
Abstract
In this study, we aimed to investigate the prevalence and molecular characteristics of Bartonella infections in small mammals from the central region of the Qinghai-Tibetan Plateau. Toward this, small mammals were captured using snap traps in Yushu City and Nangqian County, West China, and the spleen tissue was used for Bartonella culture. The suspected positive colonies were evaluated using polymerase chain reaction (PCR) amplification and by sequencing the citrate synthase (gltA) gene. We discovered that 31 out of the 103 small mammals tested positive for Bartonella, with an infection rate of 30.10%. Sex differences between the mammals did not result in a significant difference in infection rate (χ2 = 0.018, P = 0.892). However, there was a significant difference in infection rates in different small mammals (Fisher’s exact probability method, P = 0.017) and habitats (χ2 = 7.157, P = 0.028). Additionally, 31 Bartonella strains belonging to three species were identified, including B. grahamii (25), B. japonica (4) and B. heixiaziensis (2), among which B. grahamii was the dominant epidemic strain (accounting for 80.65%). Phylogenetic analyses showed that most of the B. grahamii isolates identified in this study may be closely related to the strains isolated from Japan and China. Genetic diversity analyses revealed that B. grahamii strains had high genetic diversity, which showed a certain host and geographical specificity. The results of Tajima’s test suggested that the B. grahamii followed the progressions simulated by a neutral evolutionary model in the process of evolution. Overall, a high prevalence and genetic diversity of Bartonella infection were observed in small mammals in the central region of the Qinghai-Tibetan Plateau. B. grahamii as the dominant epidemic strain may cause diseases in humans, and the corresponding prevention and control measures should be taken into consideration in this area.
Similar content being viewed by others
Introduction
Bartonella species are small, intracellular, vector-borne hemotrophic gram-negative bacteria. Thus far, there are over 40 species and subspecies have been reported to infect a wide range of mammals, including cats, dogs, rodents, bats, and so on1. Over 10 Bartonella species, including B. bacilliformis2, B. quintana3, B. henselae4, B. elizabethae5, B. clarridgeiae6, B. koehlerae7, B. vinsonii subsp. arupensis8, B. vinsonii subsp. berkhoffii9, B. grahamii10,11, B. rochalimae12, B. tamiae13, B. ancashensis14, B. washoensis15, can cause human diseases with various clinical manifestations, including periods of intermittent fever, and poly tissue inflammation involving the heart, liver, lymph nodes, and other tissues16. Small mammals, particularly rodents, are considered important reservoirs of Bartonella species, with an infection rate of 70% worldwide17. Hence, investigating the epidemiological characteristics of Bartonella in small mammals has important implications for the prevention and control of human bartonellosis.
The Qinghai-Tibetan Plateau, referred to as the "Roof of the World", is an inland plateau in Asia; the largest in China and the highest in the world. The Yushu Tibetan Autonomous Prefecture lies in the central region of the Qinghai-Tibetan Plateau and belongs to the Sanjiangyuan Region, the source of the Yangtze, Yellow, and Lantsang rivers (between 31.65° and 36.27° N, 89.40° and 102.38° E), with an average elevation of 4493 m18. It has an important ecological status, with the highest concentration of biodiversity area in the world; nearly 30 species of mammals have been reported to inhabit this area. Our team has previously detected Bartonella species infection in small mammals in some areas of the Qinghai-Tibetan Plateau, with infection rates of 18.99% and 38.61%19,20. However, investigations of Bartonella species in small mammals in the central region of the Qinghai-Tibetan Plateau have not yet been undertaken. This region’s tourism industry was greatly developed following the reconstruction work after the Yushu earthquake. This increased the probability of people being infected with natural infectious diseases. Therefore, in this study, we investigated the prevalence and genetic diversity of Bartonella species in small mammals in the Yushu Tibetan Autonomous Prefecture. Our findings provide insights into the distribution and genetic diversity of Bartonella in small mammals and the scientific basis for the control and prevention of Bartonella infection in humans in this region.
Results
Animal collection
A total of 103 small mammals were captured and categorized into 10 species based on their morphology, including Apodemus peninsulae (58), Ochotona curzoniae (16), Microtus arvalis (8), Cricetidae (7), Microtus gregalis (4), Microtus oeconomus (3), Sorex araneus Linnaeus (3), Eozapus setchuanus (2), Mustela altaica (1), and Mus musculus (1). The geographical distribution of the trapped small mammals is shown in Fig. 1.

