
Abstract
Natural attenuation, involving microbial adaptation, helps mitigating the effect of oil contamination of surface soils. We hypothesized that in soils under fluctuating conditions and receiving oil from seeps, aerobic and anaerobic bacteria as well as fungi could coexist to efficiently degrade hydrocarbons and prevent the spread of pollution. Microbial community diversity was studied in soil longitudinal and depth gradients contaminated with petroleum seeps for at least a century. Hydrocarbon contamination was high just next to the petroleum seeps but this level drastically lowered from 2 m distance and beyond. Fungal abundance and alpha-diversity indices were constant along the gradients. Bacterial abundance was constant but alpha-diversity indices were lower next to the oil seeps. Hydrocarbon contamination was the main driver of microbial community assemblage. 281 bacterial OTUs were identified as indicator taxa, tolerant to hydrocarbon, potentially involved in hydrocarbon-degradation or benefiting from the degradation by-products. These taxa belonging to lineages of aerobic and anaerobic bacteria, have specific functional traits indicating the development of a complex community adapted to the biodegradation of petroleum hydrocarbons and to fluctuating conditions. Fungi are less impacted by oil contamination but few taxa should contribute to the metabolic complementary within the microbial consortia forming an efficient barrier against petroleum dissemination.
Similar content being viewed by others

Introduction
The expansion of the oil industry in the first half of the twentieth century left behind sites where oil continues to seep to the surface. Contamination of surface soils by oil from leakage of old petroleum pumping wells or natural petroleum seeps is not necessarily a major problem because natural attenuation often limits the extent of the damage. During natural attenuation process, indigenous microorganisms degrade organic contaminants without external inputs that can promote bioremediation1. These natural phenomena occur in environments contaminated by many different compounds such as polychlorobiphenyls2 or hydrocarbons3,4. Natural attenuation reflects the fact that biota can adapt over time to resist contamination and efficiently degrade it in various environments and conditions. Most of the reported studies on microbiology of sites contaminated with hydrocarbons are focused on the short-term effect of the crude oil after a major contamination event (oil spill, oil pipe damage)4,5,6,7, but very few have studied the long-term effect of chronic crude oil seeps on the composition of microbial communities. Studies focusing on petroleum-contaminated soils in oil exploitation area have mostly tended to isolate microbes with hydrocarbon degradation properties for bioremediation application8. But there has been little in the way of attempts to analyse the composition of microbial communities in surface soils9, such as those contaminated for decades by oil seeps, to identify who are the best-adapted microbes and tend to found taxa as bioindicator of efficient petroleum degradation.
A wide diversity of microorganisms (fungi, bacteria and algae) are able to degrade and use petroleum (aliphatic and aromatic hydrocarbons) as sole carbon source using specific enzymatic machinery10,11. Although aerobic hydrocarbon biodegradation11,12 is the most energy-efficient respiration process13, the contaminated subsurface environments are often naturally anaerobic14, especially within oil plumes where the available oxygen has been depleted. Anaerobic bacterial degradation of petroleum-related compounds such as benzene15, toluene16, polycyclic aromatic hydrocarbons17 and alkanes18 has been demonstrated in aquifers, sediment and soil environments. Local consumption of oxygen19 or fluctuating conditions20 could create aerobic and anaerobic microniches in soil, then a multitude of degradation processes could coexist at a small scale, involving aerobic and anaerobic microbial functional populations21. Fungi could also be key players in degrading recalcitrant organic contaminants because of their variety of extracellular enzymes22. The fungal mobilization and degradation of contaminants could contribute to the release of bioavailable intermediates that could be further degraded by bacteria23,24. Hence, catabolic interactions among different microbial groups during biodegradation are extremely important. In this context, we therefore hypothesized that in environments under fluctuating conditions and receiving constant input of petroleum, both bacteria and fungi and, among bacteria, both aerobic and anaerobic taxa could coexist and be involved in hydrocarbon degradation processes. It is therefore essential to identify the whole microbial community adapted to chronic soil petroleum contamination and to estimate how far hydrocarbon contamination and its impact on communities extend around oil seeps.
In this study, we present a detailed spatial analysis of the microbial community structure in two forest soils which have both been impacted by petroleum seeps for at least a century but with contrasting soil characteristics. For both sites, we studied the hydrocarbon contamination longitudinal gradients away from oil seeps. Bacterial and fungal community composition was studied through high throughput sequencing of 16S and 18S rDNA and indicator taxa developing mainly next to the oil seeps and potential hydrocarbon-degraders were identified thanks to an original analysis rarely used in microbial ecology (TITAN) but well-adapted to environmental gradients.
Material and methods
Soil sampling
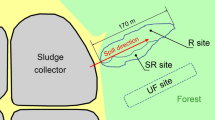
The first sampling campaign was carried out in May 2016, on two sites with contrasting soil characteristics located in the Haguenau forest (Alsace, France; Fig. 1). Both sites have been impacted by oil seeps since the end of petroleum extraction which took place in the region between the end of the eighteenth century and the middle of the twentieth century by Pechelbronn petroleum. The soil of the Gunstett site (GUN; 48° 55′ 9.8" N, 7° 48′ 6.7" E) presented a clay texture (42% clay, 40% silt, 18% sand) while that of Etangs-Verts (ETV; 48° 52′ 21.8″ N, 7° 46′ 51.6″ E) is sandy loam (15% clay, 23% silt, 62% sand). This difference was also highlighted by the different cationic exchange capacities (CEC) which were of 24.5 and 6.0 cmol kg−1 on average in GUN and ETV soils, respectively. Continuous oil seeps created ponds of 5–10 m of diameter (Fig. 1). On each site, 15 m longitudinal transects from each oil seep were carried out and 5 sampling zones were chosen at 0, 2, 4, 10 and 15 m from the oil seeps (named GUN-0, GUN-2, GUN-4, GUN-10, GUN-15 and ETV-0, ETV-2, ETV-4, ETV-10, ETV-15). Surface core soil samples (0 to 5 cm deep) were taken in triplicates (3 subsites 1 m apart from each other) in the 5 sampling zones, representing 30 samples (2 sites × 5 sampling zones × 3 replicates; Fig. 1).

Schematic map showing the location of the ETV and GUN study sites in the Haguenau forest (France), illustrations of the sampling designs for longitudinal and depth gradients and pictures (credit P. Faure) of the ponds created by oil seeps.
The second sampling campaign focused only on the Etangs-Verts (ETV) site and was carried out in May 2017. Only the first zone (0 m, ETV-0), the closest to the oil seep, was sampled (Fig. 1). Three independent soil cores of 30 cm depth were taken and were separated into 4 samples, to recover samples from 0–2 cm, 2–5 cm, 5–15 cm and 15–30 cm deep (named ETV0_P0-2, ETV0_P2-5, ETV0_P5-15 and ETV0_P15-30) representing 12 samples. The 0–2 cm depth sample was a solidified petroleum crust.
Soils (except ETV0_P0-2) were sieved at 4 mm, aliquots of fresh soil sample were kept at 4 °C until the return to the laboratory for mineralization activity measurements and estimation of the percentage of humidity. The rest was immediately frozen at -80 °C for further analyses (DNA and organic extractions). Texture (NF X 31–107), total nitrogen and organic carbon content (NF ISO 13,878 and NF ISO 10,694) and pH (in water, NF ISO 10,390) were measured at the LAS INRAE at Arras (France).
Mineralization activities under aerobic and anaerobic conditions
Two replicates of each soil samples (10 g fresh weight corresponding to 5.8 to 7.8 g dry weight) collected in May 2016 on longitudinal transects were incubated into 125 ml glass flasks sealed hermetically with Teflon septum. One replicate was incubated under aerobic condition (ambient atmosphere) and the other under anaerobic condition by flushing and replacing the atmosphere by N2/H2 (95:5) gas mix. Empty control flasks without soil (similar atmosphere than for aerobic and anaerobic tests) were run in the same way to correct CO2 measurements. Flasks were incubated at 24 °C for 7 days. Carbon dioxide (CO2) release was measured after 1, 2, 3, 4 and 7 days on a 2 ml portion of the flask atmosphere (sampled using a syringe) by infrared spectrophotometer (Binos 1004, absorption at 2325.6 cm−1)25. The mineralization activities were represented by the produced CO2 expressed as carbon mass per gram of dry weight soil per day (µg C g−1 d−1).
Organic extraction and hydrocarbon measurement
Solvent extractions were performed using an automated solvent extractor Dionex® ASE 350. Before extraction procedure, activated copper powder (1 g) and Na2SO4 (1 g) were added to 2 to 10 g of soil in the extraction cells to remove the molecular sulphur and the residual water, respectively. Extractions were performed twice at 130 °C and 100 bars with HPLC grade dichloromethane (DCM) using a static time of 5 min. Organic extracts were diluted with DCM to reach 20 ml. An aliquot (3 ml) was dried in a fume hood (36 h) to quantify the mass of the extractable organic matter (EOM)26.
