
Abstract
The cartilage aggrecan proteoglycan is crucial for both skeletal growth and articular cartilage function. A number of aggrecan (ACAN) gene variants have been linked to skeletal disorders, ranging from short stature to severe chondrodyplasias. Osteochondritis dissecans is a disorder where articular cartilage and subchondral bone fragments come loose from the articular surface. We previously reported a missense ACAN variant linked to familial osteochondritis dissecans, with short stature and early onset osteoarthritis, and now describe three novel ACAN gene variants from additional families with this disorder. Like the previously described variant, these are autosomal dominant missense variants, resulting in single amino acid residue substitutions in the C-type lectin repeat of the aggrecan G3 domain. Functional studies showed that neither recombinant variant proteins, nor full-length variant aggrecan proteoglycan from heterozygous patient cartilage, were secreted to the same level as wild-type aggrecan. The variant proteins also showed decreased binding to known cartilage extracellular matrix ligands. Mapping these and other ACAN variants linked to hereditary skeletal disorders showed a clustering of osteochondritis dissecans-linked variants to the G3 domain. Taken together, this supports a link between missense ACAN variants affecting the aggrecan G3 domain and hereditary osteochondritis dissecans.
Similar content being viewed by others

Introduction
The large proteoglycan aggrecan is vital for skeletal development and cartilage function, creating a swelling pressure in the tissue that provides resistance to compression and load dissipation over the joint surface1. Aggrecan contains an extended central region carrying keratan sulfate and chondroitin sulfate chains. This region is flanked by globular protein domains, the N-terminal G1 and G2 domains and the C-terminal G3 domain, mediating interactions with other components of the extracellular matrix2.
A complete lack of aggrecan causes severe chondrodysplasia and is incompatible with postnatal life, as observed in mice3, cattle4 and chick5. In humans, several aggrecan (ACAN) gene variants have been linked to hereditary skeletal disorders. Some of these variants are linked to the skeletal dysplasias spondyloepiphyseal dysplasia Kimberley type (SEDK)6, and spondyloepimetaphyseal dysplasia aggrecan type (SEMD)7. In recent years multiple additional ACAN gene variants have been identified, many of which are variants of unknown significance (VUS) or linked to milder phenotypes such as idiopathic short stature (Supplemental Table 1)6,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34. While heredity has not been confirmed for some of these variants, most show clear segregation with disease.
Most ACAN variants result in truncation or haploinsufficiency due to premature stop codons, or in nonsense mediated decay of the aggrecan mRNA as shown for the bovine Dexter Bulldog ACAN variant4. Less is known concerning disease mechanisms for ACAN missense variants. These have predominantly been found in the globular domains of aggrecan and are linked to idiopathic short stature. Two of the ACAN missense variants have been studied in more detail. The SEMD missense variant p.D2419N (originally referred to as p.D2267N, see Table 1 for updated variant nomenclature) affects the C-type lectin of the G3 globular domain of aggrecan7. This recessively inherited variant results in the formation of a novel N-linked glycosylation site, potentially blocking interactions of the C-type lectin. Another missense ACAN variant was linked to familial osteochondritis dissecans (fOCD) with short stature and early onset osteoarthritis8,9,10. This variant results in a single amino acid substitution p.V2455M (originally referred to as p.V2303M, see Table 1) in the C-type lectin repeat of the C-terminal G3 globular domain of the protein, affecting intermolecular interactions with other cartilage extracellular matrix components9.
