
Abstract
Among the snail genera most responsible for vectoring human-infecting schistosomes, Bulinus, Biomphalaria, and Oncomelania, the former is in many respects the most important. Bulinid snails host the most common human blood fluke, Schistosoma haematobium, responsible for approximately two-thirds of the estimated 237 million cases of schistosomiasis. They also support transmission of schistosomes to millions of domestic and wild animals. Nonetheless, our basic knowledge of the 37 Bulinus species remains incomplete, especially with respect to genome information, even including mitogenome sequences. We determined complete mitogenome sequences for Bulinus truncatus, B. nasutus, and B. ugandae, and three representatives of B. globosus from eastern, central, and western Kenya. A difference of the location of tRNA-Asp was found between mitogenomes from the three species of the Bulinus africanus group and B. truncatus. Phylogenetic analysis using partial cox1 sequences suggests that B. globosus is a complex comprised of multiple species. We also highlight the status of B. ugandae as a distinct species with unusual interactions with the S. haematobium group parasites deserving of additional investigation. We provide sequence data for potential development of genetic markers for specific or intraspecific Bulinus studies, help elucidate the relationships among Bulinus species, and suggest ways in which mitogenomes may help understand the complex interactions between Schistosoma and Bulinus snails and their relatives.
Similar content being viewed by others
Introduction
Of the world's 237 million estimated cases of human schistosomiasis1, about 85% occur in sub-Saharan Africa2,3,4, and approximately two-thirds of the patients are afflicted with urogenital schistosomiasis2,3. Like intestinal schistosomiasis, urogenital schistosomiasis causes underappreciated morbidity along with well-known pathological symptoms such as hematuria, and an association with bladder pathology including cancer5,6,7. Damage to the urogenital system, especially in females, is increasingly recognized as a factor favoring the transmission of the Human Immunodeficiency Virus (HIV)8,9. The relatively recent emergence of urinary schistosomiasis in Corsica, France10,11 has highlighted the opportunistic nature of Schistosoma haematobium, particularly with respect to a large number of recent studies suggestive of its ability to hybridize with closely related species like S. bovis or S. curassoni12,13,14.
Freshwater pulmonate snails of the genus Bulinus (Gastropoda, Planorbidae) are the obligate intermediate hosts of S. haematobium. Thirty-seven species of Bulinus are recognized and predominantly distributed on the African continent including Madagascar and associated smaller oceanic islands, several Mediterranean islands and southern continental Europe, and southwest Asia including the Arabian Peninsula15. Several species including Bulinus globosus, B. nasutus, B. truncatus, and B. africanus serve as intermediate hosts of S. haematobium. Additionally, bulinids also vector other parasites. Bulinus forskalii serves as an intermediate host for S. guineensis, and B. globosus transmits S. intercalatum, both schistosome species being etiologic agent of human intestinal schistosomiasis16. Several Bulinus species are implicated in transmission of other members of the S. haematobium group of species which cause schistosomiasis in domestic and wild animals17,18,19,20,21,22. Moreover, bulinids can also serve as intermediate hosts for other trematode species, particularly amphistomes, pathogens of livestock23,24. Clearly, Bulinus snails play a crucial role in transmission of snail-transmitted diseases in the tropical world.
Better understanding all aspects of the biology of vector snails including their population genetics, distributions, basic immunobiology including susceptibility to infection, symbionts and co-infections, and response to environmental change are critical to develop sound strategies for the future control of human schistosomiasis and other snail-transmitted diseases of concern. One of the basic, long-standing challenges is how to accurately and efficiently identify Bulinus species. Characters based solely on morphological traits are often difficult to discern and subject to eco-phenotypic variation. Molecularly-based approaches have revealed that considerable genetic variation exists within and among species and have been essential in providing a framework of objective criteria for more rigorously delineating taxa25,26,27. Molecular data including partial DNA sequences from mitochondrial cytochrome c oxidase subunit 1 (cox1) and internal transcribed spacer regions (ITS) of nuclear ribosomal DNA (rDNA) have been provided to address questions related to population genetics and phylogenetics26,28,29,30. These studies have provided much-needed insights into phylogenetic relationship among species but have not always yielded consistent results26,27,28,31. More comprehensive molecular data for Bulinus snails are needed.
Complete mitochondrial genome (mitogenome) sequences have proven to be useful in addressing a broad range of questions in evolutionary biology32,33. Mitogenomes have been reported for several molluscan species, including multiple species of Biomphalaria34,35,36 and one species of Oncomelania37. For the genus Bulinus, only a mitogenome sequence for B. truncatus has thus far been reported38. This is surprising because of the magnitude of the problems posed by S. haematobium and related species. To help fill this gap, we applied high through-put Illumina sequencing to determine six complete mitogenomes for Bulinus snails, five specimens collected in Kenya and one from laboratory-reared Bulinus truncatus, originally from Egypt. The study provides basic sequence information for designing more markers for identifying bulinid species and revealing relationships among species, highlights some specific topics deserving additional study pertaining to understanding compatibility and biogeography of snails and schistosomes, and provides new tools of use in studies of host use, transmission, and control of human schistosomiasis.
