
Abstract
The Salmonella enterica SseK1 protein is a type three secretion system effector that glycosylates host proteins during infection on specific arginine residues with N-acetyl glucosamine (GlcNAc). SseK1 also Arg-glycosylates endogenous bacterial proteins and we thus hypothesized that SseK1 activities might be integrated with regulating the intrabacterial abundance of UPD-GlcNAc, the sugar-nucleotide donor used by this effector. After searching for new SseK1 substrates, we found that SseK1 glycosylates arginine residues in the dual repressor-activator protein NagC, leading to increased DNA-binding affinity and enhanced expression of the NagC-regulated genes glmU and glmS. SseK1 also glycosylates arginine residues in GlmR, a protein that enhances GlmS activity. This Arg-glycosylation improves the ability of GlmR to enhance GlmS activity. We also discovered that NagC is a direct activator of glmR expression. Salmonella lacking SseK1 produce significantly reduced amounts of UDP-GlcNAc as compared with Salmonella expressing SseK1. Overall, we conclude that SseK1 up-regulates UDP-GlcNAc synthesis both by enhancing the DNA-binding activity of NagC and by increasing GlmS activity through GlmR glycosylation. Such regulatory activities may have evolved to maintain sufficient levels of UDP-GlcNAc for both bacterial cell wall precursors and for SseK1 to modify other bacterial and host targets in response to environmental changes and during infection.
Similar content being viewed by others


Introduction
The Gram-negative bacterium Salmonella enterica is a human and animal pathogen and one of the most common causative agents of food-borne diseases1,2. This pathogen acquired two pathogenicity islands that each encode a type III secretion system (T3SS) apparatus and numerous effector proteins that subvert host cell functions3,4,5. The SseK enzymes are T3SS effector glycosyltransferases that glycosylate target proteins on arginine residues6,7. Many Salmonella enterica genomes encode up to three SseK paralogs, SseK1, SseK2, and SseK3. The NleB enzymes from E. coli and Citrobacter rodentium are SseK orthologs. At a structural level, these enzymes are composed of three major domains, a catalytic domain that includes DXD and HEN motifs, a helix-loop-helix (HLH) domain, and a C-terminal lid domain8,9,10. These enzymes are important to bacterial virulence because they disrupt host innate immune signaling pathways by Arg-glycosylating multiple proteins, including the FAS-associated death domain-containing protein (FADD) and the tumor necrosis factor receptor (TNFR) type 1-associated death domain protein (TRADD)6,7. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), the transcriptional regulator of cellular O2 homeostasis Hif1α, and the tubulin-binding cofactor B (TBCB), are also glycosylation targets of some of the NleB/SseK orthologs11,12,13,14. Subcellular fractionation experiments facilitated the identification of the Rab GTPases Rab1, Rab5, and Rab11 as targets of SseK315. NleB2 is a bacterial Arg-glucose transferase that glucosylates RIPK116.
We and others have demonstrated that NleB/SseK not only glycosylate host cell proteins but also modify bacterial proteins to improve bacterial survival under hostile environmental conditions. For example, C. rodentium NleB Arg-glycosylates the glutathione synthase GshB, leading to enhanced glutathione synthase activity and consequently increased resistance to oxidative stress17. SseK1 Arg-glycosylates and enhances the activity of several enzymes (GloA, GloB, GloC, and YajL) involved in methylglyoxal detoxification18. It was recently described that SseK3-mediated Arg-glycosylation also plays an important role in modulating the DNA-binding activity of Salmonella PhoP, suggesting a mechanism for how Arg-glycosylation may also act as a regulator of gene transcription19.
UDP-GlcNAc is an essential precursor for cell wall biosynthesis and the nucleotide-sugar donor for SseK glycosyltransferase activity. UDP-GlcNAc synthesis is mediated through four steps catalyzed by glucosamine-6-phosphate synthase (GlmS), phosphoglucosamine mutase (GlmM), and the bi-functional enzyme glucosamine-1-phosphate acetyltransferase/N-acetylglucosamine-1-phosphate uridyltransferase (GlmU)20. UDP-GlcNAc synthesis has multiple levels of regulation, including transcriptional activation of the glmUS operon by the NagC dual regulator21, as well as post-transcriptional regulation of glmS mRNA levels through the action of the small RNAs GlmY and GlmZ22,23,24. In Bacillus subtilis, another level of GlmS regulation involves the interaction of the UDP-GlcNAc binding protein GlmR with GlmS. GlmR binding to GlmS enhances the glucosamine-6-phosphate synthase activity of GlmS25,26.
This study was performed to investigate whether UDP-GlcNAc biosynthesis is regulated by the SseK paralogs in Salmonella. Here we report that SseK1 Arg-glycosylates NagC. As a result of this glycosylation, NagC affinity to its target promoters is increased, leading to greater downregulation of the nagE-BACD operon encoding the proteins required for GlcNAc uptake and metabolism27, as well as greater activation of glmUS operon. We also identified glmR, which encodes the GlmS enhancer, GlmR (also known as YvcK) as a NagC-activated gene. GlmR is also Arg-glycosylated by SseK1 and this glycosylation improves its GlmS enhancer activity, leading to increased glucosamine-6-phosphate synthesis by GlmS. Consequently, SseK1-deficient bacteria are impaired for UDP-GlcNAc production. Thus, SseK1 regulates two aspects of UDP-GlcNAc regulation via Arg-glycosylation to maintain bacterial UDP-GlcNAc homeostasis. Combined, these results support a growing body of evidence that bacterial Arg-glycosylation plays additional regulatory roles in fine-tuning pathogen activities.