Osteochondritis dissecans (OCD) is a joint disease with partial or total focal detachment of articular cartilage and underlying subchondral bone fragments. The majority of OCD cases are sporadic with a knee OCD prevalence of 15–29 per 100,000 reported in Sweden11. The etiology of OCD remains unclear, but is likely multifactorial. Several causes have been suggested, including trauma or microfracture, ischemia, and genetic predisposition12. A rare hereditary form, fOCD, presents as multiple osteochondral lesions in knees and/or hips and/or elbows, disproportionate short stature, and early osteoarthritis10. We previously identified an association of the p.V2455M ACAN variant with fOCD9. Since then, other ACAN variants linked to fOCD have been reported8,13,14. Additional ACAN variants have also been described where only the probands present with OCD8,15,16,17,18,19,20. Interestingly, among these, the ACAN missense variant p.L2393P (originally referred to as p.L2355P, see Table 1) is located in the G3 domain of aggrecan, like the p.V2455M fOCD variant, and result in short stature with early onset osteoarthritis and with OCD observed in the proband, although heredity for the latter remains unclear8,17. In view of this, it is perhaps not surprising that there has been a call for more studies on gene variants linked to fOCD21.
In this paper we describe three novel missense ACAN gene variants in families with hereditary OCD, short stature and early onset osteoarthritis. These novel variants are missense mutations, affect different amino acid residues in the C-type lectin repeat of aggrecan, and result in decreased secretion and disturbed interactions of the variant protein.
Results
Aggrecan G3 variant nomenclature
Earlier studies have based the ACAN gene variant position numbering on different reference sequences. Unfortunately, neither of these cover the entire coding sequence of aggrecan. This is due to alternative splicing of the ACAN mRNA, where the presence or absence of exons coding for EGF or complement regulatory protein-like repeats (CRP, also known as sushi repeats) in the G3 domain results in different aggrecan isoforms. In this study, we base the variant cDNA and protein nomenclature on the recently added reference sequence for ACAN variant 3, which includes the complete G3 domain; comprising EGF1 and EGF2, C-type lectin and CRP/sushi repeats (NM_001369268.1, PRI 14-OCT-2019). Table 1 compares this nomenclature with versions used in previous publications7,8,9.
Family 1 clinical characteristics
A 19-year-old male was referred for molecular genetic analysis, due to short stature, OCD, and a family history of OCD and premature OA (Table 2). He was 155 cm tall, and he had very short 2nd metatarsal bones. He had experienced multiple arthroscopies to remove osteochondral fragments. His mother was 145 cm tall. Her surgical history includes multiple arthroscopies to remove osteochondral fragments, and unilateral hip and knee replacement. The grandfather was 135 cm tall, and had a history of OCD and premature osteoarthritis.
Family 2 clinical characteristics
A 13-year-old male was referred for molecular genetic analysis, due to short stature. He had a normal birth history and development (Table 2). Endocrine evaluation showed partial growth hormone (GH) deficiency, and the boy was treated with GH for 5 years. His height was 127.5 cm (− 3.5 SDS) at 13 years of age. At the age of 16 years, he was 148 cm in stature and still on GH treatment. He had bilateral knee pain with right-sided genu varum that was corrected with proximal tibial lateral 8-plating (Fig. 1a–c). MRI and X-ray of the knee showed OCD (Fig. 1d,e). At the age of 19 he was 155 cm tall, and GH treatment was discontinued. MRI of knees at 18 years confirmed OCD (Fig. 1f–h). Clinical skeletal examination at 16 years showed no abnormalities of the spine, elbows or hands. Family history revealed that his 46-year-old mother, and 23-year-old sister both have short stature (143 cm and 139 cm respectively) and knee OCD initially diagnosed in teenage years (Fig. 1i,j). Both his sister and mother have had multiple right (sister) or bilateral (mother) arthroscopies to remove osteochondral fragments. His mother also has a history of bilateral knee osteoarthritis since her 20s. The 82-year-old maternal grandmother was 139 cm tall, and had bilateral hip replacement for osteoarthritis in her 60s/70s, but there is no confirmed history of OCD. There is also a family history of short stature and knee joint problems, possibly related to OCD in at least two further maternal relatives but their clinical data are not available.

Clinical observations in Family 2. The proband is an 18 years old man with short stature (height 127.5 cm at 13 years old, − 3.5 SDS), normal birth history and development, and a family history of short stature. The proband was treated with GH from the age of 14. At 16 years of age, the proband was still on GH treatment and presented with bilateral knee pain and right-sided genu varum (a) that was corrected surgically (b,c). His knees at 15 (d), 16 (e) and 18 years of age (f–h) showed OCD. Both his 23-year-old sister (i,j) and his 46-year-old mother (143 cm, not shown) have short stature and OCD. Arrows indicate osteochondral lesions.