The Hydrocarbon Index (HI) was measured according to the ISO 16,703:2004 procedure using a GC-FID 7890A Agilent technologies27. The integration limits of the chromatogram area, corresponding to the n-decane (C10H22) and n-tetracontane (C40H82), were defined using a standard mixture (ASTM D5307). For external calibration an ISO 11,046 standardized oil was used. Twelve solutions of different concentrations (from 1 to 31 mg ml−1) were injected into the GC-FID in order to obtain calibration curves. The HI was estimated from integration of the area under each chromatogram and corrected by the background noise (by integrating the area chromatogram obtained with a sample of pure dichloromethane that serves as a control).
Compound molecular distribution of soil EOM and crude oil (sampled directly in the middle of the oil seep) was determined through injection in a Shimadzu gas chromatograph (GC2010) equipped with a capillary column in silica glass DB-5MS (60 m × 0.25 mm i.d. × 0.1 µm film thickness) coupled with a mass spectrometer (QP2010) operating at 70 eV in fullscan mode. The inlet temperature was set at 300 °C and injections were run in split mode (split ratio of 1:5). The oven temperature program was as followed: 70 °C for 2 min, from 70 to 130 °C at 15 °C/min, then 130–315 °C at 4 °C/min followed by an isothermal stage at 315 °C for 25 min. The carrier gas was helium at 1.5 ml.min−1 constant flow. Compound identification was confirmed by the mass spectra obtained with the MS detector in comparison with the NIST08 and Wiley database spectra. Carbon preference index (CPI) was calculated according to Colombo et al.28 with the n-alkanes areas obtained on the GC–MS extracted ion chromatogram (m/z 57) of EOM from the different sampling points, using the formula: CPI = 2 × (n − C27 + n − C29)/(n − C26 + 2 × n − C28 + n − C30). CPI is a ratio classically used for EOM source and contribution type28,29, a ratio close to one is indicator of crude oil signature and higher values (> 5) indicates biogenic contribution.
DNA extraction, bacterial and fungal abundance and quantification of known hydrocarbon-degradation genes
Genomic DNA was extracted from ca. 0.5 g of the soil samples using a Fast DNA spin kit for Soil (MP Biomedicals). Genomic DNA extracts were diluted to 5 ng µl−1 to be used as templates for further PCR and qPCR analyses.
Quantification of bacterial and fungal communities was done using real-time PCR quantification using primer sets 968F/1401R and Fung5F/FF390R for 16S and 18S rRNA gene quantification, as described previously by Cébron et al.30 and Thion et al. 31. PAH-RHD\(\mathrm{\alpha }\) genes encoding alpha subunits of ring-hydroxylating dioxygenases from Gram negative (GN) Proteobacteria and Gram-positive (GP) Actinobacteria, alkB genes encoding alkane monooxygenases32 and bssA genes encoding benzylsuccinate synthases were quantified using PAH-RHD\(\mathrm{\alpha }\) GN and GP primers, alkB-F/R primers and 7772F/8546R primers following Cébron et al.30, Gielnik et al.33 and Winderl et al.34, respectively. Real-time PCR assays were performed using iQ SybrGreen Supermix (Biorad) and CFX96 apparatus (BioRad). Data were expressed as gene copy number per gram of dry weight soil thanks to ten-time dilutions (from 108 to 102 copies/µl) of standard plasmids.
Bacterial 16S and fungal 18S rDNA sequencing
The V3/V4 region of bacterial 16S rRNA genes (ca. 450 bp) was amplified using S-D-Bact-0341-a-S-17 and S-D-Bact-0787-b-A-20 primers following a previously described dual-index strategy35 using PCR primers with an Illumina adaptor, pad and index sequences36. PCR reactions were performed on 2 μl of diluted gDNA using Phusion high-fidelity polymerase (Thermo Scientific). PCR reactions consisted of 31 cycles with touchdown annealing temperature for 18 cycles (63–54 °C with a decrease of 0.5 °C/cycle) and 13 cycles at 54 °C. Amplification products were checked on 1% agarose gel electrophoresis and purified using the UltraClean-htp 96Well PCR Clean-Up kit (Qiagen) following the manufacturer’s instructions. After Quant-iT PicoGreen ds-DNA Assay Kit (Invitrogen) quantification, an amplicon library was prepared (equimolar pool at 10 nM), purified on QIAquick PCR purification Kit Column (Qiagen) and sent for sequencing to the Genewiz platform (South Plainfield, NJ, USA) which used an Illumina MiSeq V2 Kit for 2 × 250 bp paired-end sequencing.
The V7–V8 region of fungal 18S rRNA genes was amplified (ca. 390 bp) using FR1 and FF390 primers37 fused with a partial sequencing adapter. This first PCR was performed using Phusion high-fidelity polymerase (Thermo Scientific). The PCR reactions consisted of 25 to 30 cycles (depending on the samples) with an annealing temperature of 54 °C. Amplification products were checked by 1% agarose gel electrophoresis before being sent to Microsynth AG (Balgach, Switzerland) for further steps namely second PCR, library preparation and Illumina MiSeq 2 × 250 bp paired-end sequencing.
Illumina MiSeq paired-end reads for 16S and 18S rDNA were deposited in the SRA database under BioProjects ID: PRJNA685399 and PRJNA686256, respectively.
Processing of bacterial 16S and fungal 18S rDNA sequencing data
Sequence data were analysed following the MiSeq SOP procedure available in March 2017 and described in Kozich et al.35, using Mothur v.1.36.038. Only 41 out of 42 samples were treated for 16S rDNA because sequencing failed for GUN-15 third replicate. Paired-end reads were trimmed to a minimum QScore of 20 (both 16S and 18S rRNA reads) and joined using the following criteria: no ambiguous bases and 404 bp < length < 430 bp or 348 bp < length < 359 bp for 16S or 18S rDNA paired-end reads, respectively. After, alignment of unique sequence representatives to Silva V132 fasta data (released in Dec. 2017) and pre-clustering of sequences (sequences that are within 2 nt of each other), chimeras were detected and removed using Uchime and singletons (split.abund command removing sequences represented only once among all reads) were removed. Taxonomy was assigned using the Silva V132 tax data with a cut-off = 80 and using the Wang method. Sequences affiliated to archaea, chloroplasts, unknown, mitochondria and eukaryota were removed for 16S rDNA analysis, while sequences affiliated to archaea, chloroplasts, unknown, mitochondria and bacteria were removed for 18S rDNA analysis. After uncorrected pairwise distances calculation between aligned sequences, the bacterial and fungal sequences were clustered in Operational Taxonomic Units (OTUs) at 97% similarity, and consensus taxonomy for each OTU was determined using classify.otu. Finally, datasets were rarefied to the lowest number of sequences per sample (53,422 and 14,260 reads/sample for bacterial and fungal sequences, respectively).
The number of reads for each OTU was expressed as a percentage to express the relative proportion or relative abundance of each OTU within the total community. Alpha diversity was expressed by calculating Chao1 richness, Pielou’s evenness J’ and Shannon diversity H’ indices using Mothur.
Bacterial functional trait inference
Functional traits were compared between increasing indicator taxa found in ETV0 and GUN0 samples (identified through TITAN2 analysis, see below) and the whole bacterial community inhabiting the control forest samples ETV15 and GUN15.
Inference of 19 bacterial traits (e.g. tolerance to pH, NaCl, temperature, preference to oxygen, Gram staining, width, length, shape, motility, pigment, spore production, trophic type and DNA GC %) was performed using the BactoTraits database39. The relative proportion of each trait attributes for all bacterial OTUs were calculated and attributed based on their taxonomic affiliation to get OTUs trait profiles. Based on the relative abundance of each OTU, the relative abundance of each trait attributes for the 19 traits was calculated and compared for the two different populations by Wilcoxon rank-sum test (p value < 0.05).
The potential functional pathways of the two bacterial populations were profiled using Tax4Fun40 (v 1.0.1) run on Galaxy (X. SIGENAE [http://www.sigenae.org/]). Differences in the level 1, level 2 and level 3 KEGG predicted functions of increasing indicator taxa and of control forest soil communities were compared and analyzed by Wilcoxon rank-sum test (p value < 0.05).
Statistical analysis
Statistical analyses were performed using R studio version 3.6.041. Values of soil parameters (extractable organic matter EOM, hydrocarbon index HI, total organic carbon C, nitrogen N and C/N ratio), mineralization activities, bacterial and fungal quantification (16S and 18S rRNA gene copy numbers), alpha-diversity estimators (Chao1, Shannon diversity and Pielou’s evenness indices), relative abundance of bacteria phyla and fungal divisions and the relative abundance of the bacterial functional trait attributes were compared among the 5 different zones (0, 2, 4, 10 and 15 m from the petroleum seeps) for both sites independently (GUN and ETV) and for different sampling depths at the ETV site. This was carried out through one-way analyses of variance (Anova) with p < 0.05, followed by Tukey HDS PostHoc tests.