Geographical distribution of the trapped small mammals in the central region of the Qinghai-Tibetan Plateau, China. The map was prepared in ArcGIS 10.2.2 using political boundaries from the National Geomatics Center of China (http://www.ngcc.cn/ngcc) for illustrative purposes only, these data are available free of charge.
Bartonella infections
Spleens of the small mammals were collected and used for Bartonella isolation, and the pure colonies obtained were confirmed by polymerase chain reaction (PCR) amplification of the partial citrate synthase (gltA) gene (379 bp). In total, 31 small mammals were positive for Bartonella infection, with an infection rate of 30.10% (31/103), which were classified into five species (Apodemus peninsulae (22/58), Microtus arvalis (2/8), Cricetidae (4/7), Microtus gregalis (1/4), Microtus oeconomus (2/3)). The difference of infection rate among different small mammals was significant (Fisher’s exact probability method, P = 0.017) (Table 1).
Of the 103 small mammals, 42 were male, 50 were female, and 11 had no sex information. The infection rate was 32.00% (16/50) in females and 33.33% (14/42) in males, and the difference was not significant (χ2 = 0.018, P = 0.892). Forty-nine small mammals—corresponding to nine species—were captured in farmlands, with a Bartonella infection rate of 30.61% (15/49). Forty-one small mammals—corresponding to seven species—were captured in forests with an infection rate of 39.02% (16/41). Additionally, 13 small mammals of the same species were captured in meadows with no Bartonella infection. Thus, the infection rates between different habitats were significantly different (χ2 = 7.157, P = 0.028) (Tables 2, 3).
Identification of Bartonella species
Through BLAST analysis of the gltA gene, 25 isolates were identified to be B. grahamii with 97.11–100.00% identity, including 18 isolates from A. peninsulae, 4 isolates from Cricetidae, 1 isolate from M. arvalis, 1 isolate from M. gregalis and 1 isolate from M. oeconomus; 4 isolates from A. peninsulae were B. japonica with 97.89–99.70% identity; 2 isolates were B. heixiaziensis with 98.59–99.44% identity, including 1 isolate from M. arvalis and 1 isolate from M. oeconomus (Table 1).
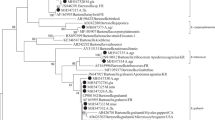
In our previous study, phylogenetic analyses of Bartonella species was performed based on the DNA sequences of the gltA, ftsZ, rpoB and ribC revealed the same results20. Of these, gltA is the most commonly used in the phylogenetic analyses of Bartonella. Therefore, in this study, we selected gltA to construct a phylogenetic tree using the maximum likelihood (ML) method. All Bartonella strains could be divided into three clusters, i.e., B. grahamii, B. heixiaziensis, and B. japonica; B. grahamii were the dominant Bartonella species in this area (Fig. 2). Bartonella was detected in small mammals from three of the four trapping sites and the distribution of Bartonella species showed slight geographical differences (Fig. 3).

Phylogenetic trees constructed based on gltA gene of 31 Bartonella isolates. The tree was constructed by using the maximum-likelihood (ML) method with the Kimura 2-parameter model, bootstrap values calculated with 1000 replicates in MEGA version 7.0 (https://www.megasoftware.net). The sequences detected in this study are indicated with black dots. Brucella abortus was used as outgroup.

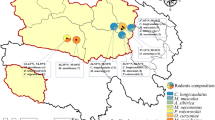
Bartonella species composition in different sampling sites in the central region of the Qinghai-Tibetan Plateau, China. The map was prepared in ArcGIS 10.2.2 using political boundaries from the National Geomatics Center of China (http://www.ngcc.cn/ngcc) for illustrative purposes only, these data are available free of charge.