Family 3 clinical characteristics
A 31-year-old woman was referred for molecular genetic analysis, because of joint pain, early-onset degenerative joint disease, and osteochondral lesions in multiple joints (Table 2). Since adolescence she has experienced arthralgia in her hips, knees, ankles, feet, back, shoulders, wrists, and hands. She has mild scoliosis, osteoarthritis, and osteochondral lesions diagnosed on X-ray and MRI studies of her knees and hips (Fig. 2a–c). Surgical history includes left and right hip arthroscopies to treat femoral-acetabular impingement and labrum tears (Fig. 2c). Distal phalanges of the thumbs appear short and broad and she has mild clinodactyly of the 5th fingers (Fig. 2d,e). She has mild myopia (− 0.50) and normal hearing. She was the first child born to non-consanguineous parents of Scotch, Canadian (Nova Scotia), English, and German ancestry, a 26-year-old mother 170 cm tall and a 34-year-old father 155 cm tall. Family history is remarkable in that multiple paternal family members present with short stature and joint problems. The middle child is a brother, currently 28 years old and 155 cm tall, who has a history of knee pain and was said to have a Grade I OCD lesion of the medial femoral condyle on X-ray when he was 11 years old. He has also been told he has issues with his trochleae. Currently he experiences knee pain when running as well as miscellaneous joint cracking. The youngest sibling is a sister, now 25 years old and 150 cm tall; she has dislocated her kneecaps in the past, is said to have trochlear problems, and was diagnosed with a soft tissue sarcoma when she was 18 years old. The father of our proband is currently 65 years old. He has a history of OCD lesions, knee problems, and has had multiple surgeries on his left knee (before the age of 30) and underwent a left total knee replacement in his early 60s. In his youth his parents brought him to the National Institutes of Health (NIH) for evaluation of growth problems. Growth hormone therapy was discussed but not pursued at that time. He was diagnosed with Alzheimer’s disease in his 60s. His mother, i.e. the paternal grandmother of the proband, is now deceased, but was said to be 150 cm tall and had a history of hip and knee problems. Other members of her side of the family were known for being short-statured. She retired early due to joint issues, and eventually underwent hip and knee replacement surgeries. Her husband, the paternal grandfather of our proband, 188 cm tall, is still living at 93 years old. That couple had two other children, a son and a daughter who are said to be well and are said to be 188 cm and 170 cm tall, respectively, and have no known joint problems.

Clinical observations in Family 3. The proband is a now 33-year-old woman, 156 cm tall, with joint pain, early onset osteoarthritis and osteochondral lesions in multiple joints, including knees (a) and hips (b). She has had hip arthroscopic treatment for femoral-acetabular impingements and labrum tears (c, arrow). Hand X-rays show short and broad distal phalanges of the thumbs (d,e). She has a family history of short stature and joint problems on the paternal side, including OCD.
Molecular genetic analysis
Targeted sequence analysis of ACAN revealed heterozygous likely pathogenic variants in two families. In family 1, we identified a missense variant: c.7156T>A, in exon 15 (p.C2386S) (Supplemental Fig. 1a) in DNA from the proband sent from Australia. The sister and mother of the proband were found to carry the p.C2386S variant (Fig. 3a). The healthy father was not a carrier.

Pedigrees of the studied families. Pedigrees are shown for (a) Family 1, from Australia, (b) Family 2, from the United Kingdom, and (c) Family 3, from New York, USA. The probands are identified by arrows. Filled symbols indicate individuals with heterozygous ACAN gene variants verified by DNA sequencing, question marks indicate no sequence information is available for the individual. The height and diagnosed osteoarthritis (OA) or osteochondritis dissecans (OCD) are indicated below symbols.