Using the Vegan R package42, non-metric multidimensional scaling (NMDS) analyses were performed (‘metaMDS’ function) based on the Bray–Curtis dissimilarity matrix generated from bacterial and fungal relative abundances to estimate the dissimilarity in structure between all samples. For bacteria, OTUs having at least 0.1% relative abundance (≥ 54 reads) were kept for this analysis. The environmental gradients of hydrocarbon content were built on NMDS ordination space using the ‘Ordisurf’ function of the Vegan R package which used generalized addictive modelling (GAM) to overlay environmental variables42,43. Permutational multivariate analyses of variance (PERMANOVA) were performed with 999 permutations using the ‘adonis’ function in Vegan R package to determine if bacterial and fungal community composition differed significantly between sites (next to the oil seeps vs. the others).
Threshold Indicator Taxa ANalysis (TITAN) was performed to identify environmental variable values (in our case hydrocarbon index values) maximizing taxa frequency and abundance using bootstrapping to identify reliable indicator taxa44. TITAN 2.4 package in R45 was used. Association was measured by taxon abundances weighted by their occurrence in each sample as for Indicator taxa determination46 and standardized as z-scores to facilitate cross-taxon comparison via permutation of samples along the predictor. The number of bootstraps and permutations performed in our TITAN2 analysis was 500 and 250, respectively. Analysis was performed at the OTU taxonomic level. Only OTUs observed more than 3 times across all sites were used and thus 3,306 and 861 OTUs were considered for bacterial and fungal analyses respectively. TITAN2 distinguishes declining and increasing Indicator Taxa along environmental gradient.
Originality-significance statement
The significance and novelty of our work is the study of bacterial and fungal diversity and the identification of indicator taxa developing next to petroleum seeps and most likely involved in the hydrocarbon degradation. The originality is that these indicator taxa are well-known aerobic and anaerobic bacteria having specific functional traits making them well-adapted to hydrocarbon-contaminated environment as well as fluctuating redox conditions.
Results and discussion
Soil characteristics, and hydrocarbon contamination levels
Soil properties were characterized for the two sampling sites and both campaigns (Table 1). At the ETV site, nitrogen content was higher next to the oil seep and its content decreased with depth, while staying constant all over the transect at GUN site. Due to high concentration of petroleum carbon, the C/N ratio was high next to the oil seeps (c.a. 40) compared to values found at a 15 m distance (c.a.15). Finally, the pH values were also higher next to the oil seeps and decreased with distance. It was previously shown that oil contaminated soils hrabored alkalin pH47,48. At the first zone next to the petroleum seep, at both sites, the average quantity of extractable organic matter (119.2 and 118.6 mg EOM g−1 in the GUN and ETV sites, respectively), and the hydrocarbon index (HI, 73.7 and 35.3 mg g−1 in the GUN and ETV sites, respectively) values were significantly higher than in the samples farthest from the seeps at a distance of 10 or 15 m which revealed high oil contamination of the soil at the vicinity of the seeps. The EOM and HI values did not decrease gradually with distance but dropped by more than 90% from the second sampling zone (a 2 m distance from the oil seeps). In opposite way, an increase in the carbon preference index (CPI) with increasing distance from the oil seep (Table 1), was shown. CPI is a ratio classically used for EOM source and contribution type28,29. The low ratio (around 1) as measured next to the oil seeps is indicator of crude oil signature and higher values (> 5) indicates a major contribution of biogenic compounds to the soil organic matter. For both sites, samples closest to the oil seeps were dominated by n-alkanes similar to those of the crude oil (Figure S1); this strong crude oil contribution in the EOM fingerprint decreased as the sampling points get further from the oil seepage. The EOM molecular distribution became dominated by n-alkanols and fatty aldehydes with even number of carbon atoms, and n-alkanes with odd number of carbon atoms (Figure S1), characteristic of biogenic samples and higher plant contribution28,29 commonly found in natural soils. At the farthest sampling points from the oil seeps (ETV15 and GUN15) petroleum signature completely disappeared (Figure S1). Samples from the second sampling campaign showed a similar gradient of hydrocarbon contamination with depth but at a smaller scale, highlighting higher contamination in the surface crust (108.9 and 42.3 mg g−1 for EOM and HI, respectively) compared to the samples taken at a depth of 15 and 30 cm (Table 1). Same gradients were observed for the total organic carbon content with distance and depth (Table 1).
Microbial activity and abundance
To evaluate the functional potential of hydrocarbon degradation, we measured the microbial activity of organic matter mineralization under aerobic and anaerobic conditions. Both aerobic and anaerobic microbial mineralization activities were detected. Mineralization activity was 3–7 times greater under aerobic than under anaerobic conditions for both sites (Table 2). Both aerobic and anaerobic mineralization activities were significantly higher next to the oil seeps at the first sampling zone compared to the rest of the transect, and decreased with depth at ETV site. Significant positive Pearson correlations (p < 10–5) were found between organic matter mineralization activities and HI values (r = 0.61 and 0.52 for aerobic and anaerobic mineralization activities, respectively). As much of the organic matter found next to the oil seep is composed of petroleum hydrocarbons we can assume that microbial activities were mainly dedicated to hydrocarbon degradation both in aerobic and anaerobic conditions.
The distance to the petroleum seep was not found to significantly influence the number of bacteria and fungi (Table 2). We quantified 5 × 1010 to 10 × 1010 copies of 16S rRNA genes per gram of soil in both soil transects. Fungal 18S rRNA gene copy number was 100 times lower than for 16S rRNA one. The quantification of bacteria showed a significant decrease with soil depth from 10 × 1010 to 2 × 1010 16S rRNA gene copy numbers per gram of soil from the surface (0–2 cm) to the 15–30 cm depth samples.
Quantification of known hydrocarbon-degradation genes
The well-known bacterial genes encoding PAH dioxygenase (PAH-RHD\(\mathrm{\alpha }\)), alkane monooxygenase (alkB) and benzylsuccinate synthase-like (bssA) were quantified to estimate the functional population potentially involved in hydrocarbon degradation in aerobic and anaerobic conditions (Table S1). No significant difference in PAH-RHD\(\mathrm{\alpha }\) and alkB gene abundance was shown across the transects even if these genes involved in aerobic hydrocarbon degradation tended to be in higher quantity next to the oil seep in samples from ETV site. At this site, the abundance of PAH-RHD\(\mathrm{\alpha }\) genes from both Gram-negative Proteobacteria and Gram-positive Actinobacteria was significantly higher in surface crust (0–2 cm) than in the deeper samples (5–30 cm). However, when expressed as percentage compared to 16S rRNA genes, PAH-RHD \(\mathrm{\alpha }\) genes represent less 0.02% while alkB genes could represent up to 1.0% next to the oil seep. Even if well-known Proteobacteria and Actinobacteria possess these functional genes10,30,32, it’s more likely that most taxa involved in hydrocarbon degradation in situ would possess other suites of genes because they were detected in really low quantity. It was previously showed that for active taxa belonging to PAH-degrading consortia identified through stable isotope probing, the ratio of PAH-RHD\(\mathrm{\alpha }\) relative to 16S rRNA genes only accounted for 1.2 to 5.9%49. Similarly, genes involved in all steps of aromatic compounds metabolism represented less than 5% of the metagenome of an active PAH-degrading consortia where most of the taxa were not involved in the first hydroxylating step but in intermediate metabolite degradation50. Targeting PAH-RHD\(\mathrm{\alpha }\) and alkB genes involved in this first step is not sufficient to evaluate the abundance of bacteria involved in all steps of hydrocarbon mineralization. When targeting bssA genes involved in anaerobic hydrocarbon degradation, a significant higher abundance of these genes was detected in DNA extracted from sites close to the oil seeps in both ETV and GUN sites and in the surface petroleum crust in ETV site (Table S2). However, these genes also represent a low percentage compared to 16S rRNA genes (less than 0.004%). Even if the identification of individual metabolic genes involved in oil degradation in situ sometime showed correlation between functional-population abundance and contamination level30,34, here it was not sufficient to conclude about functional pathways involved in oil degradation. Metagenomic analysis could be helpful in resolving this question but unknown genes and pathways (not annotated) as well as the metabolic complementarity of the taxa could prevent the analysis of such complex metagenomes, especially as there seem to be both anaerobic and aerobic processes taking place in our study sites.
Bacterial and fungal community : alpha-diversity indices and composition
To help us assess the impact of hydrocarbons on the microbial community structure and composition, 16S rDNA and 18S rDNA sequencing were performed. After quality filtering, a total of 5,187,772 and 2,485,385 paired-end sequences were retrieved from the 16S rDNA and 18S rDNA libraries, ranging from 53,422 to 122,937 reads per sample for 16S rDNA and from 4,016 to 79,581 reads per sample for 18S rDNA. After sub-sampling to 53,422 and 14,260 reads per samples for 16S and 18S rDNA libraries, respectively, both 16S and 18S rDNA data were analyzed for 41 samples out of 42 (GUN-15 third replicate failed for 16S rDNA sequencing and ETV-15 s replicate was removed for 18S rDNA analysis because of a too low number of sequences). No general trend was found for fungal alpha-diversity indicators (Table 2), either according to the distance to the oil seep or in depth. However, on both sites, bacterial richness (Chao1), Shannon diversity and Evenness indices were lower next to the oil seeps compared to all other sampling distance and also in the surface crust compared to the other depth samples (Table 2). Negative Pearson correlations were observed between EOM, HI and TOC values and the 3 alpha-diversity indices (Chao1, Shannon and Evenness) describing bacterial community composition (Table 3) for the 2 sites. Similarly, a lower bacterial taxonomic richness was observed in oil plume from Deepwater horizon spill6. This observation indicate that mostly hydrocarbon-degrading bacteria were enriched next to the oil seeps and that bacteria sensitive to petroleum contamination disappeared as shown previously following oil contamination due to pipeline rupture51.