Results
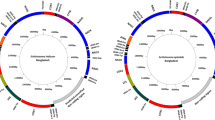
All six bulinid mitogenomes were comprised of 37 genes, including 13 protein-coding genes (PCG), 22 transfer RNA (tRNA) genes and 2 ribosomal RNA (rRNA) genes. Location of the genes in the six mitogenomes was conserved, with the only difference noted being the location of tRNA-Asp of B. truncatus relative to the other three species (B. nasutus, B. ugandae, and B. globosus) (Fig. 1). The GenBank accession numbers of the six mitogenomes generated from B. truncatus, B. nasutus, B. ugandae, and three specimens of B. globosus from eastern, central, and western Kenya, are MK414449, MK414450, MK414451, MK414452, MK414453 and MK414454, respectively. More detailed information on gene organization for all six mitogenomes is provided in the Supplementary Table S2 and Supplementary Fig. S1.

Shell morphology of six snail specimens (A) and arrangement of protein-coding genes (PCG) and tRNAs of six mitogenomes (B). In part B, diagram a shows the gene arrangements of the mitogenome of B. globosus, B. nasutus and B, ugandae whereas b shows the mitogenome of B. truncatus. Note the different position of tRNA-Asp (red arrow). The abbreviations of BuGe, BuGc and BuGw are three specimens of B. globosus that were collected from eastern, central, and western Kenya, respectively. BuT, BuN, and BuU represent B. truncatus, B. nasutus, and B. ugandae, respectively. The same abbreviations are applied to all figures below and supplementary Table 1.
Heatmap analyses revealed that the most conserved gene and its gene-product (protein) was cox1 whereas atp8 showed a relatively high degree of variation (Fig. 2).

Heatmaps showing the degree of identity of nucleotides (nt) (A) and amino acids (aa) (B) between species/specimens.
Built on the full length mitogenome sequences, a phylogenetic analysis revealed that B. globosus from eastern and western Kenya are divergent. In our samples, B. ugandae was the species most closely related to B. globosus (Fig. 3). As expected, B. truncatus, a member of B. truncatus/tropicus complex15, was most distantly related to the other three Bulinus species (all members of the B. africanus species group)15. This result was also supported by organization of gene order of the mitogenomes (Fig. 1).

A maximum likelihood (ML) phylogenetic tree with 1000 bootstrap replicates of full-length mitochondrial genome nucleotide sequences of Bulinus species. All bootstrap values are indicated at supported nodes.
Since partial cox1 sequences have been documented for many Bulinus species, we compared cox1 sequences from our samples with those previously reported. The phylogenetic analysis shows that the four species used in this study, representing two of the four generally elaborated Bulinus species groups, fit well within the framework of known species already reported (Fig. 4). This analysis further highlights the considerable diversity inherent in what might be referred to as a B. globosus species complex and the presence of a distinct, but sometimes overlooked species, B. ugandae, often associated with Lake Victoria.

A ML phylogenetic tree with 1000 bootstrap replicates generated using partial cox1 gene sequences. Bootstrap values > 60 are indicated at supported nodes. Publicly available sequences utilized in this analysis are indicated by GenBank accession number.
Discussion
Thirty-seven species of freshwater pulmonate snails of the genus Bulinus have been divided into four species groups: Bulinus forskalii group (11 species), Bulinus truncatus/tropicus group (14 species), Bulinus africanus group (10 species), and Bulinus reticulatus group (2 species)15. Definitive evolutionary relationships among the four species groups and delimitations of species within each group, especially within the africanus species group, have remained elusive.
For example, there is considerable confusion regarding differentiation of B. globosus and B. africanus15. Differences in the male copulatory organs between the two species have been noted, with the penis sheath of B. africanus being longer and broader than the preputium, as compared to B. globosus15,39. Later studies, however, suggested that such characters are not reliable for species discrimination40. Previous studies using limited molecular data have retrieved contradictory results. Morgan et al. (2002) grouped B. africanus with B. nasutus rather than with B. globosus based on analysis of partial ITS sequences41. Another study using combined cox1 and ITS sequences indicated B. africanus is clustered within a clade of B. globosus26.