In the second proband referred from UK (family 2), we identified the c.7250T>C, p.L2417P variant, located in exon 16 (Supplemental Fig. 1b). Segregation analysis showed that the p.L2417P variant was present in his affected sister, and mother (Fig. 3b).
In family 3, a third ACAN variant in exon 17 was identified by NGS at Fulgent Genetics; c.7358A>T, p.D2453V. This variant was reported to ClinVar NCBI by Fulgent Genetics, as a variant of unclear significance (VUS).Two siblings of the proband as well as the father were found to be heterozygous carriers for the p.D2453V ACAN variant by Sanger sequencing (Fig. 3c). The familial variant was not detected in the proband’s mother and a paternal aunt, both of normal stature and neither of whom have any history of orthopedic problems.
The novel missense ACAN variants affect the C-type lectin repeat of aggrecan
The novel fOCD-linked missense ACAN variants all locate in the C-type lectin repeat of the aggrecan C-terminal globular G3 domain (Fig. 4a). They affect the same aggrecan domain as the previously described fOCD-linked variant p.V2455M, and are all dominantly inherited.

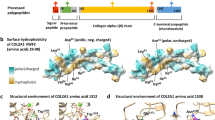
Hereditary ACAN variants segregating with skeletal disorders. (a) ACAN variants (published until October 2021) are shown schematically on the domain structure of the aggrecan core protein. Missense variants are shown below the core protein, all other types of variants above. Variants linked to familial OCD are shown in red, SEMD in blue and SEDK in pink. Variants where OCD has been reported in only one family member are shown in green. The novel fOCD variants reported in this study are underlined. (b) Locations of amino acid residues mutated in missense ACAN variants shown on the structure of the G3 domain C-type lectin repeat. Green spheres represent coordinated calcium ions. ACAN variant colouring is the same as in (a).
When visualized on the three-dimensional structure of the aggrecan C-type lectin repeat (Protein Data Bank 1TDQ)22, the novel fOCD-linked variants affect amino acid residues in the hydrophobic core of the protein or residues coordinating calcium ions critical for structure and function of ligand binding loops on the protein surface (Fig. 4b). Additional, previously reported, missense variants where the proband shows OCD (p.W2362R, p.L2393P), also affect hydrophobic core residues8,15,17. The SEMD-linked variant (p.D2419N) affects one of the calcium-coordinating residues, replacing Asp with Asn, and thus forming a novel N-glycosylation site. Variants associated with short stature, but not OCD, appear more frequently at polar or charged amino acid residues exposed on the protein surface.
Secretion of the fOCD variant aggrecan proteins is decreased
To investigate effects on aggrecan function, we produced recombinant G3 domain proteins with the novel aggrecan variants, along with previously published C-type lectin repeat variants and the corresponding wild-type protein for comparison. To cover the range of domain sizes resulting from alternative splicing, we expressed the full domain (E1E2LCt, where E stands for EGF repeat, L for C-type Lectin, C for Complement regulatory protein-like/sushi repeat, and t for a short C-terminal tail sequence), the most common (LCt) and the shortest (Lt) splice variants in human 293-c18 cells. All variants were expressed (Fig. 5d–f), but as shown in Fig. 5a–c, secretion of the different fOCD variants to the culture medium was only efficient when the C-type lectin repeat was stabilized by flanking repeats (E1E2LCt). Interestingly, the p.L2393P variant protein behaved like the fOCD variants, even though OCD was only described in the proband for this variant. In contrast, the SEMD (p.D2419N) variant proteins were secreted even as the C-type lectin repeat alone (Fig. 5c). The slower mobility of the p.D2419N variant proteins result from the attachment of a carbohydrate chain to the novel N-linked glycosylation site formed by the mutation.

