de/download/public/surfactant.
References
Gutnick, D. & Bach, H. 3.59-Biosurfactants. In Comprehensive Biotechnology 2nd edn (ed. Moo-Young, M.) 699–715 (Academic Press, 2011).
Sałek, K. & Euston, S. R. Sustainable microbial biosurfactants and bioemulsifiers for commercial exploitation. Process Biochem. 85, 143–155 (2019).
Ahmad, Z. et al. Production, functional stability, and effect of rhamnolipid biosurfactant from Klebsiella sp. . on phenanthrene degradation in various medium systems. Ecotoxicol. Environ. Saf. 207, 111514 (2021).
Giri, S. S., Ryu, E., Sukumaran, V. & Park, S. C. Antioxidant, antibacterial, and anti-adhesive activities of biosurfactants isolated from Bacillus strains. Microb. Pathog. 132, 66–72 (2019).
Santos, D., Luna, J., Rufino, R., Santos, V. & Sarubbo, L. Biosurfactants: Multifunctional materials of the XXI century. Int. J. Mol. Sci. 17, 2016 (2016).
Drakontis, C. E. & Amin, S. Biosurfactants: Formulations, properties, and applications. Curr. Opin. Colloid Interface Sci. 48, 77–90 (2020).
Araújo, H. W. et al. Sustainable biosurfactant produced by Serratia marcescens UCP 1549 and its suitability for agricultural and marine bioremediation applications. Microb. Cell Fact. 18, 1–13 (2019).
Hong, Y.-H. et al. Genome sequencing reveals the potential of Achromobacter sp. HZ01 for bioremediation. Front. Microbiol. 8, 1507 (2017).
Kumar, A. P. et al. Evaluation of orange peel for biosurfactant production by Bacillus licheniformis and their ability to degrade naphthalene and crude oil. 3 Biotech 6, 43 (2016).
Napp, A. P. et al. Comparative metagenomics reveals different hydrocarbon degradative abilities from enriched oil-drilling waste. Chemosphere 209, 7–16 (2018).
Hentati, D. et al. Production, characterization and biotechnological potential of lipopeptide biosurfactants from a novel marine Bacillus stratosphericus strain FLU5. Ecotoxicol. Environ. Saf. 167, 441–449 (2019).
Felix, A. K. N. et al. Purification and characterization of a biosurfactant produced by Bacillus subtilis in cashew apple juice and its application in the remediation of oil-contaminated soil. Colloids Surf., B 175, 256–263 (2019).
Trindade, R. C., Resende, M. A., Silva, C. M. & Rosa, C. A. Yeasts associated with fresh and frozen pulps of brazilian tropical fruits. Syst. Appl. Microbiol. 25, 294–300 (2002).
Konishi, M. et al. Biosurfactant-producing yeasts widely inhabit various vegetables and fruits. Biosci. Biotechnol. Biochem. 78, 516–523 (2014).
Endo, A. Fructophilic lactic acid bacteria inhabit fructose-rich niches in nature. Microb. Ecol. Health Dis. 23, 18563 (2012).
Ruiz Rodríguez, L. G. et al. Enhanced mannitol biosynthesis by the fruit origin strain fructobacillus tropaeoli crl 2034. Appl. Microbiol. Biotechnol. 101, 6165–6177 (2017).
Fessard, A. & Remize, F. Genetic and technological characterization of lactic acid bacteria isolated from tropically grown fruits and vegetables. Int. J. Food Microbiol. 301, 61–72 (2019).
da Silva Mazareli, R. C. et al. Metagenomic analysis of autochthonous microbial biomass from banana waste: Screening design of factors that affect hydrogen production. Biomass Bioenerg. 138, 105573 (2020).
Akbari, S., Abdurahman, N. H., Yunus, R. M., Fayaz, F. & Alara, O. R. Biosurfactants-a new frontier for social and environmental safety: a mini review. Biotechnol. Res. Innov. 2, 81–90 (2018).
Abbasi, H. et al. Biosurfactant-producing bacterium, Pseudomonas aeruginosa MA01 isolated from spoiled apples: physicochemical and structural characteristics of isolated biosurfactant. J. Biosci. Bioeng. 113, 211–219 (2012).
Kakakhel, M. A., Zaheer, U., Din, S. & Wang, W. Evaluation of the antibacterial influence of silver nanoparticles against fish pathogenic bacterial isolates and their toxicity against common carp fish. Microsc. Res. Tech. 2, 1–7 (2021).
Gutiérrez-Chávez, C., Benaud, N. & Ferrari, B. The ecological roles of microbial lipopeptides: Where are we going?. Comput. Struct. Biotechnol. J. 19, 1400–1413 (2021).
Blin, K. et al. Updates to the secondary metabolite genome mining pipeline antismash 50. Nucleic Acids Res. 47, 81–87 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Oliveira, J. S. et al. Biogeographical distribution analysis of hydrocarbon degrading and biosurfactant producing genes suggests that near-equatorial biomes have higher abundance of genes with potential for bioremediation. BMC Microbiol. 17, 1–10 (2017).
Huson, D. et al. MEGAN Community Edition–interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol., https://doi.org/10.1371/journal.pcbi.1004957 (2016).
Benson, D., Karsch-Mizrachi, I., Lipman, D., Ostell, J. & Wheeler, D. Genbank. Nucleic Acids Res. 1, D34-38 (2005).
Oliveira, J. S. et al. BioSurfDB: knowledge and algorithms to support biosurfactants and biodegradation studies. Database (Oxford)2015 (2015).
Huson, D. H. et al. Fast and simple protein-alignment-guided assembly of orthologous gene families from microbiome sequencing reads. Microbiome 5, 2 (2017).