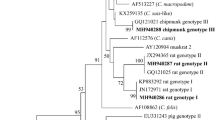
Phylogenetic analyses based on gltA sequences showed that B. grahamii was mainly grouped into four clusters, indicating that B. grahamii might have the different origins. We then obtained the gltA sequences of B. grahamii from GenBank released before July, 2021, and performed the traceability analyses. The majority of B. grahamii strains from A. peninsulae clustered with B. grahamii from A. speciosus in Japan; three strains, i.e., AP1QHYS, MA61QHYS, CR102QHYS, from A. peninsulae, M. arvalis, and Cricetidae clustered with B. grahamii from M. oeconomus in our previous study; three strains, i.e., CR34QHYS, CR36QHYS, CR103QHYS, from Cricetidae and one strain from M. gregalis (MG5QHYS) clustered separately and not with the reference strains (Fig. 4).

Traceability analyses of B. grahamii based on gltA gene. The tree was constructed by using the maximum-likelihood (ML) method with the Kimura 2-parameter model, bootstrap values calculated with 1000 replicates in MEGA version 7.0 (https://www.megasoftware.net). The sequences detected in this study are indicated with black dots.
Genetic diversity analyses
Subsequently, the genetic diversity of the gltA gene sequence (326 bp) from 77 strains of B. grahamii was analyzed, including 25 strains in this study and 52 strains from our previous studies, isolated from three regions of Qinghai-Tibetan Plateau, including Haixi Mongolian and Tibetan Autonomous Prefecture, Huangnan Tibetan Autonomous Prefecture, and Haibei Tibetan Autonomous Prefecture. We found 19 polymorphic loci (S = 19) and 15 haplotypes (H = 15). The haplotype diversity (Hd) was 0.880 ± 0.019, the mean number of nucleotide differences (κ) was 4.386, and the nucleotide diversity (π) was 0.01345 ± 0.00077. DNA polymorphism was analyzed using a sliding window with a length of 100 bp and a step size of 25 bp. It was found that fragment diversity was the highest between 151 and 250 bp (Fig. 5). The results indicated high genetic diversity in B. grahamii in this area. Tajima's D was calculated as 0.39958 (P > 0.10), suggesting that B. grahamii followed the progressions simulated by a the neutral evolutionary model in the process of evolution.

Genetic diversity of different nucleotide position in gltA gene of B. grahamii. Genetic diversity was analyzed using DNASP 6.12.03 (http://www.ub.edu/dnasp) with a sliding window interval of 25 bp.
Haplotype network analyses showed that 35 strains from Cricetulus longicaudatus contained 10 haplotypes (2 strains for Hap 1, 3 strains for Hap 5, 1 strain for Hap 7, 19 strains for Hap 8, 4 strains for Hap 9, 3 strains for Hap 10, 1 strain for Hap 11, 1 strain for Hap 12 and 1 strain for Hap 13), 18 strains from A. peninsulae contained 3 haplotypes (9 strains for Hap 1, 8 strains for Hap 2, and 1 strain for Hap 3), 12 strains from O. curzoniae contained 2 haplotypes (11 strains for Hap 14 and 1 strain for Hap 15), 4 strains from Cricetidae contained 2 haplotypes (1 strain for Hap 4 and 3 strains for Hap 5), 2 strains from Apodemus speciosus for Hap 7, 2 strains from M. musculus for Hap 9, 1 strain from M. arvalis and 2 strains from M. oeconomus for Hap 3, and 1strain from M. gregalis for Hap 6. In addition, 25 strains isolated from Yushu contained 6 haplotypes (9 strains for Hap 1, 8 strains for Hap 2, 3 strains for Hap 3, 1 strain for Hap 4, 3 strains for Hap 5, and 1 strain for Hap 6), 30 strains isolated from Haixi contained 8 haplotypes (1 strain for Hap 3, 3 strains for Hap 5, 1 strain for Hap 7, 19 strains for Hap 8, 3 strains for Hap 10, 3 strains for Hap 11, 12, 13 respectively), 15 strains isolated from Huangnan contained 4 haplotypes (2 strains for Hap 1, 2 strains for Hap 7, 6 strains for Hap 9, and 5 strains for Hap 14), and 7 strains isolated from Haibei contained 2 haplotypes (6 strains for Hap 14 and 1 strain for Hap 15) (Fig. 6, Table 4).

Median-joining networks of gltA gene for B. grahamii strains from different hosts and regions in the Qinghai-Tibetan Plateau, China. The sequences were analyzed based on a median-joining network using the Population Analysis with Reticulate Trees (PopART) software version 1.7 (http://popart.otago.ac.nz/index.shtml) with the default setting (epsilon = 0).