Results
SseK1 glycosylates NagC and GlmR
Because UDP-GlcNAc is both an essential precursor of the bacterial cell wall and the nucleotide-sugar donor for SseK1, we hypothesized that consumption of UDP-GlcNAc by SseK1 might be compensated by a regulatory mechanism involving SseK1 itself. To test this hypothesis, we first determined whether SseK1 glycosylates any of the proteins involved in UDP-GlcNAc biosynthesis and regulation, namely GlmM, GlmR, GlmS, GlmU, and NagC (Fig. 1A). We expressed His-tagged forms of these proteins in wild-type Salmonella and used Western blotting to detect their potential Arg-glycosylation. We found that NagC and GlmR, but not GlmS, GlmM, or GlmU were Arg-glycosylated in wild-type (WT) Salmonella (Fig. 1B). To determine the role of SseK enzymes in this phenotype, we evaluated Arg-glycosylation of NagC and GlmR in all potential combinations of mutants possessing or lacking the SseK1, SseK2, and SseK3 enzymes. We found that Arg-glycosylation of NagC and GlmR was completely dependent upon SseK1 (Fig. 1C). We corroborated these data using in vitro glycosylation reactions and found that NagC and GlmR were Arg-glycosylated by WT SseK1, but not by an inactive form of SseK1 (HEN mutant) (Fig. 1D).

SseK1 glycosylates NagC and GlmR. (A) Schematic of NagC and Glm pathway under investigation. (B) Western blot analysis of intra-bacterial glycosylation of NagC, GlmS, GlmM, GlmU, and GlmR. (C) In vivo glycosylation of indicated proteins in wild-type Salmonella enterica and sseK mutants. (D). Western blot analysis of in vitro glycosylation of NagC and GlmR in the presence of active or inactive (HEN) forms of SseK1. (E) Mass spectrometry analysis of NagC Arg-glycosylation by SseK1. HCD spectra of the in vivo glycosylated NagC tryptic peptides containing glycosylated R54, R35, and R25. (F) Mass spectrometry analysis of GlmR Arg-glycosylation by SseK1. HCD spectra of the in vivo glycosylated GlmR GluC derived peptides containing glycosylated R212, and R110. (G) Western blot verification of Arg-glycosylation of WT and R-to-A point mutations of NagC. (H) Western blot verification of Arg-glycosylation of WT and R-to-A point mutations of GlmR.
To further corroborate these Western blotting data and to determine on which arginine residues the glycosylation occurred, we subjected NagC and GlmR to mass spectrometry analysis. Data from these experiments indicated that NagC is glycosylated on R25, R35, and R54 (Fig. 1E), and GlmR is glycosylated on R110 and R212 (Fig. 1F). While our initial mass spectrometry analysis indicated potential NagC R59 glycosylation, manual inspection of the MS data revealed it was the R54 site that was modified. However, to be comprehensive in our in vitro and in vivo studies, we still elected to mutate this site for analysis.
We used site-directed mutagenesis to corroborate the mass spectrometry data and found that mutating R35 to alanine abolished NagC Arg-glycosylation, whereas mutating R54 and R59 partially reduced NagC Arg-glycosylation, and mutating R25 had no impact on NagC Arg-glycosylation (Fig. 1G). These experiments were performed in vivo within wild-type Salmonella that expressed each of the indicated NagC mutants. These data suggest that R35, R54, and R59 are the primary NagC residues glycosylated by SseK1. We also found that mutating either R110 or R212 abolished GlmR Arg-glycosylation, suggesting that both R110 and R212 are essential for SseK1-mediated Arg-glycosylation (Fig. 1H).
SseK1-mediated glycosylation of NagC enhances DNA binding
NagC is a transcription factor that coordinates amino-sugar metabolism in bacteria. NagC is a repressor of the nagE-BACD divergent operon involved in GlcNAc uptake and metabolism28 and is an activator of the glmUS operon which encodes the GlmU and GlmS enzymes required for UDP-GlcNAc synthesis21. In the absence of GlcNAc, the divergent nagE-BACD operon is repressed and the glmUS operon is activated. GlcNAc is transported and phosphorylated by a GlcNAc-specific phosphotransferase transporter encoded by NagE. The product GlcNAc-6P, a NagC-regulon inducer, binds to NagC and interferes with its DNA binding activity, leading to de-repression of NagC-repressed genes (nagE-BACD operon, for example) and de-activation of NagC-activated genes (glmUS operon)27,29.
To determine the significance of SseK1-mediated glycosylation of NagC in vivo, we constructed transcriptional fusions of either the nagB promoter, which is repressed by NagC, or the glmU promoter, which is activated by NagC, to the green fluorescence protein (GFP). We measured GFP levels in wild-type (WT) Salmonella and in ΔsseK1 backgrounds after bacteria were grown in M9 minimal medium supplemented or not with GlcNAc. No differences in bacterial growth rates among strains were observed. In the absence of GlcNAc, repression of the nagB-gfp fusion was more pronounced in WT Salmonella than in ΔsseK1 Salmonella (Fig. 2A). Complementation of ΔsseK1 with an active form of sseK1 but not an inactive form of sseK1[sseK1(HEN)] restored GFP expression to levels observed for the WT strain. The ΔnagC mutant was used as a positive control for these assays. Conversely, greater glmU-gfp expression was seen in WT, as compared to ΔsseK1 (Fig. 2B) and complementation of the ΔsseK1 mutant restored the expected phenotypes. Addition of GlcNAc to the medium rendered the GFP expression levels insensitive to SseK1, since a similar level of nagB-gfp de-repression and glmU-gfp de-activation was observed in both WT and ΔsseK1 strains. Note that we used M9 minimal medium for these experiments specifically so that we could study the impact of SseK1 on nag and glm operon expression. Others have shown that significant amounts of SseK1 are produced in both LB and in LPM minimal medium30. We determined by using both RT-PCR and Western blotting that, in M9 minimal medium, SseK1 is expressed and is active (data not shown).