Secretion of recombinant aggrecan variant G3 domain protein. Expression plasmids for aggrecan SEMD and fOCD gene variants were transfected into human 293-c18 cells. Recombinant protein was recovered from culture medium (a–c) and cell lysates (d–f), and analyzed by immunoblotting. Each gene variant was tested in three different constructs corresponding to aggrecan G3 domain splice-forms; full-length E1E2LCt (a,d), C-type lectin and complement regulatory/sushi repeat LCt (b,e) and C-type lectin alone Lt (c,f). The domain arrangements of the G3 splice-forms tested are shown schematically at the bottom of the figure. As shown in (d,e), all gene variants in all splice-forms tested were produced. Secretion of variant full-length G3 domain E1E2LCt was somewhat decreased compared to wild-type (a), while secretion of fOCD variants as LCt protein was strongly decreased (b). No secretion of any fOCD variant Lt protein, i.e. the C-type lectin alone, was observed (c). Note that the SEMD variant p.D2419N protein was secreted in all splice-forms, although at lower levels than wild-type protein (a–c).
We next wanted to clarify if these effects on fOCD-linked variant recombinant G3 domain secretion could be biologically relevant, i.e. if the variants also could affect secretion of the full-length aggrecan proteoglycan with the additional globular (G1, G2) interglobular and glycosaminoglycan-carrying regions present in addition to the G3 domain. In a pilot experiment, we cultured cartilage explants from an fOCD patient heterozygous for the p.V2455M variant, obtained at arthroplasty for advanced osteoarthritis. Using mass spectrometry (MRM LC-MSMS) directed at variant specific peptides, we quantified the wild-type and the variant proteins in the tissue explants (Fig. 6a) and in the conditioned culture medium (Fig. 6b). Should the presence of the entire proteoglycan counteract the effects of the variant amino acid residue in the G3 domain, we would expect to detect equal amounts of wild-type and fOCD variant proteins, since the tissue donor was heterozygous for the fOCD variant gene. The variant protein was, however, only present at low levels, both in the tissue explant (Fig. 6a) and the culture medium (Fig. 6b). Some explants were also treated with interleukin-1 and oncostatin M to stimulate cartilage protein release23. This resulted in increased levels of both wild-type and variant aggrecan, and while the ratio of variant aggrecan relative to wild-type were increased, variant aggrecan levels remained considerably lower than wild-type aggrecan.

Decreased levels of fOCD variant aggrecan protein in patient cartilage. Cartilage tissue explants from a patient heterozygous for the fOCD-linked p.V2455M ACAN variant were obtained at arthroplasty. Some explants were treated with interleukin-1 and oncostatin M to stimulate protein production. After explant culture, the tissue explants were homogenized and extracted, and conditioned medium was collected. After GluC digestion, wild type and p.V2455M variant-specific peptides were quantified by mass spectrometry (MRM LC-MSMS) in the explant extracts (a) and medium (b). Note the decreased levels of p.V2455M variant compared to wild-type aggrecan proteoglycan in explant tissue (a) as well as in the culture medium (b).
The novel ACAN variants affect aggrecan protein interactions
The aggrecan C-type lectin repeat has strong affinities for several extracellular matrix ligands24,25,26,27. In earlier studies, decreased ligand binding affinities of the p.V2455M fOCD and p.D2419N SEMD variant C-type lectins were observed7,9. We now purified the recombinantly produced aggrecan G3 domains (E1E2LCt splice form) described above, and performed BIAcore surface plasmon resonance binding assays with the known cartilage ligands tenascin-C and fibulin-1, as well as the central nervous system-specific ligand tenascin-R (Fig. 7). As shown earlier, the SEMD and p.V2455M fOCD variant proteins showed lower binding to tenascin-C and fibulin-1. Interestingly, the novel fOCD variants and the OCD variant p.L2393P all showed even further reduced binding. Interaction with the brain ligand tenascin-R was affected to a lesser degree, with retained binding of all the variants tested.