Discussion
Bartonella species are distributed throughout the world. They are highly prevalent in small mammals and are generally transmitted by bloodsucking arthropod vectors21. Previous studies have revealed that Bartonella infection varies in different regions and animals16,17. For instance, infection rate of Bartonella in rodents is 4–50% in China22,23, 6–94% in Japan24,25, 7–14% in Korea26, 2–10% in Indonesia27, 60–83% in Russia28, 6–90% in United States29,30, 20–60% in England31,32. Rodents are primary reservoir hosts for B. grahamii33, B. elizabethae34, and B. vinsonii subsp. arupensis35; domestic cats are primary reservoir hosts for B. henselae36, B. clarridgeiae37, and B. koehlerae38; dogs are primary reservoir hosts for B. henselae and B. vinsonii subsp. berkhoffii39. Therefore, it is necessary to investigate the Bartonella infections in small mammals from different areas.
In this study, we observed the prevalence and molecular characteristics of Bartonella species in small mammals from the central region of the Qinghai-Tibetan Plateau. The infection rate of Bartonella species in small mammals was 30.10%, which was similar to that of 38.61% in Qaidam Basin as determined in our previous study20, and higher than that in most areas of China40. Bartonella species were detected in five out of ten species of small mammals, including A. peninsulae, M. arvalis, Cricetidae, M. gregalis, and M. oeconomus, and we found differing infection rates among them. Additionally, the infection rate varied significantly by habitats, but not by sex, which is concurrent with the results of a previous study20.
Bartonella species are fastidious, slow-growing, facultative intracellular bacteria that are difficult and time consuming to culture. In our previous study, we used spleen, liver, and brain tissue for Bartonella culture and found that the positive rates in different tissues of small mammals did not differ significantly19,20. Here, the spleen tissue of small mammals was used for Bartonella culture, and 31 Bartonella strains were obtained. BLAST and phylogenetic analyses showed that 31 Bartonella strains corresponded to three species of Bartonella—(B. grahamii, B. japonica, and B. heixiaziensis). Importantly, 80.65% isolates (25/31) were B. grahamii and detected in all five species of the small mammals studied, suggesting that it was the dominant Bartonella strain. B. grahamii is associated with neuroretinitis and cat scratch disease (CSD) in immunocompromised individuals10,11, suggesting that Bartonella species may have the ability to cause human disease in this area. In addition, four isolates of B. japonica were isolated from A. peninsulae and two isolates of B. heixiaziensis were isolated from Microtus species, indicating specificity of infection among rodent species.
Subsequently, we performed traceability analyses on the dominant B. grahamii strains and found that B. grahamii was mainly grouped into four clusters. B. grahamii was clustered with A. speciosus in Japan, M. oeconomus in China, 3 strains from Cricetidae and 1 strain from M. gregalis clustered separately. These results indicated that B. grahamii might have different origins. Further studies are needed to determine whether the pathogenicity of B. grahamii strains differs depending on their origins.
A previous study revealed that the polymorphism within gltA gene was high in Bartonella species33. Here, 15 haplotypes were detected in 77 strains of B. grahamii based on gltA gene (Hd = 0.880, π = 0.01345), suggesting that the high genetic diversity of B. grahamii in the Qinghai-Tibetan Plateau. With the exception of C. longicaudatus, B. grahamii strains were isolated from A. peninsulae for Hap 1–3, Cricetidae for Hap 4–5, M. arvalis and M. oeconomus for Hap 3, M. musculus for Hap 9, and O. curzoniae for Hap 14–15, which suggested the haplotypes of B. grahamii showed a certain host specificity. This also indicated the haplotypes of B. grahamii had a certain geographical specificity, although, the geographical crossover existed. Additionally, B. grahamii isolated from C. longicaudatus showed complex haplotypes that intersected with many rodents, suggesting that it might be important for the evolution of B. grahamii in this area.
Conclusions
In conclusion, Bartonella infection rate was 30.10% in small mammals in the central region of the Qinghai-Tibetan Plateau, with significant differences between different animal species and habitats. B. grahamii, B. japonica, and B. heixiaziensis were detected in five rodent species, A. peninsulae, M. arvalis, Cricetidae, M. gregalis and M. oeconomus. B. grahamii was the dominant strain, and originated from the B. grahamii strains in different areas. In addition, high genetic diversity in B. grahamii was observed in this area, and the haplotypes of B. grahamii showed a certain host and geographical specificity. Our results further enrich the prevalence and molecular characteristics of Bartonella infection in small mammals in the Qinghai-Tibetan Plateau, which could provide the scientific basis for prevention and control of rodent-Bartonella species.