source for 8 h. GFP levels are expressed as RFU (relative fluorescence units)/OD600 ratio. (B) Quantification of glmU::gfp transcriptional fusions. (C) SDS-PAGE and Western blot analysis of purified NagC and NagC-GlcNAc. (C) Purification of native and Arg-glycosylated NagC combined with validation of glycosylation by using Western blotting. (D) EMSAs comparing the DNA-binding activity of NagC and NagC-GlcNAc towards nagB promoter DNA. Two nmoles of Alexa fluor-labeled and then DNA–protein complexes were resolved on 0.5% agarose gels. (E) glmU EMSAs. (F) Quantification of binding affinities of native and Arg-glycosylated NagC to nagB promoter DNA. (G) Quantification of binding affinities of native and Arg-glycosylated NagC to glmU promoter DNA. (H) EMSAs comparing the DNA-binding activity of NagC, NagC-GlcNAc, and NagC (R–K) mutant towards nagB promoter DNA. Experiments were performed as described for panel D.
Arg-glycosylation of NagC enhances its DNA binding activity. (A) Quantification of GFP expression levels of nagB::gfp transcriptional fusions in wild-type Salmonella enterica and its ΔsseK1 or ΔnagC derivatives grown in M9 minimal medium supplemented with either 0.2% glucose or 0.2% GlcNAc as the sole carbon
The increased repression of the nagB-gfp fusion and the higher activation of the glmU-gfp fusion in WT Salmonella in the absence of GlcNAc might be explained if SseK1-mediated Arg-glycosylation of NagC increases NagC affinity to its cognate promoters. To test this hypothesis, native and Arg-glycosylated forms of NagC were purified after their co-expression in E. coli BL21(DE3) cells (Fig. 2C) and their DNA-binding affinities were quantified by using Electrophoretic Mobility Shift Assays (EMSAs). Alexa fluor-labeled DNA duplexes corresponding to either the nagB or glmU promoters were incubated with NagC and complexes were resolved on agarose gels. The sopB promoter was used as a negative control to assess any non-specific DNA binding by NagC. Consistent with the transcriptional fusion data, Arg-glycosylated NagC-GlcNAc showed ~ 2 to threefold stronger affinity to the nagB and glmU promoters, as compared to the unglycosylated form of NagC (Figs. 2D, E). Unglycosylated NagC bound with 45.3 nM affinity to the nagB promoter; Arg-glycosylation of NagC improved the affinity to 15.1 nM (Fig. 2F). Unglycosylated NagC bound with 74 nM affinity to the glmU promoter; Arg-glycosylation of NagC improved the affinity to 42 nM (Fig. 2G). To determine the role of basic character of the amino acids targeted by SseK1 in mediating affinity to the nagB promoter, we generated and purified a NagC mutant in which we converted the R25, R35, R54, and R59 residues to lysines. We found that this NagC (R-K) mutant had significantly reduced affinity for the nagB promoter (Fig. 2H).
NagC activates glmR expression
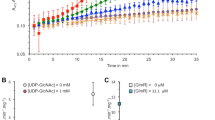
As compared with glmU, less is known regarding glmR regulation. Because this gene encodes a UDP-GlcNAc binding protein that is important for GlmS activity26, we hypothesized that this gene might be regulated by NagC. We found in the glmR promoter region a DNA motif that is similar to the cognate NagC-binding site in glmU, consisting of a 23 base pair pseudo-palindrome with a GC-rich central region flanked by the characteristic T/T and A/A motifs at the −11/−10 and +10/+11 positions respectively, as well as an external AT-rich region (Fig. 3A). To determine whether NagC regulates glmR expression, we constructed a glmR-gfp fusion and measured GFP expression in WT Salmonella in the presence or absence of GlcNAc (NagC inducer). We found that reduced glmR-gfp expression was seen in the ΔsseK1 mutant as compared to WT Salmonella. glmR-gfp expression levels were increased in the absence of GlcNAc, suggesting that NagC is an activator of glmR (Fig. 3B). Consistent with the other EMSAs (Fig. 2) and these transcriptional fusion data, Arg-glycosylated NagC-GlcNAc showed ~ threefold stronger affinity to the glmR promoter, as compared to the unglycosylated form of NagC (Fig. 3C). Unglycosylated NagC bound with 36 nM affinity to the glmR promoter; Arg-glycosylation of NagC improved the affinity to 10 nM (Fig. 3D). The sopB promoter was again used as a negative control to assess any non-specific DNA binding by NagC.

source for 8 h. GFP levels are expressed as RFU (relative fluorescence units)/ OD600 ratio. (C) EMSAs comparing the DNA-binding activity of NagC and NagC-GlcNAc towards glmR promoter DNA. (D) Quantification of binding affinities.