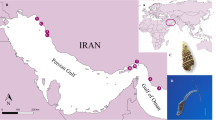
In the present study, we collected B. globosus samples from eastern, central, and western Kenya (Fig. 5). At the beginning of the work, we suspected the sample from Asao stream, a perennially flowing stream in western Kenya might be B. africanus. As stated by Brown et al. (1981), B. africanus is widely distributed in Kenya and associated with perennially flowing streams and permanently filled dams42. However, our phylogenetic trees based on cox1 sequences clearly showed the west Kenya sample from Asao stream closely grouped with B. globosus samples collected from Kisumu, about 15 km away26 (Fig. 4). In addition, cox1 sequence of a snail sampled from the eastern coastal area of Kenya clustered with sequences reported as B. globosus by others from eastern Kenya (Fig. 4). The sample from central Kenya was more closely related to those from west Kenya and Tanzania. We observed samples from eastern and western Kenya to be divergent, in agreement with findings based on microsatellite data31,43. Notably, our study also supports that B. globosus from Angola, the type locality of B. globosus15, is divergent from B. globosus from other localities31 (Fig. 4). According to these molecular data, it is likely that there are multiple disparate lineages represented as “B. globosus”, one from the type locality, and conservatively, at least two additional related lineages represented in Kenya alone. Pennance (personal communication), who has collected additional relevant mitogenome sequences, has also confirmed the notion of a B. globosus “species complex” (see also31,43). Whether these lineages should be classified into different formally named species or subspecies, or simply highlight the existence of a single broadly distributed and highly variable species is an interesting question deserving resolution. It bears directly on the issue of defining the full spectrum of snail host species for S. haematobium as B. globosus in its various guises is repeatedly implicated as an excellent host for S. haematobium and several other related schistosomes15,20. More molecular evidence such as mitogenomes collected from a wide range of geographic locations will help resolve the question.

Localities of snail samples used for the study. The maps were drawn by the first author S-MZ and photos were taken by S-MZ.
Bulinus ugandae is found in Lake Victoria and associated backwaters, and north to South Sudan and Ethiopia. It appears to be refractory to infection with S. haematobium42, but is host for S. bovis, a parasite infecting livestock44. Our sample collected from the Kisumu shoreline of Lake Victoria is more closely related to B. globosus than to the other species we sampled. This agrees with a previous ITS sequence-based study suggesting that B. ugandae is closely related to B. globosus41.
Bulinus nasutus, a species repeatedly implicated in S. haematobium transmission in coastal East Africa, including Kenya45, was collected from a reservoir in southern Mombasa, a coastal city of eastern Kenya. This specimen and those reported from eastern Kenya and Tanzania by others are well clustered. As a member of the B. africanus group, B. nasutus also grouped with B. globosus and B. ugandae (africanus group) (Fig. 4).
Bulinus nasutus, another member of the B. africanus group, also as supported by Figs. 3 and 4, has been repeatedly implicated in S. haematobium transmission in coastal East Africa, including Kenya45. Our cox1 analysis is suggestive of variation within this taxon as well, that may also have implications with respect to S. haematobium transmission.
Bulinus truncatus originating from Egypt has been maintained with NIH support for decades, most recently at the Biomedical Research Institute (BRI), Maryland, USA, and is used for routine maintenance of the life cycle of S. haematobium (www.afbr-bri.com). Unlike the preceding three taxa we examined which were all from Kenya and belong to the Bulinus africanus group, the B. truncatus specimen is classified to the Bulinus truncatus/tropicus group, supported by both the analysis of the whole mitogenome sequences (Fig. 3) and the cox1 phylogenetic analysis (Fig. 4). In addition, the position of tRNA-Asp revealed in this study is also different from that of B. africanus group (Fig. 1B).
The mitogenome sequence characterized by us is equal in length (13,767 bp) to the sequence characterized independently from B. truncatus also obtained from BRI38. The two mitogenomes generated by two different sequencing methods (Nanopore38 vs Illumina) differ by 7 nucleotides (0.05%); resulting in 5 non-synonymous replacements (one each in nad5 and nad3, three in atp6), one synonymous replacement in the start codon of nad4 and a single pyrimidine (T, C) transition in the 16S rDNA sequence. These differences indicate a modest level of genetic diversity in the BRI stock of B. truncatus.
It is noteworthy that the B. truncatus nad4 gene does not have a regular TAA stop codon observed from nad4 in other Bulinus species, nor does it have an incomplete stop codon punctuated by a downstream tRNA sequence. Secondary structures, however, may mark gene boundaries in polycistronic mitochondrial RNA46. Annotation indicated that the last complete trinucleotide codon of the 3’ terminus of the nad4 gene sequence is followed by a single T nucleotide and, immediately downstream, by an inverted repeat. This TAACAGAATTCTGTTA sequence likely yields a hairpin structure in the 3’ end of the gene transcript to define an incomplete stop codon (T—), that is completed by polyadenylation of the mRNA.