Materials and methods
Animal collection
Small mammals were captured using snap traps in July 2019 in Yushu City (32.68°–33.77° N, 95.68°–97.73° E) and Nangqian County (31.53°–32.72° N, 95.35°–97.12° E) of Qinghai Province, which were identified morphologically.
Bartonella culture
Spleens were harvested under sterile conditions from each animal after euthanasia. Approximately 20 mg of each spleen sample was homogenized by adding 200 μL sterilized trypsin soy broth (BD Biosciences, Franklin Lakes, NJ, USA), plated onto two trypsin soy agars containing 5% (vol/vol) defiber sheep blood (BD Biosciences), and incubated at 37 °C in an atmosphere containing 5% CO2. Pure Bartonella colonies were obtained using a protocol described in previous studies19.
DNA extraction, PCR amplification and DNA sequencing
Crude DNA was extracted using a previously reported method19. PCR was performed to detect the Bartonella gltA gene. DNA amplification was performed in 50 μL mixtures containing 25 μL 2 × TransTaq-T PCR SuperMix (Beijing TransGen Biotech Co., Ltd., Beijing, China), 22 μL double-distilled H2O, 1 μL (10 μmol/L) of each primer (BhCS781.p: GGGGACCAGCTCATGGTGG; BhCS1137.n: AATGCAAAAAGAACAGTAAACA41), and 1 μL of DNA template. gltA amplification was performed under the following conditions: one cycle for 5 min at 94 °C; 33 cycles for 30 s at 94 °C, 30 s at 53 °C, and 20 s at 72 °C; and a final extension for 7 min at 72 °C. Next, 5 μL of each PCR product was run on 1% agarose gels, stained with ethidium bromide, and visualized using a gel imaging system (Bio-Rad, Hercules, CA, USA). The expected PCR products were purified using the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocols, and then sequenced on an Applied Biosystems 3730 xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Phylogenetic analyses
The sequences generated in this study have been submitted to GenBank (accession numbers MZ126613-MZ126643). The nucleotide sequences of the isolated sequences were compared against the Bartonella species sequences hosted on GenBank using BLAST at the National Center for Biotechnology Information Website (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The gltA sequences of B. grahamii hosted on GenBank released before July, 2021 were collected for traceability analyses. Furthermore, one strain isolated from the same host in the same laboratory at the same time was selected randomly as the reference strain. A phylogenetic tree was created using the maximum-likelihood method with the Kimura 2-parameter model in MEGA version 7.0, and bootstrap values were calculated with 1000 replicates42,43. Brucella abortus was used as the outgroup.
Genetic diversity analyses
The polymorphism of nucleotide sequences, including the number of polymorphic sites (S), the number of haplotypes (H), the nucleotide diversity (π), the mean number of nucleotide differences (κ), and the haplotype diversity (Hd), was analyzed using DNASP 6.12.03. A sliding window interval of 25 bp was used to determine which segment of the target gene sequence had the highest nucleotide diversity (π) by analyzing 100 bp each time. Tajima’s test was performed to determine whether the target gene sequence followed the progressions simulated by a neutral evolutionary model in the process of evolution. Then, the sequences were analyzed based on a median-joining network using the Population Analysis with Reticulate Trees (PopART) software version 1.7 with the default setting (epsilon = 0).
Statistical analysis
The positive rates of Bartonella in different habitats and sexes of small mammals were analyzed using the Chi-square test. The infection rates of Bartonella in different mammals were analyzed using the Fisher’s exact probability method. All data were analyzed using SPSS 22.0 (SPSS, Inc., Chicago, IL, USA). Significance was set at P < 0.05.
Ethical approval
This study was approved by the Ethics Committee of Chinese Center for Disease Control and Prevention (No: ICDC-2015001). All animals were treated according to the ARRIVE guidelines44, the Guidelines of Regulations for the Administration of Laboratory Animals (Decree No. 2 of the State Science and Technology Commission of the People’s Republic of China, 1988) and the Guidelines for Treating Animals Kindly from Ministry of Science and Technology of the People’s Republic of China. All efforts were made to minimize discomfort to the animals.