Overall no significant bacterial community composition difference (Wilcoxson test, p = 0.543) was detected between GUN and ETV sites at the phylum level. The distance to the petroleum seep positively or negatively influenced many bacterial phyla (Fig. 2A). The relative abundance of Actinobacteria, Chloroflexi, Firmicutes and Kiritimatiellaoeota members was significantly higher next to the oil seep and decreased with distance on GUN site (p ≤ 0.003). For the two first phyla the same tendency was observed on ETV site along the longitudinal gradient (p ≤ 0.016) and with depth with a significantly higher proportion (p < 0.0001) of these phyla in the surface petroleum crust than at 5–30 cm depth. It was previously shown that Actinobacteria and Firmicutes dominated the bacterial community in soil after oil pipeline rupture52 and that Chloroflexi and Firmicutes relative abundance correlated with diesel-contamination level53. Chloroflexi can degrade petroleum hydrocarbons by anaerobic respiration. The relative abundance of Verrucomicrobia, Alpha-Proteobacteria, Planctomycetes, Elusimicrobia and Fibrobacteres (the two latter classified in the group “others”) members (Fig. 2A) increased significantly when moving away from the oil seep for both sites (p ≤ 0.047). Although no trend was found in the literature at the phylum level, some members of these phyla are most likely sensitive to oil contamination. For both GUN and ETV sites, the bacterial community structures at the OTU level were found to be changed according to the petroleum contamination of soil samples as confirmed using NMDS analysis where the hydrocarbon concentration gradient was overlapped (Fig. 3A). On the first component, samples were separated according to the hydrocarbon content and the two sites were separated on the 2nd component. At OTU level, the bacterial community composition was significantly different among the different sampling areas (Permanova, p = 0.001). Previous studies have shown such structuration of the bacterial communities according to hydrocarbon contamination level54,55.

Changes in relative abundance of bacterial phyla (A) and fungal sub-divisions (B) in GUN and ETV sites according to the distance from the petroleum seeps (0, 2, 4, 10 and 15 m) and depth (P0-2, P0-5, P2-5, P5-15 and P15-30 cm) at ETV site for the 0 m distance. Data are means (n = 3). Different classes of Proteobacteria (plot A) and sub-divisions of Mucoromycota, Basidiomycota and Ascomycota (plot B) were shown. The group “Others” (plot A) includes phyla below 0.5% (267 reads) of relative abundance and affiliated to Armatimonadetes, BCR1, Caldiserica, Calditrichaeota, Cyanobacteria, Deferribacteres, Deinococcus-Thermus, Elusimicrobia, Entotheonellaeota, FBP, FCPU426, Fibrobacteres, Firestonebacteria, Fusobacteria, GAL15, Halanaerobiaeota, Hydrogenedentes, Kiritimatiellaeota, Lentisphaerae, Margulisbacteria, Nitrospinae, Omnitrophicaeota, Schekmanbacteria, Spirochaetes, TA06, Tenericutes, unclassified_bacteria, WPS_2, WS2, WS4 and Zixibacteria.

Ordisurf model showing the distribution of hydrocarbon index (HI) level over the NMDS ordination of bacterial (A) and fungal (B) community structures at the OTU level. The hydrocarbon gradient is visualized through green curves. ETV and GUN sites are differentiated through circles and diamonds, respectively. Different sampling zones are visualized thanks to colour gradients from dark red (0 m, close to the oil seep), red (2 m), orange (4 m), light orange (10 m) and yellow (15 m).
Overall no significant fungal community composition difference (Wilcoxson test, p = 0. 861) was detected between GUN and ETV sites at the division level. Fewer changes were observed for the fungal community composition at the division and sub-division levels along longitudinal and depth gradients (Fig. 2B). Although the relative abundance of members of the Taphrinomycotina sub-division (Ascomycota) was very low (< 0.3%), a significant increase was observed next to the oil seep (p = 0.014) and in the petroleum surface crust (p = 0.042). Similarly, the relative abundance of Ustilaginomycotina, Pucciniomycotina (both Basidiomycota) and Pezizomycotina (Ascomycota) members was significantly higher in the surface petroleum crust and decreased with depth (p ≤ 0.032). Pezizomycotina was found to be dominant56 and were isolated57 in diesel-contaminated soil, maybe because of their hydrocarbon-degradation pathways58. Modification of the fungal community structure at the OUT level was less pronounced than for bacteria, but a similar gradient was observed according to the hydrocarbon content (Fig. 3B). The lower impact of on fungal community can suggest that hydrocarbons are less toxic to fungi, that fungi can cope with hydrocarbon contamination or that they can benefit from hydrocarbon compounds for their growth being well adapted to the degradation of complex carbon substrates. Fungi with their multiple enzymatic activities (hydrolytic enzymes or oxidases such as peroxidases or phenol oxidases) are able to degrade organic pollutants such as aliphatic hydrocarbons and PAHs59.
Bacterial and fungal indicator taxa
As bacterial and fungal community composition are shaped by petroleum contamination, indicator taxa (IT) relative to the hydrocarbon level were sought to further understand which microorganisms are tolerant/sensitive to oil contamination, potentially involved in hydrocarbon degradation or benefiting of degradation metabolites. Data from both sampling sites were combined and indicator taxa were sorted using TITAN2, a well-adapted tool for gradient studies. Bacterial and fungal indicator taxa increasing (Fig. 4) or decreasing (Figure S2) along the hydrocarbon contamination gradient (HI values) were identified. As both bacterial and fungal indicator taxa were identified, we confirmed our hypothesis that different groups of bacteria and fungi with complementary metabolic properties may coexist and potentially cooperate at the vicinity of the oil seeps for oil degradation60. The presence of all these microorganisms with varied metabolisms should form an efficient barrier against dissemination of the petroleum contamination under fluctuating environmental conditions.

Relative abundance of increasing bacterial (A) and fungal (B) indicator taxa (summed at the class and sub-division levels for bacteria and fungi, respectively) determined using TITAN2 along the gradient of oil contamination for the combination of both ETV and GUN sites. ETV and GUN sites are symbolised by pentagon and triangle symbols, respectively, and distance to the oil seep is shown with colour gradient (from red to green). At the OTU level, 281 and 61 bacterial and fungal indicator taxa were sorted with purity and reliability parameters both > 0.95. For the complete list of bacterial and fungal OTU found as indicator taxa see Table S2 and S3, respectively. The relative abundances of OTU belonging to similar classes were summed. The group “Others” comprise classes having less than 3 representative OTUs and were affiliated to Chlamydiae, Melainanabacteria (Cyanobacteria phylum), Dadabacteriia, Babeliae (Dependentiae phylum), Gemmatimonadetes, Latescibacteria, Nitrospira, Saccharimonadia (Patescibacteria phylum) and Rokubacteria NC10 classe. The group “other_Acidobacteria” comprise OTU affiliated to Holophagae, subgroup_5 and subgroup_22 classes. The group “other_Actinobacteria” comprise OTU affiliated to Thermoleophilia and Coriobacteriia classes. The group “other_Chloroflexi” comprise OTU affiliated to Chloroflexia, TK10 and JG30-KF-CM66 classes. The group “other_Firmicutes” comprise OTU affiliated to Bacilli and BRH-c20a classes.