NagC activates glmR. (A) Identification of a potential NagC binding site in the glmR promoter based on similarity to the glmU promoter. (B) Quantification of GFP expression levels of glmR::gfp transcriptional fusions in wild-type Salmonella enterica and its ΔsseK1 or ΔnagC derivatives grown in M9 minimal medium supplemented with either 0.2% glucose or 0.2% GlcNAc as the sole carbon
GlmR Arg-glycosylation enhances its GlmS enhancer activity
In Bacillus subtilis, GlmR interacts with GlmS when UDP-GlcNAc concentrations are low26. This interaction is crucial to enhancing the D-fructose-6-phosphate aminotransferase activity of GlmS26. Since we identified GlmR as an SseK1 glycosylation target, we next assessed the impact of GlmR glycosylation on GlmS activity. To measure GlmS activity, we conducted an assay in which the GlmS product GlcN6P is acetylated by the yeast GlcN6P N-acetyltransferase 1, GNA-1, to produce GlcNAc6P and CoASH31. GlmS, GNA-1, GlmR and GlmR-GlcNAc were purified and the Arg-glycosylation of purified GlmR-GlcNAc was confirmed by using Western blotting (Fig. 4A). We observed that the Arg-glycosylated form of GlmR significantly increased GlmS activity, as compared to the unglycosylated form of GlmR (Fig. 4B).

Arg-glycosylation of GlmR improves its GlmS enhancer activity. (A) SDS-PAGE and Western blot analysis of purified proteins used for GlmS activity assays. (B) GlmS activity was measured in the presence of absence of either native or Arg-glycosylated GlmR by measuring the amount of CoASH produced in an enzyme-coupled assay. The GlmS product GlcN6P was acetylated by the yeast GlcN6P N-acetyltransferase 1, GNA-1, to produce GlcNAc6P and CoASH which was measured at 412 nm.
UDP-GlcNAc levels are higher in WT than ΔsseK1 Salmonella
To evaluate the consequence of NagC and GlmR Arg-glycosylation by SseK1 on UDP-GlcNAc levels in Salmonella, we measured the UDP-GlcNAc levels in WT and the ΔsseK1 mutant. Cell lysates from WT or ΔsseK1 Salmonella were incubated in vitro with SseK1 to hydrolyze UDP-GlcNAc into UDP and GlcNAc. The generated UDP was then converted into ATP for use in luciferase assays (Fig. 5). WT Salmonella produced significantly higher amounts of UDP-GlcNAc than the ΔsseK1 mutant. WT levels of UDP-GlcNAc were partially restored upon complementation with an active form of SseK1 but not with the inactive HEN mutant.

Quantification of UDP-GlcNAc levels. Wild-type Salmonella, ΔsseK1, and complemented ΔsseK1 strains were grown overnight in M9 medium. Cell lysates were incubated for 2 h at room temperature with 100 mM SseK1 to hydrolyze UDP-GlcNAc. The UDP was quantified by using a UDP detection reagent (Promega) that converts UDP into ATP to generate light in a luciferase reaction.
Discussion
SseK1 is a T3SS effector that glycosylates target proteins with GlcNAc on arginine residues. Within the host, SseK1-mediated glycosylation of target proteins interferes with the proper function of adaptor proteins in signaling pathways, leading to reduced host inflammatory response against the pathogen. Within the bacterium, SseK1-mediated glycosylation of target proteins leads to, in addition to the phenotypes we show here, enhanced resistance to methylglyoxal18. Here we found that to promote UDP-GlcNAc production, SseK1 Arg-glycosylates two proteins that regulate different aspects of UDP-GlcNAc biosynthesis (Fig. 6). First, by enhancing the ability of NagC to regulate the glmUS operon and the glmR gene; second, by improving the ability of GlmR to enhance GlmS activity. This regulatory mechanism may allow Salmonella to maintain sufficient levels of UDP-GlcNAc for cell well synthesis and for the glycosylation of other bacterial and host proteins (Fig. 6).

Working Model. SseK1 Arg-glycosylates NagC to enhance transcriptional activation of glmUS and glmR. SseK1 also glycosylates GlmR to increase GlmS activity, leading to increased levels of UDP-GlcNAc.
SseK1 glycosylates arginine residues that are in or near the NagC HTH domain that is responsible for DNA binding, suggesting that the significance of targeting NagC by SseK1 is to modulate its DNA binding affinity towards target gene promoters. Upon Arg-glycosylation, NagC bound with higher affinity to the nagB, glmU, and glmR promoters. Since NagC is a pleiotropic regulator that controls the expression of multiple genes involved in several metabolic pathways, its glycosylation by SseK1 adds an additional layer of gene regulation to this pathogen for the fine-tuning of gene expression in response to bacterial needs.
Several pathogens have evolved mechanisms to modulate GlmR activity. For example, in Bacillus subtilis, GlmR interacts with either GlmS or YvcJ depending on UDP-GlcNAc availability26. In the presence of non-glycolytic carbon sources such as intermediates of the Krebs cycle, GlmR binds to GlmS and enhances its catalytic activity. In the presence of glycolytic carbon sources such as glucose, UDP-GlcNAc concentrations are sufficient and GlmR instead binds to YvcJ to avoid excessive stimulation of GlmS and unnecessary production of UDP-GlcNAc26. In Listeria monocytogenes, GlmR (YcvK) is phosphorylated by the serine protein kinase PkrA; regulation of GlmR phosphorylation is critical for virulence and cytosolic survival32. In this study, we found that in Salmonella enterica, which lacks YcvJ or PrkA homologs, SseK1 glycosylates GlmR to enhance GlmS activity. Future analysis of how GlmR Arg-glycosylation affects its affinity for GlmS may provide a mechanistic explanation for this observation. We also desire in the future to conduct experiments to assess the impact of GlmR glycosylation and/or mutation on Salmonella virulence. We have not yet performed any specific experiments to assess cell-wall related phenotypes in the sseK1 mutant, some of which might be postulated by the differential levels of UDP-GlcNAc we observed. However, we note that the growth rates between the WT and the sseK1 mutant were not significantly different (data not shown), suggesting that there is not a globally-significant impact to the bacterial cell wall due to SseK1 activity, at least under these specific experimental conditions.