Altschul, S. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Parks, D. H., Tyson, G. W., Hugenholtz, P. & Beiko, R. G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124 (2014).
Zhang, H. et al. Enterobacteriaceae predominate in the endophytic microbiome and contribute to the resistome of strawberry. Sci. Total Environ. 727, 138708 (2020).
Roy, N., Choi, K., Khan, R. & Lee, S.-W. Culturing simpler and bacterial wilt suppressive microbial communities from tomato rhizosphere. Plant Pathol. J. 35, 362 (2019).
Andrés-Barrao, C. et al. Complete genome sequence analysis of Enterobacter sp. SA187, a plant multi-stress tolerance promoting endophytic bacterium. Front. Microbiol. 8, 2023 (2017).
Abdelfattah, A., Malacrino, A., Wisniewski, M., Cacciola, S. O. & Schena, L. Metabarcoding: A powerful tool to investigate microbial communities and shape future plant protection strategies. Biol. Control 120, 1–10 (2018).
Nitschke, M. et al. Oil wastes as unconventional substrates for rhamnolipid biosurfactant production by Pseudomonas aeruginosa LBI. Biotechnol. Prog. 21(5), 1562–1566 (2005).
Balachandran, C., Duraipandiyan, V., Balakrishna, K. & Ignacimuthu, S. Petroleum and polycyclic aromatic hydrocarbons (PAHs) degradation and naphthalene metabolism in Streptomyces sp. (ERI-CPDA-1) isolated from oil contaminated soil. Bioresour. Technol. 112, 83–90 (2012).
Gong, Z. et al. Construction and optimization of escherichia coli for producing rhamnolipid biosurfactant. Chin. J. Biotechnol. 31, 1050–1062 (2015).
Yadav, T. C., Pal, R. R., Shastri, S., Jadeja, N. B. & Kapley, A. Comparative metagenomics demonstrating different degradative capacity of activated biomass treating hydrocarbon contaminated wastewater. Biores. Technol. 188, 24–32 (2015).
Patel, K. & Patel, M. Improving bioremediation process of petroleum wastewater using biosurfactants producing Stenotrophomonas sp S1VKR-26 and assessment of phytotoxicity. Bioresour. Technol. 315, 123861 (2020).
Williams, W. & Trindade, M. Metagenomics for the discovery of novel biosurfactants. In Functional Metagenomics: Tools and Applications 95–117 (Springer, 2017).
Franco Marcelino, P. R. et al. Biosurfactants produced by Scheffersomyces stipitis cultured in sugarcane bagasse hydrolysate as new green larvicides for the control of Aedes aegypti, a vector of neglected tropical diseases. PLoS ONE 12, e0187125 (2017).
Thomas, N., Dionysiou, D. D. & Pillai, S. C. Heterogeneous fenton catalysts: A review of recent advances. J. Hazard. Mater. 404, 124082 (2021).
Finking, R. & Marahiel, M. A. Biosynthesis of nonribosomal peptides. Annu. Rev. Microbiol. 58, 453–488 (2004).
Jozala, A. Fermentation processes (BoD–Books on Demand, 2017).
Jasim, B., Sreelakshmi, K., Mathew, J. & Radhakrishnan, E. Surfactin, iturin, and fengycin biosynthesis by endophytic Bacillus sp. from Bacopa monnieri. Microb. Ecol. 72, 106–119 (2016).
Dubern, J.-F., Lugtenberg, B. J. & Bloemberg, G. V. The ppuI-rsaL-ppuR quorum-sensing system regulates biofilm formation of Pseudomonas putida PCL1445 by controlling biosynthesis of the cyclic lipopeptides putisolvins I and II. J. Bacteriol. 188, 2898–2906 (2006).
Dubern, J.-F. & Bloemberg, G. V. Influence of environmental conditions on putisolvins i and ii production in pseudomonas putida strain pcl1445. FEMS Microbiol. Lett. 263, 169–175 (2006).
Van Hamme, J. D., Singh, A. & Ward, O. P. Physiological aspects: Part 1 in a series of papers devoted to surfactants in microbiology and biotechnology. Biotechnol. Adv. 24, 604–620 (2006).
Buchfink, B., Xie, C. & Huson, D. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Acknowledgements
We acknowledge hardware support by the High Performance and Cloud Computing Group at the Zentrum für Datenverarbeitung of the University of Tübingen, the state of Baden-Württemberg through bwHPC, the German Research Foundation (DFG) through grant no. INST 37/935-1 FUGG. We also acknowledge support of the BMBF-funded de.NBI Cloud within the German Network for Bioinformatics Infrastructure (de.NBI) (031A532B, 031A533A, 031A533B, 031A534A, 031A535A, 031A537A, 031A537B, 031A537C, 031A537D, 031A538A). This work was also supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - finance code 001. The authors acknowledge infrastructural support by the cluster of Excellence EXC2124 Controlling Microbes to Fight Infection (CMFI), project ID 390838134. Furthermore, we acknowledge support from the Open Access Publishing Fund of University of Tübingen.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
G.F.S., I.C.S.D., T.P.D., and V.M.O. conceived and planned the experiments. G.F.S., A.G. and D.H.H. conceived and carried out the bioinformatics analysis. G.F.S., A.G. and D.H.H. wrote the manuscript. All authors provided critical feedback and helped shape the research, analysis and manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Silva, G.F.d., Gautam, A., Duarte, I.C.S. et al. Interactive analysis of biosurfactants in fruit-waste fermentation samples using BioSurfDB and MEGAN. Sci Rep 12, 7769 (2022). https://doi.org/10.1038/s41598-022-11753-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-11753-0
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