Impaired interactions of fOCD variant aggrecan G3 domains. Biacore surface plasmon resonance sensorgrams showing binding and dissociation of recombinant aggrecan G3 domains to immobilized cartilage ligands fibulin-1 (a) and tenascin-C (b) and the central nervous system ligand tenascin-R (c). Recombinant aggrecan G3 variants (E1E2LCt splice-form) injected at 50, 100 or 200 nM over the immobilised ligand surfaces. Duplicate injections of each concentration are shown in the graphs, with the start and end of injections marked by arrows under each curve. Note pronounced loss of binding of fOCD variants to fibulin-1 and tenascin-C (a,b). The SEMD-ACAN variant p.D2419N shows less affected binding, whereas as an ACAN variant linked to idiopathic short stature with OCD in the proband (p.L2393P) shows binding similar to the fOCD variants. Interaction with the central nervous system ligand tenascin-R (c) is less affected than the cartilage ligands, with residual binding of all aggrecan variants.
Discussion
The aggrecan proteoglycan is an extracellular matrix component fundamental for the structure and function of all cartilages, including both in the growth plate during endochondral ossification and in adult articular cartilage. Disrupted aggrecan expression or function can thus cause developmental as well as adult phenotypes, or a combination of these. Growth plate phenotypes are evident in different hereditary disorders due to ACAN gene variants, with a phenotypic spectrum ranging from mild idiopathic short stature to severe chondrodysplasia. Variants affecting articular cartilage function appear less common, but variants linked to hereditary OCD or early onset osteoarthritis (in addition to short stature) may reflect different etiologies than variants affecting skeletal growth alone.
Most ACAN variants linked to growth plate phenotypes are autosomal dominant inherited nonsense or frameshift mutations and may affect any domain of aggrecan. These variants cause premature termination of translation and likely nonsense-mediated decay of the transcript, and result in decreased levels of aggrecan proteoglycan. Naturally, the decreased aggrecan levels caused by these variants may also affect articular cartilage function.
An interesting exception is the recessively inherited SEMD-linked missense mutation p.D2419N, which causes severe chondrodysplasia when present on both alleles7. This may reflect the combined reduction in aggrecan level resulting from both alleles being mutated, since individuals heterozygous for the variant had mild, proportionate short stature, with height around 150 cm7. Indeed, another case of SEMD was shown to result from compound heterozygosity for two different ACAN missense variants in the extended chondroitin sulfate chain attachment region28. The considerably milder phenotype of the compound heterozygous patient, however, suggests that effects on the G3 domain is an important component in the p.D2419N SEMD pathogenesis.
In contrast to nonsense and frameshift variants, the severity of missense mutations is difficult to predict and these are often classified as variants of unknown significance (VUS). In ACAN, missense mutations appear concentrated to the exons coding for folded, globular domains of the aggrecan core protein (Table 3). This may simply reflect that a missense mutation in any of the glycosaminoglycan attachment sites of the non-folded parts of the core protein would only result in the loss of one of approximately a hundred glycosaminoglycan chains. This marginal effect on aggrecan function would likely not result in an overt phenotype.
The aggrecan globular domains, on the other hand, mediate interactions important for cartilage extracellular matrix assembly and organization. The fOCD-linked missense variants described in this paper obliterate interactions with known cartilage extracellular matrix ligands, as was shown earlier for the fOCD-linked p.V2455M variant9. Interestingly, the residues mutated in the fOCD-linked variants or the p.L2393P and p.W2263R variants (where the probands showed OCD) are not exposed on the surface of the folded protein (Fig. 4b). Indeed, neither of the mutated residues are part of the intermolecular binding surface of the aggrecan C-type lectin and tenascin-R22, suggesting that the loss of ligand interactions result from misfolding of the domain. Supporting this, we found that the secretion of fOCD variant proteins is decreased, both in cell culture models and in fOCD patient cartilage. This may be related to ER-stress and an unfolded protein response, as suggested by experiments on chondrocytes differentiated from mesenchymal stromal cells or induced pluripotent stem cells derived from a p.V2455M fOCD patient29. It seems reasonable to assume that the reduced aggrecan production contributes to the short stature due to effects in the growth plate. The impaired extracellular matrix interactions of fOCD variant aggrecan proteoglycans may contribute to poorer function or stability of cartilage tissue by affecting the molecular organization of the extracellular matrix. In addition, variant aggrecan proteolytic fragments lacking the G1 domain, and thus not anchored to hyaluronan, may be lost from the articular cartilage by diffusion to the joint space due to their failed G3 domain interactions, further decreasing aggrecan content of the tissue.