A reliable taxonomy of the genus Bulinus and useful genetic markers for species identification are a fundamental prerequisite for fully understanding the epidemiology of Bulinus-transmitted schistosomiasis in humans and animals. Our study provides basic data to design primers for further in-depth studies: markers derived from different mitogenome regions can be developed for studies with objectives ranging from species differentiation (cox1) to sequences like atp8 which may be appropriate for studies of intraspecific variation. As next generation sequencing (NGS) becomes even more cost-effective further comparative analysis of complete mitogenomes will follow47,48 including, as noted by Pennance (personal communication) to allow novel insights into relationships and evolution among Bulinus species.
The perspective offered by more mitogenomes will be particularly important for the gastropod family Planorbidae because, in addition to containing two genera of medically relevant snails (i.e., Bulinus and Biomphalaria), a third genus, Indoplanorbis, hosts the representatives of the Asian Schistosoma indicum group of veterinary significance49,50. Indoplanorbis is generally considered the sister genus to Bulinus27,41 and given the support from the fossil record for an African origin for Bulinus50, it has been considered that Bulinus-like snails originating in Africa may have dispersed to Asia and given rise to Indoplanorbis which increasing evidence suggests is actually a complex of as many as five cryptic species49,50,51,52. This is turn supports the notion that Bulinus-transmitted Schistosoma may have recolonized Asia using Indoplanorbis as their snail host50,52. A greater representation of mitogenomes from Bulinus, along with needed mitogenome sequences from the various cryptic species of Indoplanorbis would provide further insight into the Bulinus-Indoplanorbis connection and its relation to today’s distribution of species of Schistosoma.
Additionally, the relationships between the Schistosoma mansoni species group (transmitted by Biomphalaria) and the S. haematobium species group (transmitted by Bulinus) in sub-Saharan Africa remain enigmatic, particularly given that the two snail genera involved are not close relatives within the Planorbidae, and the arrival of Biomphalaria to Africa is believed to have been relatively recent, long after Bulinus had originated there41. More precise knowledge of host use, systematic clarification of the snail groups involved, and provision of more genomic information, to which this study contributes, will help to us appreciate the manner in which African Schistosoma diversified.
It has been noted that gastropods display an unusually large variety of gene orders among their mitogenomes46,53,54. Within the order Hygrophila, complete mitogenomes have been published for representatives of only three of eight families, the Planorbidae34,35,36,38,55, Lymnaeidae56,57,58, and Physidae59. For Bulinus, recognized by Bouchet et al. (2017) as a member of a separate family60, the Bulinidae, mitochondrial gene order is almost identical to representatives of the Planorbidae for which mitogenomes are available. Several phylogenetic studies also group Bulinus within the Planorbidae, and a role in transmission of Schistosoma parasites is also suggestive of inclusion of Bulinus in the Planorbidae27,41,61. Planorbidae mitochondrial gene order is similar to the Lymnaeidae except for differences in location of a few tRNAs, whereas gene order is quite different in the Physidae60. Further studies are needed to characterize more gastropod mitogenomes, which will shed light on their evolution and diversification in mollusks and at the same time, provide more genetic markers for population genetics, evolutionary and phylogenetic studies.
Materials and methods
Specimens
Bulinus nasutus was collected from Nimbodze dam (03° 28.32ʺ S, 39° 27.08ʺ E), southern Mombasa, Kenya. B. ugandae was sampled from the car-wash site in Kisumu city, Kenya (00° 05′ 45.00ʺ S, 34° 44′ 57.69ʺ E). Three B. globosus snails were collected from Mazeras quarry dam (03° 54ʹ 0.58ʺ S, 39° 31ʹ 0.72ʺ E) (northern Mombasa, eastern Kenya), Kwa Katiwa Dam (01° 12ʹ 0.00ʺ S, 37° 16ʹ 0.41ʺ E) (central Kenya), and Asao (00° 19′ 5.50ʺ S, 35° 0′ 24.99ʺ E) in western Kenya, in 2013 (Figs. 1 and 5). Bulinus truncatus was obtained from Biomedical Research Institute (BRI); it had originally been collected from Egypt decades ago (http://www.afbrbri.com/schistosomiasis/materials-offered/) (Figs. 1 and 5). The field collected specimens were placed in 90% ethanol and stored at 4 °C until use. This study was undertaken with the approvals of the National Commission for Science, Technology, and Innovation (permit number P/15/9609/4270 and P/21/9648), National Environmental Management Authority (permit numbers NEMA/AGR/46/2014 and NEMA/AGR/149/2021), and Kenya Wildlife Service (permit numbers KWS 0004754 and KWS-0045-03-21).
Extraction of DNA
After removing the shell, the whole body of a single live snail was ground to a fine powder using mortar and pestle in liquid nitrogen. The powder was transferred to 1.5 ml tubes for subsequent DNA extraction using a Qiagen Miniprep kit-based mtDNA enrichment method36,62. For ethanol-preserved samples, the ethanol was replaced by distilled water. The water was changed every 12 h for 4 times. After removing shells, individual snails were placed in a 1.5 ml tube and ground using a pestle and a CTAB method was used for DNA extraction63.