TITAN2 sorted 281 and 664 bacterial OTUs as indicator taxa which were found to increase and decrease respectively with HI values (the complete list is available in Table S2). The relative abundance of these indicator taxa reached 30–40% when getting closer to or further from the oil seeps for increasing and decreasing taxa respectively. Among the bacterial IT increasing next to the oil seeps, we found 110 OTUs belonging to Proteobacteria, 51 to Acidobacteria, 39 to Actinobacteria, 16 to Chloroflexi, 16 to Firmicutes and 14 to Bacteroidetes (Fig. 4A). Various IT belonging to lineages of anaerobic or facultative anaerobic bacteria, had their relative abundance increasing next to the oil seeps. Members of the Acidithiobacillaceae family were identified. In this family, bacterial strains belonging to the Acidithiobacillus genera are chemolithoautrophes that can oxidize elemental sulphur or oxidize minerals containing ferrous iron61. Among the Rhodocyclaceae family, OTUs belonging to Dechloromonas and Sulfuritalea genera were identified as increasing IT. These genera were previously detected in hydrocarbon-polluted sites62 and could degrade a wide variety of aromatic compounds under anaerobic nitrate-reducing conditions63,64,65 through potentially original pathways66. Moreover, Sulfuritalea are also well recognized as chemolithoautotrophic sulfur-oxidizing bacteria67. Some OTUs affiliated to the Chloroflexi and belonging to the Anaerolineae were also identified as increasing IT. This lineage is composed of bacteria known as strictly anaerobic fermentative and sulphate-reducing bacteria, previously found in hydrocarbon-rich sludge samples68 and in anaerobic alkane-degrading culture69. Other IT mostly known as aerobic or facultative aerobic taxa had their relative abundance increasing next to the oil seeps. OTUs belonging to Gamma-Proteobacteria were identified, they are affiliated to the Porticoccaceae family and to Immundisolibacter, Rhodanobacter and Pseudoxanthomonas genera. Members of Porticoccaceae family have been described as aromatic-hydrocarbon degraders70 and alkane-degraders71, but they also possess in their genomes a high number of secondary alkane-degrading genes72. Immundisolibacter strain have been isolated from aerobic bioreactor for its capacity to degrade high-molecular-weight PAHs73. Even if Rhodanobacter was previously identified as a member of a PAH-degrading consortium74 it would more likely degrade intermediate metabolites75 because no PAHs degradation gene have been detected in sequenced genomes76. The Pseudoxanthomonas species have been found to degrade different PAHs77,78 and BTEX compounds79 and some strains could produce a rhamolipid surfactant that enhances the hydrocarbon degradation80. Two types of Alpha-Proteobacteria were also identified as aerobe increasing IT. The Methyloligellaceae members that are well-known methylotrophs81, and the Parvibaculum genus possessing genes involved in alkane utilisation82 that was detected in situ83 and in aerobic enrichment culture degrading crude oil84. Well known aerobic alkanes and PAHs degraders, such as the Actinobacteria belonging to Williamsia85,86, Cellulomonas87,88, Agromyces89 and Mycobacterium90 genera, were almost undetected in the forest soil but became abundant next to the oil seeps and identified as increasing IT. These Actinobacteria are equipped with hydroxylase enzymes such as monooxygenases and dioxygenases, to initiate the aerobic oxidation of hydrocarbon molecules10,91. In addition, some of these genera can also produce exopolysaccharides (EPS) which serve as bioemulsifiers such as glycolipides or curdlan-like EPS produced by Cellulomonas spp.88,92. Finally, two types of Acidobacteria appeared as increasing IT, the members of Solibacterales order with undemonstrated hydrocarbon-degradation capacities but that were previously found in oil contaminated soil and sediments93,94 and the members of the Acidobacteriales order that were identified as aerobic benzene-degraders using stable isotope probing technique95.
As shown before, the fungal community structure was less impacted by petroleum contamination and thus a low number of OTUs indicator taxa was found. TITAN2 sorted 61 and 40 fungal OTUs as indicator taxa which were found to increase (Fig. 4B) and decrease (Figure S2) respectively with hydrocarbon index values (complete list is available in Table S3). The relative abundance of the indicator taxa which increased with the gradient (Fig. 4B) was highly variable from one sample to another (1 to 42%). However, two fungal OTUs have higher relative abundance close to the oil seep, one belonging to Lactarius genus (Basidiomycota, Agaricomycetina) and another belonging to Cryptomycota LKM11. Lactarius genera are well-known forest soil symbiotroph ectomycorrhizal fungi but can also be facultative saprotroph96. Lactarius species increased up to 33.8% close to the oil seep indicating an essential role in the biodegradation of hydrocarbons. Indeed, the role of Agaricomycetes in bioremediation of crude oil-contaminated soil was demonstrated97 and the in vitro degradation of various hydrocarbons was reported for many Basidiomycetes98. Cryptomycota LKM11 represented 1.8 to 12.7% at the sampling sites close to the oil seeps. This fungal sub-division was recently found in PAH contaminated soils99, anoxic and hydrocarbon-enriched oil sand tailings ponds100 or creosote polluted site101.
Functional traits of the increasing bacterial indicator taxa
Functional traits were predicted from BactoTraits39 (morphological and physiological traits and affiliation to functional groups) and Tax4Fun40 (KEGG functional pathways) tools to better characterize the bacterial indicator taxa increasing and dominant next to the oil seeps (Fig. 5). We hypothesized that increasing bacterial IT possess specific functional traits making them tolerant to hydrocarbon contamination and giving them a functional advantage to survive and grow next the oil seeps.

Functional traits of the increasing bacterial indicator taxa (detected in ETV0 and GUN0, in red) compared to control forest soil bacteria (detected in ETV15 and GUN15, in green) based on Bactotraits (A) and Tax4Fun (B) analyses. Only traits with significant differences between the 2 populations were represented (except for few functional pathways at the Level2 on plot B) and labelled with a * meaning that Wilcoxon rank sum tests had a p value < 0.05. Trait attributes are expressed as mean percentage (n = 6 for increasing Indicator taxa, and n = 5 for forest bacterial community) and ± standard deviation. (A) Morphological (Gram staining: the rest are Gram negatives, and motility: the rest are motile bacteria possessing flagella) and physiological (trophic type as lithotroph, i.e. bacteria that obtains its energy (electron donor) from inorganic compounds (iron, sulfur…) in opposition to organotroph, and oxygen preference: all attribute classes are shown) and affiliation to functional groups (5 functional groups were defined, no difference was found for stress-tolerant group) were assessed using BactoTraits (Cébron et al. 2021). (B) Genomic traits, i.e. KEGG functional pathways shown at Level 1, Level 2 and Level 3. Only functions representing more than 0.1% were represented. M: Metabolism, GIP: Genetic information processing, EIP: Environmental Information Processing and CP: Cellular Processes.
Among the bacterial IT increasing next to the oil seeps, a higher proportion of Gram positive and non-motile bacteria were found (Fig. 5A) and confirmed by predicted functions showing that functional pathways related to cell motility (bacterial chemotaxis and flagellar assembly) were detected in lower proportion (Fig. 5B). Bacteria characterized as “competitors” and “colonizers” were more abundant while the “stress-sensitives” and “mesophiles” were in lower proportion among the bacterial IT next to the oil seeps than in the control forest soil bacterial community (Fig. 5A). We previously described that competitors possessed traits improving the access to resources even when limited such as in oligotrophic environments and that Gram-positive and non-motile bacterial traits characterized this functional group. In contrast, colonizers were bacteria with high phenotypic plasticity with the ability to grow in anaerobic or facultative anaerobic conditions39. The higher proportion of bacteria belonging to these two functional groups among the IT could confirm the presence of two complementary populations for efficient hydrocarbon degradation next to the oil seeps.
There was also a higher proportion of known lithotrophs and of anaerobic or facultative anaerobic bacteria close to the oil seeps (Fig. 5A). Chemolithotropich bacteria close to the oil seeps could provide Fe(III) as electron acceptor to hydrocarbon-degraders102. Moreover, among bacterial IT, both aerobic and anaerobic/facultative anaerobic taxa were identified representing 62.5 and 33.1% respectively, while control bacterial community was mainly composed of aerobic bacteria. This finding agrees with higher rates of both aerobic and anaerobic hydrocarbon mineralization that were measured next to the oil seep (Table 2). These findings could indicate a zone of mixing between aerobic bacteria from surface soil and anaerobic bacteria brought to the surface soil with the oil seep as anaerobic hydrocarbon degraders prevail in sub-surface oil samples and contaminated petroleum groundwater103,104. The co-occurrence of different metabolisms has the advantage of reducing the concentrations of both toxic contaminants and of degradation metabolites105. Aerobic and anaerobic populations could alternatively be active depending on fluctuating redox conditions in the soil or could cooperate for hydrocarbon degradation such as in syntrophic metabolisms21 by substrates sharing104,106. Joint anaerobic and aerobic metabolisms were rarely suggested107, but recent evidence of the co-transcription of genes involved in both aerobic and anaerobic benzene degradation in continuous culture108 would explain our findings as well as previous ones of co-existence of aerobic and anaerobic bacteria in oil contaminated desert soils55.
At level 1 of KEGG functional categories, functions related to “Metabolism” and “Genetic information processing GIP” were detected in higher proportion while “Environmental information processing” and “Cellular processes” were detected in lower proportion for bacterial IT next to the oil seeps than for the control communities (Fig. 5B). Within the Metabolism category, “carbohydrate metabolism” and “nucleotide metabolism” were over-represented, with higher proportion of functions involved in glycolysis/gluconeogenesis, pentose phosphate pathway, and galactose, starch, sucrose, propanoate, C5-branched dibasic acid, and purine metabolisms. Some functions involved in the metabolism of lipids, cofactors and vitamins and terpenoids and polyketides were also detected in higher proportion. Similarly, Zhang et al. 109 showed that in sediment supplemented with pyrene, genes involved into almost every metabolic pathways showed a dramatic increase. Our finding indicates the possible availability of simple carbon sources besides hydrocarbons because these functions are dedicated to optimize the use of available resources and underline the large genetic investment of bacteria in the processing of environmental information important in these oil polluted environments110.