O-linked glycosylation of eukaryotic transcription factors is relatively common and has been studied for decades33. Such glycosylation can both increase or decrease gene expression33. For example, the MORC family CW-type zinc finger 2 protein (MORC2), a chromatin-remodeling enzyme involved in DNA-damage response, exhibits higher transcription activation activity upon O-GlcNAc transferase (OGT)-mediated glycosylation at T55634. Another study describes a direct correlation between glycosylation of the Hedgehog pathway transcription factors GLI1 and GLI2 and their transcriptional activity35.The pancreatic/duodenal homeobox-1 protein (PDX1), which is required for pancreatic function and development is glycosylated by OGT at high glucose concentrations, leading to increased DNA-binding affinity and consequently greater insulin secretion36. OGT also glycosylates the NF-κB c-Rel subunit on S350, a process required for c-Rel DNA binding and transactivation functions37. However, in most cases, transcription factor glycosylation also affects their nuclear translocation, and there are relatively few direct measurements of the impact of transcription factor glycosylation on affinity for target gene promoters. One the few examples where both nuclear localization and DNA affinity of a glycosylated transcription factor were assessed is illustrated by the impact on the NF-κB p65 subunit by OGT-mediated glycosylation, leading to aggravated TNF-α-stimulated inflammation both in vitro and in vivo38.
By contrast, our study links Arg-glycosylation directly to differential DNA-binding affinity. We observed that Arg-glycosylated NagC bound with higher affinity to the nagB, glmU, and glmR promoters than did the unglycosylated form of NagC. To some extent, these results are counter-intuitive, because one might reasonably expect that glycosylating a basic amino acid would tend to reduce protein affinity for DNA. We also note that, in a related system, the recent analysis of SseK3 glycosylation of PhoP concluded that there was a slight reduction in PhoP affinity for DNA as a function of PhoP R215 glycosylation, although the binding affinities were not calculated19. However, it is not clear from these studies whether the investigators used phosphorylated PhoP or recombinant, non-phosphorylated PhoP for their EMSAs, a variable which might affect the interpretation of these data.
The work described here provides the first evidence that Arg-glycosylation of the bacterial transcription factor NagC by SseK1 increases NagC affinity for DNA. These data also represent, to our knowledge, along with the recently described work regarding Arg-glycosylation of PhoP by SseK319, the first example of a T3SS effector directly regulating gene expression by modifying a transcription factor. Future experiments aiming to solve the co-crystal structure of an Arg-GlcNac-NagC-DNA complex may permit a molecular understanding of how Arg-glycosylation of transcription factors alters their interactions with DNA.
Materials and methods
Plasmids, strains, and cloning
The plasmids and strains used in this study are listed in Tables 1 and 2, respectively. Wild type sseK1 (Salmonella enterica) and its derivative H244A E255A N256A, were cloned into pET42a. nagC, glmR, glmU, glmS and glmM were cloned in pTac using ABC cloning40. nagC deletions were constructed using lambda red recombination with the pKD3 and pKD119 plasmids41. Mutants were screened on LB medium supplemented with 10 µg/ml chloramphenicol and mutations were confirmed by PCR and DNA sequencing. Protein purification was performed as described previously12. For the purification of glycosylated substrates, His-tagged substrates of SseK1 were co-expressed (or not) with FLAG-tagged SseK1 and purified against the His-epitope, as described previously18.
In vitro glycosylation assays
Assays were performed as described previously12. Briefly, 200 nM of SseK1 was incubated with 1 μM of either wild type or mutant forms of NagC or GlmR in buffer containing 50 mM Tris–HCl pH 7.4, 1 mM UDP-GlcNAc, 10 mM MnCl2, and 1 mM DTT. After 2 h incubation at room temperature, samples were subjected to western blotting using an anti-R-GlcNAc monoclonal antibody (Abcam).
Digest of gel-separated proteins
SDS-PAGE separated affinity-purified proteins were fixed and visualized with Coomassie staining. Bands of interest were excised and then destained of Coomassie with 50 mM NH4HCO3, 50% ethanol for 20 min at room temperature with shaking at 750 rpm. Destained samples were then dehydrated with 100% ethanol, dried by vacuum-centrifugation for 20 min and then rehydrated in 50 mM NH4HCO3, 10 mM DTT. Samples were reduced for 1 h at 56 °C with shaking. Following reduction, the reduction buffer was removed, and the gel bands were washed twice in 100% ethanol for 10 min to remove residual DTT. Dehydrated gel bands were then rehydrated with 55 mM iodoacetamide in 50 mM NH4HCO3 and allowed to alkylate in the dark for 45 min at room temperature. The alkylation buffer was removed and gel samples washed with 50 mM NH4HCO3, followed by two rounds of 100% ethanol and vacuum dried. Alkylated samples were then rehydrated with either 20 ng/µl of trypsin (Promega) for NagC or 20 ng/ul of Glu-C (Promega) for GlmR in 40 mM NH4HCO3 at 4 °C for 1 h. Excess protease was removed, gel pieces were covered in 40 mM NH4HCO3 and incubated overnight at 37 °C. Peptides were collected, desalted using homemade R3/C18 stage tips as previously described42 before analysis by LC–MS.