The novel ACAN variants described in this study were identified from families with hereditary OCD. Like the previously reported fOCD-linked variant p.V2455M9, the novel variants all segregate with OCD and idiopathic short stature, and at least for two of the variants with early onset osteoarthritis. In other studies, a further ten missense variants in the G3 domain have been linked to idiopathic short stature (Fig. 4)8,13,15,17,18,30,31,32,33. The probands of two of these families presented with OCD, but no heredity for OCD was reported for these families8,15,17. Thus, both fOCD and OCD variants are over-represented in the aggrecan G3 domain (Table 3). While the latter could simply reflect the normal prevalence of sporadic OCD, further OCD cases may yet appear in these families. Indeed, this is supported by the similarities in decreased secretion (Fig. 5) and impaired ligand interactions (Fig. 7) between the p.L2393P idiopathic short stature variant (with proband only showing OCD) and the fOCD variants. It is interesting to note that both these non-familial OCD missense variants (p.L2393P and p.W2362R) affect hydrophobic amino acid residues buried in the core of the folded domain, like the familial OCD variants p.C2386S, p.L2417P and p.V2455M (Fig. 4b). Based on this, we speculate that fOCD results from such variants disrupting the folding of the C-type lectin repeat in the aggrecan G3 domain, whereas variants affecting charged or polar residues exposed on the protein surface may be less detrimental.
Taken together, the fOCD and OCD linked to ACAN missense variants investigated in this study may constitute a subgroup of the SSOAOD classification. This may be considered as phenotypic variations of the same disorder or possibly reflect a different pathogenetic mechanism, although this would require further study. At present, the fOCD-linked missense variants, along with short stature and osteoarthritis-linked aggrecan variants have been classified together into one group (OMIM 165800, Short stature and advanced bone age, with or without early-onset osteoarthritis and/or osteochondritis dissecans). Previously, this group was termed “short stature, osteoarthritis and osteochondritis dissecans, SSOAOD”34,35. In the latest nosology of genetic skeletal disorders, this group is referred to as “short stature and advanced bone age, sometimes with osteochondritis dissecans”36. However, the inclusion of advanced bone age as a group criterion may have been premature, since later studies have shown that reduced or unchanged bone age frequently occur in idiopathic short stature associated with ACAN variants9,11,15,16,17,20,21,26,28,30,31.
To our knowledge, no other gene than ACAN has been linked to hereditary OCD. Interestingly, an additional fOCD missense ACAN variant has been reported affecting the aggrecan G1 globular domain8,13. This suggests that the aggrecan globular domains, and perhaps the G3 domain in particular, may be key to understanding the etiology of hereditary OCD and possibly also give insight into mechanisms behind sporadic OCD.
Methods
Ethical approval
All participants or their legal guardians gave informed consent for research and publication.
The study was first approved by the Regional Ethical Review Board at Umeå University, Sweden (Dnr 01-244 and 07-109M), and later by the Regional Ethical Review Board at Uppsala University, Sweden (Dnr 2018/189), and all research was performed in accordance with the relevant guidelines and regulations.
Molecular genetic analysis
Genomic DNA from two index patients (family 1 and 2) were analyzed with an ABI Prism 3170 DNA sequencer at the Department of Clinical Genetics, Umeå University Hospital Umeå, Sweden. Bidirectional sequencing of exons and intron–exon boundaries of the ACAN gene (NM_00135.2, NM_013227.2) were performed as previously described9. The variants in ACAN were validated using Sanger sequencing on DNA from the index patient, and family members. A sample from a third index patient (family 3) was analyzed with NGS at Fulgent Diagnostics, Temple City, USA. Sanger sequencing was performed in family members for the variant identified in the index patient.