Moreover, within the “xenobiotic biodegradation and metabolism” category, functions involved in polycyclic aromatic hydrocarbon degradation as well as drug metabolism were significantly more present in bacterial IT increasing next to the oil seeps than in the control communities (Fig. 5B). All these traits indicate greater and different metabolic capacities of the bacterial IT identified, as well adapted to hydrocarbon contamination. Finally, within the GIP category, functions related to “Folding, sorting and degradation”, “Transcription” and “Translation” were also over-represented. These regulatory functions may be related to the adaptation of microorganism to this highly hydrocarbon-contaminated environment. Similarly, through genome analysis of the oil-degrading Pseudomonas aeruginosa N002 strain, authors found among other gene categories, a very high abundance of genes for transcription111.
Conclusions
At this study site, where petroleum seeps have been contaminating the soil for at least a century, we were able to demonstrate the natural attenuation phenomenon. Indeed, the presence of microbial communities, that are well adapted to oil and its degradation, limits the extent of the contamination to a few meters around the seeps. We identified bacterial taxa that are well known for their capacity to biodegrade hydrocarbons, which it would be interesting to isolate to study their metabolism and use them for bioremediation purposes. Amongst the indicator taxa, which are highly favoured in the vicinity of oil seeps, we have shown the coexistence and possible cooperation of fungi as well as aerobic and anaerobic bacteria having specific functional traits that make them well adapted to the hydrocarbon contamination and to fluctuating redox conditions.
Change history
07 July 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41598-022-15752-z
References
Sarkar, D., Ferguson, M., Datta, R. & Birnbaum, S. Bioremediation of petroleum hydrocarbons in contaminated soils: Comparison of biosolids addition, carbon supplementation, and monitored natural attenuation. Environ. Pollut. 136(1), 187–195 (2005).
Vergani, L. et al. Bacteria associated to plants naturally selected in a historical PCB polluted soil show potential to sustain natural attenuation. Front. Microbiol. 8, 1385 (2017).
Kachienga, L., Jitendra, K. & Momba, M. Metagenomic profiling for assessing microbial diversity and microbial adaptation to degradation of hydrocarbons in two South African petroleum-contaminated water aquifers. Sci. Rep. 8(1), 1–6 (2018).
Ferguson, D. K. et al. Natural attenuation of spilled crude oil by cold-adapted soil bacterial communities at a decommissioned High Arctic oil well site. Sci. Total Environ. 722, 1358 (2020).
Kleikemper, J. et al. Activity and diversity of sulfate-reducing bacteria in a petroleum hydrocarbon-contaminated aquifer. Appl. Environ. Microbiol. 68(4), 1516–1523 (2002).
Hazen, T. C. et al. Deep-sea oil plume enriches indigenous oil-degrading bacteria. Science 330(6001), 204–208 (2010).
Kostka, J. E. et al. Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the Deepwater Horizon oil spill. Appl. Environ. Microbiol. 77(22), 7962–7974 (2011).
Spini, G. et al. Molecular and microbiological insights on the enrichment procedures for the isolation of petroleum degrading bacteria and fungi. Front. Microbiol. 9, 2543 (2018).
Higashioka, Y., Kojima, H., Sato, S. & Fukui, M. Microbial community analysis at crude oil-contaminated soils targeting the 16S ribosomal RNA, xylM, C23O, and bcr genes. J. Appl. Microbiol. 107(1), 126–135 (2009).
Habe, H. & Omori, T. Genetics of polycyclic aromatic hydrocarbon metabolism in diverse aerobic bacteria. Biosci. Biotechnol. Biochem. 67(2), 225–243 (2003).
Abbasian, F., Lockington, R., Mallavarapu, M. & Naidu, R. A comprehensive review of aliphatic hydrocarbon biodegradation by bacteria. Appl. Biochem. Biotechnol. 176(3), 670–699 (2015).
Liu, Q. et al. Aerobic degradation of crude oil by microorganisms in soils from four geographic regions of China. Sci. Rep. 7(1), 1–12 (2017).
Ghosal, D., Ghosh, S., Dutta, T. K. & Ahn, Y. Current state of knowledge in microbial degradation of polycyclic aromatic hydrocarbons (PAHs): a review. Front. Microbiol. 7, 1369 (2016).
Aitken, C. M., Jones, D. M. & Larter, S. R. Anaerobic hydrocarbon biodegradation in deep subsurface oil reservoirs. Nature 431(7006), 291–294 (2004).
Burland, S. M. & Edwards, E. A. Anaerobic benzene biodegradation linked to nitrate reduction. Appl. Environ. Microbiol. 65(2), 529–533 (1999).
Ahad, J. M. et al. Carbon isotope fractionation during anaerobic biodegradation of toluene: implications for intrinsic bioremediation. Environ. Sci. Technol. 34(5), 892–896 (2000).
Chang, B. V., Shiung, L. C. & Yuan, S. Y. Anaerobic biodegradation of polycyclic aromatic hydrocarbon in soil. Chemosphere 48(7), 717–724 (2002).
Coates, J. D. et al. Anaerobic degradation of polycyclic aromatic hydrocarbons and alkanes in petroleum-contaminated marine harbor sediments. Appl. Environ. Microbiol. 63(9), 3589–3593 (1997).
Cozzarelli, I. M., Herman, J. S. & Baedecker, M. J. Fate of microbial metabolites of hydrocarbons in a coastal plain aquifer: the role of electron acceptors. Environ. Sci. Technol. 29(2), 458–469 (1995).
Cravo-Laureau, C. et al. Role of environmental fluctuations and microbial diversity in degradation of hydrocarbons in contaminated sludge. Res. Microbiol. 162(9), 888–895 (2011).
Gieg, L. M., Fowler, S. J. & Berdugo-Clavijo, C. Syntrophic biodegradation of hydrocarbon contaminants. Curr. Opin. Biotechnol. 27, 21–29 (2014).
Fernandez-Luqueno, F. et al. Microbial communities to mitigate contamination of PAHs in soil-possibilities and challenge: a review. Environ. Sci. Pollut. R. 18, 12–30 (2010).
Cerniglia, C. E. & Sutherland, J. B. Relative roles of bacteria and fungi in polycyclic aromatic hydrocarbon biodegradation and bioremediation. Fungi Biogeochem. Cycles 24(182), 1 (2006).
Jacques, R. J. et al. Microbial consortium bioaugmentation of a polycyclic aromatic hydrocarbons contaminated soil. Biores. Technol. 99(7), 2637–2643 (2008).
Cébron, A. et al. Experimental increase in availability of a PAH complex organc contamination from an aged contaminated soil: Consequences on biodegradation. Environ. Pollut. 177, 98–105 (2013).
Biache, C. et al. Effects of thermal desorption on the composition of two coking plant soils: Impact on solvent extractable organic compounds and metal bioavailability. Environ. Pollut. 156, 671–677 (2008).
Usman, M., Hanna, K. & Faure, P. Remediation of oil-contaminated harbor sediments by chemical oxidation. Sci. Total Environ. 634, 1100–1107 (2018).
Colombo, J. C., Pelletier, E., Brochu, C. & Khalil, M. Determination of hydrocarbon sources using n-alkane and polyaromatic hydrocarbon distribution indexes. Case study: Rio de La Plata estuary. Argentina. Environ. Sci. Technol. 23, 888–894 (1989).
Bray, E. E. & Evans, E. D. Distribution of n-paraffins as a clue to recognition of source beds. Geochim. Cosmochim. Acta 22, 2–15 (1961).
Cébron, A., Norini, M. P., Beguiristain, T. & Leyval, C. Real-Time PCR quantification of PAH-ring hydroxylating dioxygenase (PAH-RHDα) genes from Gram positive and Gram negative bacteria in soil sediment samples. J. Microbiol. Methods 73, 148–159 (2008).
Thion, C., Cébron, A., Beguiristain, T. & Leyval, C. Long-term in situ dynamics of the fungal communities in a multi-contaminated soil are mainly driven by plants. FEMS Microbiol. Ecol. 82(1), 169–181 (2012).
Powell, S. M., Ferguson, S. H., Bowman, J. P. & Snape, I. Using real-time PCR to assess changes in the hydrocarbon-degrading microbial community in Antarctic soil during bioremediation. Microb. Ecol. 52(3), 523–532 (2006).
Gielnik, A. et al. Functional potential of sewage sludge digestate microbes to degrade aliphatic hydrocarbons during bioremediation of a petroleum hydrocarbons contaminated soil. J. Environ. Manag. 280, 1148 (2021).
Winderl, C., Anneser, B., Griebler, C., Meckenstock, R. U. & Lueders, T. Depth-resolved quantification of anaerobic toluene degraders and aquifer microbial community patterns in distinct redox zones of a tar oil contaminant plume. Appl. Environ. Microbiol. 74(3), 792–801 (2008).
Kozich, J. J. et al. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79(17), 5112–5120 (2013).
Thomas, F. & Cébron, A. Short-term rhizosphere effect on available carbon sources, phenanthrene degradation, and active microbiome in an aged-contaminated industrial soil. Front. Microbiol. 7, 92 (2016).