Reverse phase LC–MS/MS
Peptides were resuspended in Buffer A* (2% MeCN, 0.1% TFA) and separated using a two-column chromatography set on a Dionex Ultimate 3000 UHPLC (Thermo Fisher Scientific). Samples were first concentrated on a PepMap100 C18 20 mm × 75 μm trap at 5 μl/min for 5 min with Buffer A (0.1% formic acid, 2% DMSO) and then separated on a PepMap C18 500 mm × 75 μm analytical column (Thermo Fisher Scientific). Separated peptide were infused into a Q-Exactive plus Mass Spectrometer (Thermo Fisher Scientific) at 300 nl/minute for 119 min by altering the buffer composition from 2% Buffer B (0.1% formic acid, 77.9% acetonitrile, 2% DMSO) to 28% B over 90 min, then from 28% B to 4% B over 10 min, then from 40% B to 80% B over 5 min. The composition was held at 100% B for 5 min, and then dropped to 2% B over 1 min before being held at 2% B for another 9 min. The Q-Exactive plus Mass Spectrometer was operated in a data-dependent mode, acquiring one full precursor scan (resolution 70,000; 375–1800 m/z, AGC target of 1 × 106) followed by 5 data-dependent HCD MS–MS events (using three collision energies of 28, 32, and 38; resolution 35 k AGC target of 2 × 105 with a maximum injection time of 110 ms).
Mass spectrometry data analysis
Identification of Arg-glycosylation events was accomplished using MaxQuant (v1.6.17.0)43. The predicted amino acid sequences for GlmR and NagC were combined into a database with the Salmonella typhimurium SL1344 proteome (Uniprot accession: UP000008962) and searched, allowing carbamidomethylation of cysteine set as a fixed modification and the variable modifications of oxidation of methionine and Arg-GlcNAcylation (H13C8NO5; 203.0793 Da to Arginine). Searches were performed with either Trypsin or GluC cleavage specificity depending on the protease used for digestion, allowing 2 miscleavage events with a maximum false discovery rate (FDR) of 1.0% set for protein and peptide identifications. The resulting modified peptide output was processed within the Perseus (v1.4.0.6)44 analysis environment to remove reverse matches and common protein contaminants. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE45 partner repository with the dataset identifier PXD030710.
GFP reporter assay
A low-copy number plasmid (pHG165) carrying nagB, glmU, or glmR promoter transcriptional fusions to gfp was electroporated into Salmonella. Two hundred µl of M9 minimal medium supplemented with either 0.2% glucose or 0.2% GlcNAc was used to grow the transformed bacteria in 96 well plates. GFP expression levels were measured after 8 h of growth and GFP data were presented as an average of RFU (relative fluorescence units)/OD600 ratio.
EMSAs
Two nmoles of 5’ Alexa-fluor labeled DNA corresponding to nagB, glmU, or glmR promoters were incubated for 10 min at room temperature in the presence of either NagC or NagC-GlcNAc in a buffer containing 50 mM HEPES, 100 mM K glutamate (pH 8.0), and 0.5 mg/ml BSA. Samples (10 µl) samples were loaded on 0.5% agarose gels and subjected to electrophoresis in 0.5X TBE buffer. DNA–protein complexes were visualized by using a Li-COR Odyssey. Dissociation constant estimates were calculated by fitting the EMSA data (% bound and unbound DNA) using non-linear regression in GraphPad Prism.
GlmS activity assay
GlmS activity assays were performed as previously described26. Briefly, reactions were performed in a buffer containing 50 mM Tris–HCl pH 7.4, 1 mM EDTA, 2 mM F6P, 2 mM L-Gln, 0.5 mM Ac-CoA, 0.5 mM DTNB, 10 µg GNA-1, and 1 µM GlmS, in the presence of absence of 1 µM native or Arg-glycosylated GlmR. GlmS activity was measured for 2 h at 37 °C by monitoring CoA production at 412 nm.
Quantification of UDP-GlcNAc
Salmonella strains were grown overnight in M9 medium and cell lysates were incubated for 2 h at room temperature with 100 mM SseK1 to hydrolyze UDP-GlcNAc. The released UDP was quantified using a UDP detection reagent (Promega) that converts UDP into ATP to generate light in a luciferase reaction.
Statistical analyses
Fluorescence and luminescence data were analyzed statistically using Dunn’s multiple comparisons. EMSA and enzyme assay data were analyzed statistically using Kruskal–Wallis tests. p-value < 0.05 were considered significant.
References
Kirk, M. D. et al. World health organization estimates of the global and regional disease Burden of 22 Foodborne bacterial, protozoal, and viral diseases, 2010: A data synthesis. PLoS Med. 12(12), e1001921. https://doi.org/10.1371/journal.pmed.1001921 (2015).
Majowicz, S. E. et al. The global burden of nontyphoidal Salmonella gastroenteritis. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 50(6), 882–889. https://doi.org/10.1086/650733 (2010).
Hensel, M. et al. Simultaneous identification of bacterial virulence genes by negative selection. Science 269(5222), 400–403. https://doi.org/10.1126/science.7618105 (1995).
Mills, D. M., Bajaj, V. & Lee, C. A. A 40 kb chromosomal fragment encoding Salmonella typhimurium invasion genes is absent from the corresponding region of the Escherichia coli K-12 chromosome. Mol. Microbiol. 15(4), 749–759. https://doi.org/10.1111/j.1365-2958.1995.tb02382.x (1995).
Shea, J. E., Hensel, M., Gleeson, C. & Holden, D. W. Identification of a virulence locus encoding a second type III secretion system in Salmonella typhimurium. Proc. Nat. Acad. Sci. USA 93(6), 2593–2597. https://doi.org/10.1073/pnas.93.6.2593 (1996).