After extraction, DNA samples were treated with RNAse A (Invitrogen) at 37 °C for 30 min and then 70 °C for 10 min. DNA samples were further purified using SPRselect Beads (Beckman Coulter). Quality and quantity of DNA were measured using a Nanodrop spectrophotometer and Qubit fluorometer (Invitrogen).
Preparation, amplification, and sequencing of Illumina libraries
A genomic library for each sample was prepared (KAPA Hyper Prep Kit, KAPA Biosystems, www.kapabiosystems.com). Each snail DNA was barcoded with an adaptor. The libraries were sequenced (150 nucleotide (nt) × 2 paired-end) on the Illumina NextSeq500 platform at the UNM Biology Department’s Molecular Biology Facility (http://ceti.unm.edu/core-facilities/molecular-biology.html)36.
Assembly and annotation of mitogenomes
Two complementary methods were used to assemble mitochondrial genomes, reads baiting and iterative mapping assembly using MITOBIM64 and de novo assembly using SPAdes65. MITOBIM is a tool developed to map reads to a related reference mitogenome and then use these to recursively find additional reads to build the mitogenome of the target. The complete Biomphalaria glabrata mitochondrion genome (NCBI accession NC_005439.1) was used to bait Bulinus mitochondrial reads for genome assembly and extension until a saturation status was reached, where no further reads were found to extend the genome assembly. The longest contig generated with SPAdes with BLASTN e-value < 10–5 against the reference mitogenome of the closely related snail Biomphalaria glabrata (Bulinus and Biomphalaria belong to Planorbidae)36was selected as the mitogenome assembly. Output from the two methods were aligned and manually checked for consistency to generate the mitogenome sequence for each sample. To check the read support consistency, reads were mapped to the final assembled mitogenomes and visualized using Integrated Genome Viewer66.
The annotation of mitogenomes was conducted using MITOS2 that also includes an updated protein identification model47,67. Both RefSeq 63 Metazoa and RefSeq 81 Metazoa, were used as reference for verification and confirmation of gene predictions. Otherwise, MITOS2 default settings were applied (E-value exponent: 2; Final maximum overlap: 50; fragment quality factor: 100). Moreover, we re-checked mitogenome sequences manually using the ExPASY translate tool (http://web.expasy.org/translate/) and did BLAST to confirm the reading frames of protein coding genes. Final annotation was based on criteria from Fourdrilis et al. (2018) that incorporate unique aspects of mitogenome biology, including transcription as polycistronic RNA, the punctuation model and completion of stop codons by polyadenylation of mRNA transcripts68.
Genetic and phylogenetic analysis
Sequence alignments and percent identity of nucleotides (nt) and amino acids (aa) were determined using Clustal Omega69. Heat-maps of pair-wise sequence identities were generated using R package ggplot270. All the intermediate data organization and filtering were done with in-house bash and Perl scripts and Microsoft Excel.
Phylogenetic analyses for full mitochondrial genomes and partial cox1 were done using the maximum likelihood method and conducted with MEGAX71,72. Biomphalaria pfeifferi sequences were utilized as the outgroup for both analyses. For both datasets the GTR + G + I model was selected by Akaikeʼs information criterion in MEGAX72. Both analyses were conducted with 1000 bootstrap replicates.
Data availability
GenBank accession numbers of the mitogenomes for B. truncatus, B. nasutus, B. ugandae, and three specimen of B. globosus from eastern, central, and western Kenya, are MK414449, MK414450, MK414451, MK414452, MK414453, and MK414454, respectively.
References
World Health Organization (WHO). Schistosomiasis. Fact sheet. (2021).
Van Der Werf, M. J. et al. Quantification of clinical morbidity associated with schistosome infection in sub-Saharan Africa. Acta Trop. 86, 125–139 (2003).
Hotez, P. J. & Kamath, A. Neglected tropical diseases in sub-Saharan Africa: Review of their prevalence, distribution, and disease burden. PLoS Negl. Trop. Dis. 3, 2–11 (2009).
Adenowo, A. F., Oyinloye, B. E., Ogunyinka, B. I. & Kappo, A. P. Impact of human schistosomiasis in sub-Saharan Africa. Brazilian J. Infect. Dis. 19, 196–205 (2015).
Berry, A., Iriart, X., Fillaux, J. & Magnaval, J. F. Urinary schistosomiasis and cancer. Bull. Soc. Pathol. Exot. 110, 68–75 (2017).
Dematei, A. et al. Angiogenesis in Schistosoma haematobium -associated urinary bladder cancer. APMIS 125, 1056–1062 (2017).
Santos, L. L. et al. Urogenital schistosomiasis—history, pathogenesis, and bladder cancer. J. Clin. Med. 10, 1–11 (2021).