Consent to publish
All the authors consent to publish the article in its present form.
Data availability
The data supporting the conclusions of this article are included within the article.
References
Huang, K. et al. Molecular detection of Bartonella spp. in China and St. Kitts. Can. J. Infect. Dis. Med. Microbiol. 2019, 3209013. https://doi.org/10.1155/2019/3209013 (2019).
Minnick, M. F. et al. Oroya fever and verruga peruana: Bartonelloses unique to South America. PLoS Negl. Trop. Dis. 8, e2919. https://doi.org/10.1371/journal.pntd.0002919 (2014).
Arvand, M., Raoult, D. & Feil, E. J. Multi-locus sequence typing of a geographically and temporally diverse sample of the highly clonal human pathogen Bartonella quintana. PLoS One 5, e9765. https://doi.org/10.1371/journal.pone.0009765 (2010).
Chaudhry, R. et al. Bartonella henselae infection in diverse clinical conditions in a tertiary care hospital in north India. Indian J. Med. Res. 147, 189–194. https://doi.org/10.4103/ijmr.IJMR_1932_16 (2018).
Daly, J. S. et al. Rochalimaea elizabethae sp. Nov. isolated from a patient with endocarditis. J. Clin. Microbiol. 31, 872–881. https://doi.org/10.1128/JCM.31.4.872-881.1993 (1993).
Kordick, D. L. et al. Bartonella clarridgeiae, a newly recognized zoonotic pathogen causing inoculation papules, fever, and lymphadenopathy (cat scratch disease). J. Clin. Microbiol. 35, 1813–1818. https://doi.org/10.1128/JCM.35.7.1813-1818.1997 (1997).
Avidor, B. et al. Bartonella koehlerae, a new cat-associated agent of culture-negative human endocarditis. J. Clin. Microbiol. 42, 3462–3468. https://doi.org/10.1128/JCM.42.8.3462-3468.2004 (2004).
Fenollar, F., Sire, S. & Raoult, D. Bartonella vinsonii subsp. arupensis as an agent of blood culture-negative endocarditis in a human. J. Clin. Microbiol. 43, 945–947. https://doi.org/10.1128/JCM.43.2.945-947.2005 (2005).
Breitschwerdt, E. B. et al. Bartonella vinsonii subsp. berkhoffii and Bartonella henselae bacteremia in a father and daughter with neurological disease. Parasit. Vectors 3, 29. https://doi.org/10.1186/1756-3305-3-29 (2010).
Kerkhoff, F. T., Bergmans, A. M., van Der Zee, A. & Rothova, A. Demonstration of Bartonella grahamii DNA in ocular fluids of a patient with neuroretinitis. J. Clin. Microbiol. 37, 4034–4038. https://doi.org/10.1128/JCM.37.12.4034-4038.1999 (1999).
Oksi, J. et al. Cat scratch disease caused by Bartonella grahamii in an immunocompromised patient. J. Clin. Microbiol. 51, 2781–2784. https://doi.org/10.1128/JCM.00910-13 (2013).
Chomel, B. B. et al. Dogs are more permissive than cats or guinea pigs to experimental infection with a human isolate of Bartonella rochalimae. Vet. Res. 40, 27. https://doi.org/10.1051/vetres/2009010 (2009).
Colton, L., Zeidner, N., Lynch, T. & Kosoy, M. Y. Human isolates of Bartonella tamiae induce pathology in experimentally inoculated immunocompetent mice. BMC Infect. Dis. 10, 229. https://doi.org/10.1186/1471-2334-10-229 (2010).
Mullins, K. E. et al. Whole-genome analysis of Bartonella ancashensis, a novel pathogen causing Verruga Peruana, Rural Ancash Region, Peru. Emerg. Infect. Dis. 23, 430–438. https://doi.org/10.3201/eid2303.161476 (2017).
Kosoy, M., Murray, M., Gilmore, R. D. Jr., Bai, Y. & Gage, K. L. Bartonella strains from ground squirrels are identical to Bartonella washoensis isolated from a human patient. J. Clin. Microbiol. 41, 645–650. https://doi.org/10.1128/jcm.41.2.645-650.2003 (2003).