Li, S. et al. Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature 501(7466), 242–246. https://doi.org/10.1038/nature12436 (2013).
Pearson, J. S. et al. A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature 501(7466), 247–251. https://doi.org/10.1038/nature12524 (2013).
Ding, J. et al. Structural and functional insights into host death domains inactivation by the bacterial arginine GlcNAcyltransferase effector. Mol. Cell. 74(5), 922–35.e6. https://doi.org/10.1016/j.molcel.2019.03.028 (2019).
Esposito, D. et al. Structural basis for the glycosyltransferase activity of the Salmonella effector SseK3. J. Biol. Chem. 293(14), 5064–5078. https://doi.org/10.1074/jbc.RA118.0017 (2018).
Park, J. B. et al. Structural basis for arginine glycosylation of host substrates by bacterial effector proteins. Nat. Commun. 9(1), 4283. https://doi.org/10.1038/s41467-018-06680-6 (2018).
Araujo-Garrido, J. L., Baison-Olmo, F., Bernal-Bayard, J., Romero, F. & Ramos-Morales, F. Tubulin folding cofactor TBCB is a target of the salmonella effector protein SseK1. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21093193 (2020).
El Qaidi, S. et al. NleB/SseK effectors from Citrobacter rodentium, Escherichia coli, and Salmonella enterica display distinct differences in host substrate specificity. J. Biol. Chem. 292(27), 11423–11430. https://doi.org/10.1074/jbc.M117.790675 (2017).
Gao, X. et al. NleB, a bacterial effector with glycosyltransferase activity, targets GAPDH function to inhibit NF-kappaB activation. Cell Host Microbe. 13(1), 87–99. https://doi.org/10.1016/j.chom.2012.11.010.PubMedPMID:23332158;PMCID:PMC3553500 (2013).
Xu, C. et al. A pathogen-derived effector modulates host glucose metabolism by arginine GlcNAcylation of HIF-1alpha protein. PLoS Pathog. 14(8), e1007259. https://doi.org/10.1371/journal.ppat.1007259.PubMedPMID:30125331;PMCID:PMC6117090 (2018).
Gan, J. et al. The Salmonella effector SseK3 targets small Rab GTPases. Front. Cell Infect. Microbiol. 10, 419. https://doi.org/10.3389/fcimb.2020.00419 (2020).
Giogha, C. et al. NleB2 from enteropathogenic Escherichia coli is a novel arginine-glucose transferase effector. PLoS Pathog. 17(6), e1009658. https://doi.org/10.1371/journal.ppat.1009658 (2021).
El Qaidi, S. et al. An intra-bacterial activity for a T3SS effector. Sci. Rep. 10(1), 1073. https://doi.org/10.1038/s41598-020-58062-y (2020).
El Qaidi, S., Scott, N. E. & Hardwidge, P. R. Arginine glycosylation enhances methylglyoxal detoxification. Sci. Rep. 11(1), 3834. https://doi.org/10.1038/s41598-021-83437-0 (2021).
Xue, J. et al. Arginine GlcNAcylation and activity regulation of PhoP by a type III secretion system effector in salmonella. Front. Microbiol. 12, 825743. https://doi.org/10.3389/fmicb.2021.825743 (2021).
Mengin-Lecreulx, D. & van Heijenoort, J. Identification of the glmU gene encoding N-acetylglucosamine-1-phosphate uridyltransferase in Escherichia coli. J. Bacteriol. 175(19), 6150–6157. https://doi.org/10.1128/jb.175.19.6150-6157.1993 (1993).
Plumbridge, J. Co-ordinated regulation of amino sugar biosynthesis and degradation: the NagC repressor acts as both an activator and a repressor for the transcription of the glmUS operon and requires two separated NagC binding sites. EMBO J. 14(16), 3958–3965 (1995).
Reichenbach, B., Maes, A., Kalamorz, F., Hajnsdorf, E. & Görke, B. The small RNA GlmY acts upstream of the sRNA GlmZ in the activation of glmS expression and is subject to regulation by polyadenylation in Escherichia coli. Nucl. Acids Res. 36(8), 2570–2580. https://doi.org/10.1093/nar/gkn091 (2008).
Sachla, A. J. & Helmann, J. D. Resource sharing between central metabolism and cell envelope synthesis. Curr. Opin. Microbiol. 60, 34–43. https://doi.org/10.1016/j.mib.2021.01.015 (2021).
Urban, J. H., Papenfort, K., Thomsen, J., Schmitz, R. A. & Vogel, J. A conserved small RNA promotes discoordinate expression of the glmUS operon mRNA to activate GlmS synthesis. J. Mol. Biol. 373(3), 521–528. https://doi.org/10.1016/j.jmb.2007.07.035 (2007).
Foulquier, E. & Galinier, A. YvcK, a protein required for cell wall integrity and optimal carbon source utilization, binds uridine diphosphate-sugars. Sci. Rep. 7(1), 4139. https://doi.org/10.1038/s41598-017-04064-2 (2017).
Foulquier, E., Pompeo, F., Byrne, D., Fierobe, H. P. & Galinier, A. Uridine diphosphate N-acetylglucosamine orchestrates the interaction of GlmR with either YvcJ or GlmS in Bacillus subtilis. Sci. Rep. 10(1), 15938. https://doi.org/10.1038/s41598-020-72854-2 (2020).
Plumbridge, J. A. Repression and induction of the nag regulon of Escherichia coli K-12: The roles of nagC and nagA in maintenance of the uninduced state. Mol. Microbiol. 5(8), 2053–2062. https://doi.org/10.1111/j.1365-2958.1991.tb00828.x (1991).