Mbabazi, P. S. et al. Examining the relationship between urogenital schistosomiasis and HIV infection. PLoS Negl. Trop. Dis. 5, 1–8 (2011).
Bustinduy, A. et al. HIV and schistosomiasis co-infection in African children. Lancet Infect. Dis. 14, 640–649 (2014).
Rothe, C. et al. Developing endemicity of schistosomiasis, Corsica France. Emerg. Infect. Dis. 27, 319–321 (2021).
Gillardie, M. L. et al. Molecular approach to the epidemiology of urinary schistosomiasis in France. PLoS Negl. Trop. Dis. 15, 1–19 (2021).
Rollinson, D., Southgate, V. R., Vercruysse, J. & Moore, P. J. Observations on natural and experimental interactions between Schistosoma bovis and S. curassoni from West Africa. Acta Trop. 47, 101–114 (1990).
Platt, R. N. et al. Ancient hybridization and adaptive introgression of an invadolysin gene in schistosome parasites. Mol. Biol. Evol. 36, 2127–2142 (2019).
Savassi, B. A. E. S. et al. Mastomys natalensis (Smith, 1834) as a natural host for Schistosoma haematobium (Bilharz, 1852) Weinland, 1858 x Schistosoma bovis Sonsino, 1876 introgressive hybrids. Parasitol. Res. 120, 1755–1770 (2021).
Brown, D. Freshwater snails of Africa and their medical importance. (Taylor & Francis, 1994).
Pagès J. R., Jourdane J., Southgate V. R. & Tchuem Tchuenté L. A. Reconnaissance de deux espèces jumelles au sein du taxon Schistosoma intercalatum Fisher, 1934, agent de la schistosomose humaine rectale en Afrique. Description de Schistosoma guineensis n. sp. In Taxonomy, Ecology and Evolution of Metazoan Parasites (eds. Combes C, Jourdane J.) 139–146 (Perpignan: Presses Universitaires de Perpignan, 2003).
Southgate, V. R. & Knowles, R. J. The intermediate hosts of Schistosoma bovis in Western Kenya. Trans. R. Soc. Trop. Med. Hyg. 69, 356–357 (1975).
Moné, H., Mouahid, G. & Morand, S. The distribution of Schistosoma bovis Sonsino, 1876 in relation to intermediate host mollusc-parasite relationships. Adv. Parasitol. 44, 99–138 (1999).
Pfukenyi, D. M., Mukaratirwa, S., Willingham, A. L. & Monrad, J. Epidemiological studies of Schistosoma mattheei infections in cattle in the highveld and lowveld communal grazing areas of Zimbabwe. Onderstepoort J. Vet. Res. 73, 179–191 (2006).
Rollinson, D., Stothard, J. R. & Southgate, V. R. Interactions between intermediate snail hosts of the genus Bulinus and schistosomes of the Schistosoma haematobium group. Parasitology 123(Suppl), S245–S260 (2001).
Pennance, T. et al. Occurrence of Schistosoma bovis on Pemba Island, Zanzibar: Implications for urogenital schistosomiasis transmission monitoring. Parasitology 145, 1727–1731 (2018).
Southgate, V. R., Brown, D. S., Rollinson, D., Ross, G. C. & Knowles, R. J. Bulinus tropicus from Central Kenya acting as a host for Schistosoma bovis. Z. Parasitenkd. 71, 61–69 (1985).
Laidemitt, M. R. et al. Loads of trematodes: discovering hidden diversity of paramphistomoids in Kenyan ruminants. Parasitology 144, 131–147 (2017).
Pfukenyi, D. M. & Mukaratirwa, S. Amphistome infections in domestic and wild ruminants in East and Southern Africa: A review. Onderstepoort J. Vet. Res. 85, e1–e13 (2018).
Jones, C. S. et al. Molecular evolution of freshwater snail intermediate hosts within the Bulinus forskalii group. Parasitology 123(Suppl), S277–S292 (2001).
Kane, R. A., Stothard, J. R., Emery, A. M. & Rollinson, D. Molecular characterization of freshwater snails in the genus Bulinus: A role for barcodes?. Parasit. Vectors 1, 1–15 (2008).
Jørgensen, A. et al. A molecular phylogenetic analysis of Bulinus (Gastropoda: Planorbidae) with conserved nuclear genes. Zool. Scr. 40, 126–136 (2011).
Nalugwa, A., Jørgensen, A., Nyakaana, S. & Kristensen, T. K. Molecular phylogeny of Bulinus (Gastropoda: Planorbidae) reveals the presence of three species complexes in the Albertine Rift freshwater bodies. Int. J. Genet. Mol. Biol. 2, 130–139 (2010).
Tumwebaze, I. et al. Molecular identification of Bulinus spp. intermediate host snails of Schistosoma spp. in crater lakes of western Uganda with implications for the transmission of the Schistosoma haematobium group parasites. Parasit. Vectors 12, 565 (2019).