Vainio, E. J. & Hantula, J. Direct analysis of wood-inhabiting fungi using denaturing gradient gel electrophoresis of amplified ribosomal DNA. Mycol. Res. 104(8), 927–936 (2000).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75(23), 7537–7541 (2009).
Cébron, A. et al. BactoTraits–A functional trait database to evaluate how natural and man-induced changes influence the assembly of bacterial communities. Ecol. Indicat. 130, 108047 (2021).
Aßhauer, K. P. et al. Tax4Fun: Predicting functional proles from metagenomic 16S rRNA data. Bioinformatics 31, 2882–2884 (2015).
R Core Team R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria (2019).
Oksanen, J., et al. Vegan: Community Ecology Package. R package version 2.5–6. (2019).
Marra, G. & Wood, S. N. Practical variable selection for generalized additive models. Comput. Stat. Data Anal. 55(7), 2372–2387 (2011).
Baker, M. E. & King, R. S. A new method for detecting and interpreting biodiversity and ecological community thresholds. Methods Ecol. Evol. 1(1), 25–37 (2010).
Baker, M. E., King, R. S., Kahle, D., & Kahle, M. D. Package ‘TITAN2’. (2015).
Dufrêne, M. & Legendre, P. Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol. Monogr. 67(3), 345–366 (1997).
Wang, X., Feng, J. & Zhao, J. Effects of crude oil residuals on soil chemical properties in oil sites, Momoge Wetland, China. Environ. Monit. Assess. 161(1), 271–280 (2010).
Sun, W. et al. Microbial communities inhabiting oil-contaminated soils from two major oilfields in Northern China: Implications for active petroleum-degrading capacity. J. Microbiol. 53(6), 371–378 (2015).
Cébron, A. et al. Root exudates modify bacterial diversity of phenanthrene degraders in PAH-polluted soil but not phenanthrene degradation rates. Environ. Microbiol. 13(3), 722–736 (2011).
Thomas, F., Corre, E. & Cébron, A. Stable isotope probing and metagenomics highlight the effect of plants on uncultured phenanthrene-degrading bacterial consortium in polluted soil. ISME J. 13(7), 1814–1830 (2019).
Yang, S. et al. Hydrocarbon degraders establish at the costs of microbial richness, abundance and keystone taxa after crude oil contamination in permafrost environments. Sci. Rep. 6(1), 1–13 (2016).
Yang, S., Wen, X., Zhao, L., Shi, Y. & Jin, H. Crude oil treatment leads to shift of bacterial communities in soils from the deep active layer and upper permafrost along the China-Russia crude oil pipeline route. PLoS ONE 9(5), e9652 (2014).
Sutton, N. B. et al. Impact of long-term diesel contamination on soil microbial community structure. Appl. Environ. Microbiol. 79(2), 619–630 (2013).
Paissé, S. et al. Structure of bacterial communities along a hydrocarbon contamination gradient in a coastal sediment. FEMS Microbiol. Ecol. 66(2), 295–305 (2008).
Abed, R. M., Al-Kindi, S. & Al-Kharusi, S. Diversity of bacterial communities along a petroleum contamination gradient in desert soils. Microb. Ecol. 69(1), 95–105 (2015).
Cury, J. C. et al. Microbial diversity and hydrocarbon depletion in low and high diesel-polluted soil samples from Keller Peninsula, South Shetland Islands. Antarct. Sci. 27(3), 263–273 (2015).
Ferrari, B. C., Zhang, C. D. & Van Dorst, J. Recovering greater fungal diversity from pristine and diesel fuel contaminated sub-Antarctic soil through cultivation using both a high and a low nutrient media approach. Front. Microbiol. https://doi.org/10.3389/fmicb2011.00217 (2011).
Moktali, V. et al. Systematic and searchable classification of cytochrome P450 proteins encoded by fungal and oomycete genomes. BMC Genom. 13, 1. https://doi.org/10.1186/1471-2164-13-525 (2012).
Daccò, C. et al. Key fungal degradation patterns, enzymes and their applications for the removal of aliphatic hydrocarbons in polluted soils: A review. Int. Biodeteriorat. Biodegrad. 147, 1066 (2020).
Liu, P. W. G. et al. Bioremediation of petroleum hydrocarbon contaminated soil: Effects of strategies and microbial community shift. Int. Biodeterior. Biodegradation 65(8), 1119–1127 (2011).
Kelly, D. P. & Wood, A. P. The family Acidithiobacillaceae. The prokaryotes. Gammaproteobacteria 4, 15–25 (2014).
Yagi, J. M. et al. Subsurface ecosystem resilience: long-term attenuation of subsurface contaminants supports a dynamic microbial community. ISME J. 4(1), 131–143 (2010).
Chakraborty, R., O’Connor, S. M., Chan, E. & Coates, J. D. Anaerobic degradation of benzene, toluene, ethylbenzene, and xylene compounds by Dechloromonas strain RCB. Appl. Environ. Microbiol. 71(12), 8649–8655 (2005).
Keller, A. H., Kleinsteuber, S. & Vogt, C. Anaerobic benzene mineralization by nitrate-reducing and sulfate-reducing microbial consortia enriched from the same site: comparison of community composition and degradation characteristics. Microb. Ecol. 75(4), 941–953 (2018).
Sperfeld, M., Diekert, G. & Studenik, S. Anaerobic aromatic compound degradation in Sulfuritalea hydrogenivorans sk43H. FEMS Microbiol. Ecol. 95(1), 199 (2019).
Salinero, K. K. et al. Metabolic analysis of the soil microbe Dechloromonas aromatica str. RCB: Indications of a surprisingly complex life-style and cryptic anaerobic pathways for aromatic degradation. BMC Genom. 10(1), 351 (2009).
Watanabe, T., Kojima, H. & Fukui, M. Complete genomes of freshwater sulfur oxidizers Sulfuricella denitrificans skB26 and Sulfuritalea hydrogenivorans sk43H: genetic insights into the sulfur oxidation pathway of betaproteobacteria. Syst. Appl. Microbiol. 37(6), 387–395 (2014).
Roy, A. et al. Petroleum hydrocarbon rich oil refinery sludge of North-East India harbours anaerobic, fermentative, sulfate-reducing, syntrophic and methanogenic microbial populations. BMC Microbiol. 18(1), 151 (2018).
Tan, B., Dong, X., Sensen, C. W. & Foght, J. Metagenomic analysis of an anaerobic alkane-degrading microbial culture: potential hydrocarbon-activating pathways and inferred roles of community members. Genome 56(10), 599–611 (2013).
Gutierrez, T., Nichols, P. D., Whitman, W. B. & Aitken, M. D. Porticoccus hydrocarbonoclasticus sp. nov., an aromatic hydrocarbon-degrading bacterium identified in laboratory cultures of marine phytoplankton. Appl. Environ. Microbiol. 78(3), 628–637 (2012).
Knapik, K., Bagi, A., Krolicka, A., & Baussant, T. Discovery of functional gene markers of bacteria for monitoring hydrocarbon pollution in the marine environment - a metatranscriptomics approach. bioRxiv, 857391 (2019).
Ribicic, D. et al. Microbial community and metagenome dynamics during biodegradation of dispersed oil reveals potential key-players in cold Norwegian seawater. Mar. Pollut. Bull. 129(1), 370–378 (2018).
Corteselli, E. M., Aitken, M. D. & Singleton, D. R. Description of Immundisolibacter cernigliae gen. nov., sp. nov., a high-molecular-weight polycyclic aromatic hydrocarbon-degrading bacterium within the class Gammaproteobacteria, and proposal of Immundisolibacterales ord. nov. and Immundisolibacteraceae fam. Nov.. Int. J. Syst. Evol. Microbiol. 67(4), 925 (2017).
Kanaly, R. A., Harayama, S. & Watanabe, K. Rhodanobacter sp. strain BPC1 in a benzo[a]pyrene-mineralizing bacterial consortium. Appl. Environ. Microbiol. 68(12), 5826–5833 (2002).
Dominguez, J. J. A., Inoue, C. & Chien, M. F. Hydroponic approach to assess rhizodegradation by sudangrass (Sorghum x drummondii) reveals pH-and plant age-dependent variability in bacterial degradation of polycyclic aromatic hydrocarbons (PAHs). J. Hazard. Mater. 387, 1295 (2020).
Roslund, M. I. et al. Half-lives of PAHs and temporal microbiota changes in commonly used urban landscaping materials. PeerJ 1, 1–27 (2018).
Nayak, A. S., Sanjeev Kumar, S., Anjaneya, O. & Karegoudar, T. B. A catabolic pathway for the degradation of chrysene by Pseudoxanthomonas sp.: PNK-04. FEMS Microbiol. Lett. 320(2), 128–134 (2011).
Patel, V., Cheturvedula, S. & Madamwar, D. Phenanthrene degradation by Pseudoxanthomonas sp DMVP2 isolated from hydrocarbon contaminated sediment of Amlakhadi canal, Gujarat, India. J. Hazard. Mater. 201, 43–51 (2012).