Alvarez-Fernandez, A., Breitschwerdt, E. B. & Solano-Gallego, L. Bartonella infections in cats and dogs including zoonotic aspects. Parasit. Vectors 11, 624. https://doi.org/10.1186/s13071-018-3152-6 (2018).
Gutierrez, R. et al. Bartonella infection in rodents and their flea ectoparasites: An overview. Vector Borne Zoonot. Dis. 15, 27–39. https://doi.org/10.1089/vbz.2014.1606 (2015).
Liu, X., Yang, H., Tang, B., Liu, Y. & Zhang, L. Health status of adolescents in the Tibetan plateau area of western China: 6 years after the Yushu earthquake. Health Qual. Life Outcomes 15, 152. https://doi.org/10.1186/s12955-017-0727-4 (2017).
Rao, H. X. et al. Bartonella species detected in the Plateau Pikas (Ochotona curzoiae) from Qinghai Plateau in China. Biomed. Environ. Sci. 28, 674–678. https://doi.org/10.3967/bes2015.094 (2015).
Rao, H. et al. Genetic diversity of Bartonella species in small mammals in the Qaidam Basin, western China. Sci. Rep. 11, 1735. https://doi.org/10.1038/s41598-021-81508-w (2021).
Kosoy, M., Hayman, D. T. & Chan, K. S. Bartonella bacteria in nature: Where does population variability end and a species start?. Infect. Genet. Evol. 12, 894–904. https://doi.org/10.1016/j.meegid.2012.03.005 (2012).
Ying, B., Kosoy, M. Y., Maupin, G. O., Tsuchiya, K. R. & Gage, K. L. Genetic and ecologic characteristics of Bartonella communities in rodents in southern China. Am. J. Trop. Med. Hyg. 66, 622–627. https://doi.org/10.4269/ajtmh.2002.66.622 (2002).
Liu, Q. et al. Detection of bartonella species in small mammals from Zhejiang Province, China. J. Wildl. Dis. 46, 179–185. https://doi.org/10.7589/0090-3558-46.1.179 (2010).
Inoue, K. et al. Prevalence and genetic diversity of Bartonella species isolated from wild rodents in Japan. Appl. Environ. Microbiol. 74, 5086–5092. https://doi.org/10.1128/AEM.00071-08 (2008).
Kabeya, H. et al. Bartonella species in wild rodents and fleas from them in Japan. J. Vet. Med. Sci. 73, 1561–1567. https://doi.org/10.1292/jvms.11-0134 (2011).
Kim, C. M. et al. Detection of Bartonella species from ticks, mites and small mammals in Korea. J. Vet. Sci. 6, 327–334 (2005).
Winoto, I. L. et al. Bartonella species in rodents and shrews in the greater Jakarta area. Southeast Asian J. Trop. Med. Public Health 36, 1523–1529 (2005).
Mediannikov, O. et al. Molecular screening of Bartonella species in rodents from the Russian Far East. Ann. N Y Acad. Sci. 1063, 308–311. https://doi.org/10.1196/annals.1355.049 (2005).
Kosoy, M. Y. et al. Distribution, diversity, and host specificity of Bartonella in rodents from the SouthcentralUnited States. Am. J. Trop. Med. Hyg. 57, 578–588. https://doi.org/10.4269/ajtmh.1997.57.578 (1997).
Gundi, V. A., Billeter, S. A., Rood, M. P. & Kosoy, M. Y. Bartonella spp. in rats and zoonoses, Los Angeles, California, USA. Emerg. Infect. Dis. 18, 631–633. https://doi.org/10.3201/eid1804.110816 (2012).
Birtles, R. J. et al. Longitudinal monitoring of the dynamics of infections due to Bartonella species in UK woodland rodents. Epidemiol. Infect. 126, 323–329. https://doi.org/10.1017/s095026880100526x (2001).
Telfer, S. et al. Contrasting dynamics of Bartonella spp. in cyclic field vole populations: The impact of vector and host dynamics. Parasitology 134, 413–425. https://doi.org/10.1017/S0031182006001624 (2007).