Chibwana, F. D., Tumwebaze, I., Mahulu, A., Sands, A. F. & Albrecht, C. Assessing the diversity and distribution of potential intermediate hosts snails for urogenital schistosomiasis: Bulinus spp. (Gastropoda: Planorbidae) of Lake Victoria. Parasit. Vectors 13, 418 (2020).
Allan, F. et al. Mapping freshwater snails in north-western Angola: Distribution, identity and molecular diversity of medically important taxa. Parasit. Vectors 10, 460 (2017).
Crawford, A. R., Bassam, B. J., Drenth, A., Maclean, D. J. & Irwin, J. A. G. Evolutionary relationships among Phytophthora species deduced from rDNA sequence analysis. Mycol. Res. 100, 437–443 (1996).
Zardoya, R. Recent advances in understanding mitochondrial genome diversity. F1000Research 9, 270 (2020).
DeJong, R. J., Emery, A. M. & Adema, C. M. The mitochondrial genome of Biomphalaria glabrata (Gastropoda: Basommatophora), intermediate host of Schistosoma mansoni. J. Parasitol. 90, 991–997 (2004).
Jannotti-Passos, L. K. et al. Phylogenetic analysis of Biomphalaria tenagophila (Orbigny, 1835) (Mollusca: Gastropoda). Mem. Inst. Oswaldo Cruz 105, 504–511 (2010).
Zhang, S.-M. et al. Complete mitochondrial and rDNA complex sequences of important vector species of Biomphalaria, obligatory hosts of the human- infecting blood fluke, Schistosoma mansoni. Sci. Rep. 8, (2018).
Attwood, S. W., Ibaraki, M., Saitoh, Y., Nihei, N. & Janies, D. A. Comparative phylogenetic studies on Schistosoma japonicum and its snail intermediate host Oncomelania hupensis: origins, dispersal and coevolution. PLoS Negl. Trop. Dis. 9, e0003935 (2015).
Young, N. D. et al. Mitochondrial genome of Bulinus truncatus (Gastropoda: Lymnaeoidea): implications for snail systematics and schistosome epidemiology. Curr. Res. Parasitol. Vector-Borne Dis. 1, 100017 (2021).
Mandahl-Barth, G. Intermediate hosts of Schistosoma; African Biomphalaria and Bulinus. Monogr. Ser. World Health Organ. 57, 1–131 (1958).
Raahauge, P. & Kristensen, T. K. A comparison of Bulinus africanus group species (Planorbidae; Gastropoda) by use of the internal transcribed spacer 1 region combined by morphological and anatomical characters. Acta Trop. 75, 85–94 (2000).
Morgan, J. A. T. et al. A phylogeny of planorbid snails, with implications for the evolution of Schistosoma parasites. Mol. Phylogenet. Evol. 25, 477–488 (2002).
Brown, D. S., Jelnes, J. E., Kinoti, G. K. & Ouma, J. Distribution in Kenya of intermediate hosts of Schistosoma. Trop. Geogr. Med. 33, 95–103 (1981).
Nyakaana, S. et al. Bulinus globosus (Planorbidae; Gastropoda) populations in the Lake Victoria basin and coastal Kenya show extreme nuclear genetic differentiation. Acta Trop. 128, 226–233 (2013).
Malek, E. A. Studies on bovine schistosomiasis in the Sudan. Ann. Trop. Med. Parasitol. 63, 501–513 (1969).
Kariuki, H. C. et al. Distribution patterns and cercarial shedding of Bulinus nasutus and other snails in the Msambweni area, Coast Province Kenya. Am. J. Trop. Med. Hyg. 70, 449–456 (2004).
Ghiselli, F. et al. Molluscan mitochondrial genomes break the rules. Philos. Trans. R. Soc. B Biol. Sci. 376, 1 (2021).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Harrison, R. G. Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol. Evol. 4, 6–11 (1989).
Devkota, R., Brant, S. V. & Loker, E. S. The Schistosoma indicum species group in Nepal: Presence of a new lineage of schistosome and use of the Indoplanorbis exustus species complex of snail hosts. Int. J. Parasitol. 45, 857–870 (2015).
Jones, B. P. et al. Divergence across mitochondrial genomes of sympatric members of the Schistosoma indicum group and clues into the evolution of Schistosoma spindale. Sci. Rep. 10, 1–14 (2020).
Pickford, M. Freshwater and terrestrial mollusca from the early miocene deposits of the Northern Sperrgebiet Namibia. Mem. Geol. Surv. Namibia 20, 65–74 (2008).
Gauffre-Autelin, P., Von Rintelen, T., Stelbrink, B. & Albrecht, C. Recent range expansion of an intermediate host for animal schistosome parasites in the Indo-Australian Archipelago: Phylogeography of the freshwater gastropod Indoplanorbis exustus in South and Southeast Asia. Parasit. Vectors 10, 1–15 (2017).