Choi, E. J. et al. Comparative genomic analysis and benzene, toluene, ethylbenzene, and o-, m-, and p-xylene (BTEX) degradation pathways of Pseudoxanthomonas spadix BD-a59. Appl. Environ. Microbiol. 79(2), 663–671 (2013).
Nayak, A. S., Vijaykumar, M. H. & Karegoudar, T. B. Characterization of biosurfactant produced by Pseudoxanthomonas sp. PNK-04 and its application in bioremediation. Int. Biodeteriorat. Biodegrad. 63(1), 73–79 (2009).
Knief, C. Diversity and habitat preferences of cultivated and uncultivated aerobic methanotrophic bacteria evaluated based on pmoA as molecular marker. Front. Microbiol. 6, 1346 (2015).
Schleheck, D. et al. Complete genome sequence of Parvibaculum lavamentivorans type strain (DS-1 T). Stand Genom. Sci. 5(3), 298–310 (2011).
Alonso-Gutiérrez, J. et al. Bacterial communities from shoreline environments (Costa da Morte, Northwestern Spain) affected by the Prestige oil spill. Appl. Environ. Microbiol. 75(11), 3407–3418 (2009).
Lai, Q. et al. Parvibaculum indicum sp. nov., isolated from deep-sea water. Int. J. Syst. Evol. Microbiol. 61(2), 271–274 (2011).
Obuekwe, C. O., Al-Jadi, Z. K. & Al-Saleh, E. S. Hydrocarbon degradation in relation to cell-surface hydrophobicity among bacterial hydrocarbon degraders from petroleum-contaminated Kuwait desert environment. Int. Biodeterior. Biodegradation 63(3), 273–279 (2009).
Blanco-Enríquez, E. G. et al. Characterization of a microbial consortium for the bioremoval of polycyclic aromatic hydrocarbons (PAHs) in water. Int. J. Environ. Res. Public Health 15(5), 975 (2018).
Radwan, S. S., Al-Awadhi, H., Sorkhoh, N. A. & El-Nemr, I. M. Rhizospheric hydrocarbon-utilizing microorganisms as potential contributors to phytoremediation for the oil Kuwaiti desert. Microbiol. Res. 153(3), 247–251 (1998).
Arli, S. D., Trivedi, U. B. & Patel, K. C. Curdlan-like exopolysaccharide production by Cellulomonas flavigena UNP3 during growth on hydrocarbon substrates. World J. Microbiol. Biotechnol. 27(6), 1415–1422 (2011).
Parab, V. & Phadke, M. Co-biodegradation studies of naphthalene and phenanthrene using bacterial consortium. J. Environ. Sci. Health Part A 1, 1–13 (2020).
Kim, S. J. et al. Genomic analysis of polycyclic aromatic hydrocarbon degradation in Mycobacterium vanbaalenii PYR-1. Biodegradation 19(6), 859–881 (2008).
Van Beilen, J. B. & Funhoff, E. G. Alkane hydroxylases involved in microbial alkane degradation. Appl. Microbiol. Biotechnol. 74(1), 13–21 (2007).
Arino, S., Marchal, R. & Vandecasteele, J. P. Production of new extracellular glycolipids by a strain of Cellulomonas cellulans (Oerskovia xanthineolytica) and their structural characterization. Can. J. Microbiol. 44(3), 238–243 (1998).
Abbasian, F., Lockington, R., Megharaj, M. & Naidu, R. The biodiversity changes in the microbial population of soils contaminated with crude oil. Curr. Microbiol. 72(6), 663–670 (2016).
Neethu, C. S. et al. Oil-spill triggered shift in indigenous microbial structure and functional dynamics in different marine environmental matrices. Sci. Rep. 9(1), 1–13 (2019).
Xie, S., Sun, W., Luo, C. & Cupples, A. M. Novel aerobic benzene degrading microorganisms identified in three soils by stable isotope probing. Biodegradation 22(1), 71–81 (2011).
Lindahl, B. D. & Tunlid, A. Ectomycorrhizal fungi–potential organic matter decomposers, yet not saprotrophs. New Phytol. 205(4), 1443–1447 (2015).
Mohammadi-Sichani, M. M. et al. Bioremediation of soil contaminated crude oil by Agaricomycetes. J. Environ. Health Sci. Eng. 15, 8 (2017).
Treu, R. & Falandysz, J. Mycoremediation of hydrocarbons with basidiomycetes—a review. J. Environ. Sci. Health B 52(3), 148–155 (2017).
Lemmel, F., Maunoury-Danger, F., Leyval, C., & Cébron, A. Altered fungal communities in contaminated soils from French industrial brownfields. J. Hazard. Mater. 124296 (2020).
Aguilar, M. et al. Next-generation sequencing assessment of eukaryotic diversity in oil sands tailings ponds sediments and surface water. J. Eukaryot. Microbiol. 63(6), 732–743 (2016).
Czaplicki, L. M., et al. Investigating the mycobiome of the Holcomb Creosote Superfund Site. Chemosphere, 126208 (2020).
Hamamura, N., Olson, S. H., Ward, D. M. & Inskeep, W. P. Diversity and functional analysis of bacterial communities associated with natural hydrocarbon seeps in acidic soils at Rainbow Springs, Yellowstone National Park. Appl. Environ. Microbiol. 71(10), 5943–5950 (2005).
Mbadinga, S. M. et al. Microbial communities involved in anaerobic degradation of alkanes. Int. Biodeterior. Biodegradation 65(1), 1–13 (2011).
Kleinsteuber, S., Schleinitz, K. M. & Vogt, C. Key players and team play: anaerobic microbial communities in hydrocarbon-contaminated aquifers. Appl. Microbiol. Biotechnol. 94(4), 851–873 (2012).
Bauer, R. D. et al. Mixing-controlled biodegradation in a toluene plume—results from two-dimensional laboratory experiments. J. Contam. Hydrol. 96(1–4), 150–168 (2008).
Vogt, C. et al. Stable isotope probing approaches to study anaerobic hydrocarbon degradation and degraders. J. Mol. Microbiol. Biotechnol. 26(1–3), 195–210 (2016).
da Cruz, G. F. et al. Could petroleum biodegradation be a joint achievement of aerobic and anaerobic microorganisms in deep sea reservoirs?. AMB Express 1(1), 47 (2011).
Atashgahi, S. et al. A benzene-degrading nitrate-reducing microbial consortium displays aerobic and anaerobic benzene degradation pathways. Sci. Rep. 8(1), 1–12 (2018).
Zhang, S., Hu, Z. & Wang, H. Metagenomic analysis exhibited the co-metabolism of polycyclic aromatic hydrocarbons by bacterial community from estuarine sediment. Environ. Int. 129, 308–319 (2019).
Mukherjee, A. et al. Bioinformatic approaches including predictive metagenomic profiling reveal characteristics of bacterial response to petroleum hydrocarbon contamination in diverse environments. Sci. Rep. 7(1), 1–22 (2017).
Das, D. et al. Complete genome sequence analysis of Pseudomonas aeruginosa N002 reveals its genetic adaptation for crude oil degradation. Genomics 105(3), 182–190 (2015).
Acknowledgements
We would like to thank the CONPET project consortium for their involvement during sampling dates, C. Lorgeoux and D. Faure-Catteloin (GeoRessources) for hydrocarbon analyses and M. E. Baker and R. S. King for their help in using the TITAN2 package they developed. We also thank R. Dickinson (Inist-CNRS translation unit) for his careful proofreading of the manuscript.
Funding
This work was supported by the French national programme EC2CO (CONPET Project) and is included in the scientific program of the GISFI research consortium dedicated to the knowledge and the development on remediation technologies for degraded and polluted lands (Groupement d'Intérêt Scientifique sur les Friches Industrielles—http://www.gisfi.univ-lorraine.fr.) is part of the GISFI research project (scientific interest group on brownfields).
Author information
Authors and Affiliations
Contributions
A.C.: Conceptualization; Data acquisition; Data curation; Formal analysis; Statistical analysis; Investigation; Methodology; Supervision; Validation; Writing original draft; Writing—review & editing. A.B.: Formal analysis; Writing—review & editing. T.B.: Conceptualization; Formal analysis; Methodology; Writing—review & editing. C.B.: Conceptualization; Formal analysis; Methodology; Writing—review & editing. P.F.: Conceptualization; Methodology; Data acquisition; Funding acquisition; Project administration; Writing—review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained an error in Table 2, where the data in column ‘Anaerobic mineralization CO2 production (µg C-CO2 g−1 d−1)’ was a duplication of the data from column ‘Aerobic mineralization CO2 production (µg C-CO2 g−1 d−1)’.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cébron, A., Borreca, A., Beguiristain, T. et al. Taxonomic and functional trait-based approaches suggest that aerobic and anaerobic soil microorganisms allow the natural attenuation of oil from natural seeps. Sci Rep 12, 7245 (2022). https://doi.org/10.1038/s41598-022-10850-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-10850-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