Plumbridge, J. Regulation of PTS gene expression by the homologous transcriptional regulators, Mlc and NagC, in Escherichia coli (or how two similar repressors can behave differently). J. Mol. Microbiol. Biotechnol. 3(3), 371–380 (2001).
Plumbridge, J. & Kolb, A. DNA loop formation between Nag repressor molecules bound to its two operator sites is necessary for repression of the nag regulon of Escherichia coli in vivo. Mol. Microbiol. 10(5), 973–981. https://doi.org/10.1111/j.1365-2958.1993.tb00969.x (1993).
Kujat Choy, S. L. et al. SseK1 and SseK2 are novel translocated proteins of Salmonella enterica serovar typhimurium. Infect. Immun. 72(9), 5115–5125. https://doi.org/10.1128/IAI.72.9.5115-5125.2004 (2004).
Li, Y. et al. An enzyme-coupled assay for amidotransferase activity of glucosamine-6-phosphate synthase. Anal. Biochem. 370(2), 142–1466. https://doi.org/10.1016/j.ab.2007.07.031 (2007).
Pensinger, D. A. et al. The Listeria monocytogenes PASTA Kinase PrkA and its substrate YvcK are required for cell wall homeostasis, metabolism, and virulence. PLoS Pathog. 12(11), e1006001. https://doi.org/10.1371/journal.ppat.1006001 (2016).
Comer, F. I. & Hart, G. W. O-GlcNAc and the control of gene expression. Biochem. Biophys. Acta. 1473(1), 161–171. https://doi.org/10.1016/s0304-4165(99)00176-2 (1999).
Liu, Y. Y. et al. O-GlcNAcylation of MORC2 at threonine 556 by OGT couples TGF-β signaling to breast cancer progression. Cell Death Differ. https://doi.org/10.1038/s41418-021-00901-0 (2022).
Das, S. et al. O-GlcNAcylation of GLI transcription factors in hyperglycemic conditions augments Hedgehog activity. Lab. Invest. J. Tech. Methods Pathol. 99(2), 260–270. https://doi.org/10.1038/s41374-018-0122-8 (2019).
Gao, Y., Miyazaki, J. & Hart, G. W. The transcription factor PDX-1 is post-translationally modified by O-linked N-acetylglucosamine and this modification is correlated with its DNA binding activity and insulin secretion in min6 beta-cells. Arch. Biochem. Biophys. 415(2), 155–163. https://doi.org/10.1016/s0003-9861(03)00234-0 (2003).
Ramakrishnan, P. et al. Activation of the transcriptional function of the NF-κB protein c-Rel by O-GlcNAc glycosylation. Sci. Signal. 6(290), ra75. https://doi.org/10.1126/scisignal.2004097 (2013).
Kim, H. B. et al. O-linked N-acetylglucosamine glycosylation of p65 aggravated the inflammation in both fibroblast-like synoviocytes stimulated by tumor necrosis factor-α and mice with collagen induced arthritis. Arth. Res. Therapy 17(1), 248. https://doi.org/10.1186/s13075-015-0762-7 (2015).
El Qaidi, S. et al. High-Throughput Screening for Bacterial Glycosyltransferase Inhibitors. Front. Cell Infect. Microbiol. 8, 435. https://doi.org/10.3389/fcimb.2018.00435 (2018).
Qaidi, S. E. & Hardwidge, P. R. ABC cloning: An efficient, simple, and rapid restriction/ligase-free method. MethodsX. 6, 316–321. https://doi.org/10.1016/j.mex.2019.02.007 (2019).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97(12), 6640–6645. https://doi.org/10.1073/pnas.120163297 (2000).
Rappsilber, J., Mann, M. & Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2(8), 1896–1906. https://doi.org/10.1038/nprot.2007.261 (2007).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26(12), 1367–1372. https://doi.org/10.1038/nbt.1511 (2008).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11(12), 2301–2319. https://doi.org/10.1038/nprot.2016.136 (2016).
Csordas A, Ovelleiro D, Wang R, Foster JM, Rios D, Vizcaino JA, Hermjakob H. PRIDE: quality control in a proteomics data repository. Database (Oxford). 2012;2012:bas004. Epub 2012/03/22. doi: https://doi.org/10.1093/database/bas004. PubMed PMID: 22434838; PMCID: PMC3308160.
Acknowledgements
The project described was supported by Grant Numbers AI127973 and AI153202 from the National Institute of Allergy and Infectious Diseases (NIAID), by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health under award number P20GM130448 (PRH), and by a pilot project (SE) from P20GM130448. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. N.E.S is supported by an Australian Research Council Future Fellowship (FT200100270) and an ARC Discovery Project Grant (DP210100362). We thank the Melbourne Mass Spectrometry and Proteomics Facility of The Bio21 Molecular Science and Biotechnology Institute for access to MS instrumentation. We also acknowledge the constructive advice provided by Dr. Jacqueline Plumbridge, Institut de Biologie Physico-Chimique, Paris, France.
Author information
Authors and Affiliations
Contributions
N.E.S. Performed the mass spectrometry experiments, S.E. conceived of the project, performed the other experiments and wrote the manuscript. M.P.H. provided technical assistance and project resources. P.R.H. coordinated the project and edited the manuscript. All authors reviewed the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
El Qaidi, S., Scott, N.E., Hays, M.P. et al. Arginine glycosylation regulates UDP-GlcNAc biosynthesis in Salmonella enterica. Sci Rep 12, 5293 (2022). https://doi.org/10.1038/s41598-022-09276-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-09276-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