Li, D. M. et al. High prevalence and genetic heterogeneity of rodent-borne Bartonella species on Heixiazi Island, China. Appl. Environ. Microbiol. 81, 7981–7992. https://doi.org/10.1128/AEM.02041-15 (2015).
Tay, S. T., Kho, K. L., Wee, W. Y. & Choo, S. W. Whole-genome sequence analysis and exploration of the zoonotic potential of a rat-borne Bartonella elizabethae. Acta Trop. 155, 25–33. https://doi.org/10.1016/j.actatropica.2015.11.019 (2016).
Schulte Fischedick, F. B. et al. Identification of Bartonella Species Isolated from Rodents from Yucatan, Mexico, and Isolation of Bartonella vinsonii subsp. yucatanensis subsp nov. Vector Borne Zoonot. Dis. 16, 636–642. https://doi.org/10.1089/vbz.2016.1981 (2016).
Jaffe, D. A. et al. Bartonella henselae in small Indian mongooses (Herpestes auropunctatus) from Grenada, West Indies. Vet. Microbiol. 216, 119–122. https://doi.org/10.1016/j.vetmic.2018.02.009 (2018).
Bai, Y. et al. Coexistence of Bartonella henselae and B. clarridgeiae in populations of cats and their fleas in Guatemala. J. Vector. Ecol. 40, 327–332. https://doi.org/10.1111/jvec.12171 (2015).
Chomel, B. B., Kasten, R. W., Henn, J. B. & Molia, S. Bartonella infection in domestic cats and wild felids. Ann. N Y Acad. Sci. 1078, 410–415. https://doi.org/10.1196/annals.1374.080 (2006).
Muller, A. et al. Bartonella vinsonii subsp. berkhoffii and B. henselae in dogs. Epidemiol. Infect. 146, 1202–1204. https://doi.org/10.1017/S0950268818001127 (2018).
Liu, Q., Eremeeva, M. E. & Li, D. Bartonella and Bartonella infections in China: From the clinic to the laboratory. Comp. Immunol. Microbiol. Infect. Dis. 35, 93–102. https://doi.org/10.1016/j.cimid.2012.01.002 (2012).
Norman, A. F., Regnery, R., Jameson, P., Greene, C. & Krause, D. C. Differentiation of Bartonella-like isolates at the species level by PCR-restriction fragment length polymorphism in the citrate synthase gene. J. Clin. Microbiol. 33, 1797–1803. https://doi.org/10.1128/jcm.33.7.1797-1803.1995 (1995).
Bai, Y. et al. Global distribution of Bartonella infections in domestic bovine and characterization of Bartonella bovis strains using multi-locus sequence typing. PLoS One 8, e80894. https://doi.org/10.1371/journal.pone.0080894 (2013).
Huang, R. et al. Bartonella quintana infections in captive monkeys, China. Emerg. Infect. Dis. 17, 1707–1709. https://doi.org/10.3201/eid1709.110133 (2011).
Percie du Sert, N. et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 18, e3000410. https://doi.org/10.1371/journal.pbio.3000410 (2020).
Acknowledgements
We wish to express our gratitude to Yushu City Center for Disease Control and Prevention and Nangqian County Center for Disease Control and Prevention employees, who participated in the specimen collection.
Funding
The present work was supported by Natural Science Foundation of Shanxi Province of China (201901D111326, 20210302124299), Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi (2020L0376, 2020L0377), Academic Technology Leader Project of Changzhi Medical College (XS202103), Undergraduate Innovation and Entrepreneurship Training Program (20210584), Major Infectious Diseases Such as AIDS and Viral Hepatitis Prevention and Control Technology Major Projects (2017ZX10303404), Doctor research startup foundation of Changzhi Medical College (BS201912, BS201921).
Author information
Authors and Affiliations
Contributions
D.M.L. and H.X.R. conceived and designed the experiments. Q.D.L., D.M.L. and X.P.S. performed the experiments. J.Y., H.X.R. and D.M.L. analyzed the data. H.X.R., S.J.L. and L.L. contributed samples. L.L. identified rodents. J.Y., H.X.R. and D.M.L. wrote the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, J., Li, Q., Lu, L. et al. Detection and genetic diversity of Bartonella species in small mammals from the central region of the Qinghai-Tibetan Plateau, China. Sci Rep 12, 6996 (2022). https://doi.org/10.1038/s41598-022-11419-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-11419-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