Grande, C., Templado, J. & Zardoya, R. Evolution of gastropod mitochondrial genome arrangements. BMC Evol. Biol. 8, 61 (2008).
new insights from increased taxa sampling. Dayrat B, Conrad M, Balayan S, White TR, Albrecht C, Golding R, Gomes SR, Harasewych MG, M. A. Phylogenetic relationships and evolution of pulmonate gastropods (Mollusca). Mol Phylogenet Evol. 59, 425–437 (2011).
Schultz, J. H. et al. The mitochondrial genome of the planorbid snail Planorbella duryi. Mitochondrial DNA Part B Resour. 3, 972–973 (2018).
Feldmeyer, B., Greshake, B., Funke, E., Ebersberger, I. & Pfenninger, M. Positive selection in development and growth rate regulation genes involved in species divergence of the genus Radix. BMC Evol. Biol. 15, 164 (2015).
Schell, T. et al. An annotated draft genome for Radix auricularia (Gastropoda, Mollusca). Genome Biol. Evol. 9, 585–592 (2017).
Qin, D. M. et al. Complete mitochondrial genome of the radicine pond snail Radix plicatula (Gastropoda: Lymnaeidae). Mitochondrial DNA Part B Resour. 4, 2861–2862 (2019).
Nolan, J. R., Bergthorsson, U. & Adema, C. M. Physella acuta: atypical mitochondrial gene order among panpulmonates (Gastropoda). J. Molluscan Stud. 80, 388–399 (2014).
Bouchet, P. et al. Revised classification, nomenclator and typification of gastropod and monoplacophoran families. Malacologia 61, 1–526 (2017).
Albrecht, C., Kuhn, K. & Streit, B. A molecular phylogeny of Planorboidea (Gastropoda, Pulmonata): Insights from enhanced taxon sampling. Zool. Scr. 36, 27–39 (2007).
Quispe-Tintaya, W., White, R. R., Popov, V. N., Vijg, J. & Maslov, A. Y. Fast mitochondrial DNA isolation from mammalian cells for next-generation sequencing. Biotechniques 55, 133–136 (2013).
Winnepenninckx, B., Backeljau, T. & De Wachter, R. Extraction of high molecular weight DNA from molluscs. Trends Genet. 9, 407 (1993).
Hahn, C., Bachmann, L. & Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 41, e129 (2013).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
high-performance genomics data visualization and exploration. Thorvaldsdóttir, H., Robinson, J. T., Mesirov, J. P. & Thorvaldsdóttir H, Robinson JT, M. J. Integrative genomics viewer (IGV). Brief. Bioinforma. 14, 178–192 (2013).
Al Arab, M. et al. Accurate annotation of protein-coding genes in mitochondrial genomes. Mol. Phylogenet. Evol. 106, 209–216 (2017).
Fourdrilis, S., de Frias Martins, A. M. & Backeljau, T. Relation between mitochondrial DNA hyperdiversity, mutation rate and mitochondrial genome evolution in Melarhaphe neritoides (Gastropoda: Littorinidae) and other Caenogastropoda. Sci. Rep. 8, 17964 (2018).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Wickham, H. ggplot2: elegant graphics for data analysis. Springer (2016).
Nei, M. & Kumar, S. Molecular Evolution and Phylogenetics. (Oxford University Press, 2000).
Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39, 783–791r (1985).
Acknowledgements
We thank Martin Mutuku assisting collection of the specimens in the field. Bulinus truncatus was provided by the NIAID Schistosomiasis Resource Center of the Biomedical Research Institute (Rockville, MD) through NIH-NIAID Contract HHSN272201700014I for distribution through Biodefense and Emerging Infections Research Repository Resources (BEI). The authors are grateful to the University of New Mexico (UNM) Biology Department’s Molecular Biology Facility (MBF) for Illumina Sequencing and UNM Center for Advanced Research Computing, supported in part by the National Science Foundation, for providing the high-performance computing and large-scale storage resources used in this work.
Funding
This work was supported by NIH R37 AI101438 (ESL) and NIH AI132953 (S-MZ).
Author information
Authors and Affiliations
Contributions
S.-M.Z. and E.S.L. conceived the project; S.-M.Z. collected samples, maintained snail colonies, and conducted the experiment; S.-M.Z., L.B., L.L., C.B., and C.M.A. analyzed the data; S.-M.Z., C.M.A., and E.S.L. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, SM., Bu, L., Lu, L. et al. Comparative mitogenomics of freshwater snails of the genus Bulinus, obligatory vectors of Schistosoma haematobium, causative agent of human urogenital schistosomiasis. Sci Rep 12, 5357 (2022). https://doi.org/10.1038/s41598-022-09305-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-09305-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