Recombinant protein expression vector construction
An expression plasmid for recombinant wild-type aggrecan G3 domain fragments was constructed by inserting the cDNA into plasmid pCEP4-TAGzyme37, in frame and downstream of a BM40 signal peptide sequence followed by a hexa-histidine affinity tag.
Expression plasmids for aggrecan sequence variants were constructed through site-directed mutagenesis of the wild-type aggrecan expression plasmid, using the QuikChange mutagenesis system (Stratagene). The correct sequence of all constructs was verified by DNA sequencing of the entire inserts.
Aggrecan variant recombinant protein secretion in cell culture
Human 293-c18 cells (ATCC, CRL-10852) in monolayer culture were transfected with plasmids expressing wild-type, SEMD-linked (p.D2419N) or different fOCD- or OCD-linked (p.V2455M, p.C2386S, p.L2393P, p.L2417P or pD2453V) aggrecan variants, using FuGene6 (Roche). After 48 h, the culture medium was collected and the cells harvested by trypsination, resuspended in PBS with soy bean trypsin inhibitor, washed by centrifugation and resuspended in PBS. The cell pellet was finally resuspended in 150 μl PBS with 2 mM MgCl2 and protease inhibitors (Complete w/o EDTA, Roche) and lysed with 20 U/μl Benzonase (Sigma) for 2 h at 37 °C. His-tagged proteins were enriched from conditioned media using NiNTA-coupled magnetic Dynabead precipitation (Invitrogen). The media and cell lysate proteins were separated by 4–20% SDS-PAGE gels (Novex), transferred onto PVDF membranes and probed with a rabbit antiserum directed to the C-type lectin repeat of the aggrecan G3 domain26, using chemiluminescent detection and documentation (Bio-Rad ChemiDoc MP). The images were processed by linear contrast enhancement, exported in TIF format, and the TIF files assembled to a figure and annotated using Affinity Design 1.7 software. The uncropped image files are shown in Supplemental Fig. S2.
Aggrecan variant protein levels in FOCD patient cartilage
Knee cartilage from the tibial plateau was obtained at total knee replacement surgery, due to advanced osteoarthritis, from a 47 years old male patient heterozygous for the fOCD-linked p.V2455M aggrecan variant, with informed consent. Macroscopically normal cartilage was dissected and cultured as explants (in DMEM with 100ug/ml BSA, 50 ug/ml ascorbate, Penicillin/Streptomycin, Fungizone medium), with or without 10 ng/ml oncostatin M + 1 ng/ml Il-1 addition, as previously described38. Conditioned medium was collected and frozen at − 80 °C after 2 days in culture. The cartilage explant was snap-frozen in liquid nitrogen, pulverized and extracted with guanidine hydrochloride, as previously described39. After GluC digestion of the medium and cartilage extracts, the wild-type (DCVVMIWHE) and p.V2455M variant (DCMVMIWHE) peptides were quantified by multiple reaction monitoring (MRM) mass spectrometry using the top3 transitions (q1/q3) for each peptide (wild-type: (y7) = 594.76++/913.46+, (y6) = 594.76++/814.39+, (b3) = 594.76++/375.13+ and for V2455M: (y6) = 610.75++/814.39+, (y5) = 610.75++/715.32+ , (b3) = 610.75++/407.11+). The sample preparation and separation conditions were as previously described40.
Recombinant protein production
Recombinant proteins were produced using the 293 Freestyle mammalian expression system (Invitrogen). Briefly, the expression plasmids were transiently transfected into human 293F cells using polyethylene imide “Max” (linear, MW 25 000, Polysciences Inc.)41, grown in suspension culture in Freestyle 293 medium in 8% CO2 atmosphere. The medium was harvested 7 days after transfection and recombinant proteins purified through nickel chelation affinity chromatography, followed by MonoQ ion exchange chromatography and gel filtration on a Superdex S200 Increase column using an ÄKTA chromatography system (Pharmacia, Uppsala, Sweden). The final gel filtration step was done with BIAcore running buffer (10 mM HEPES-HCl, 150 mM NaCl, 2 mM CaCl2, and 0.005% P20 [pH 7.5]) as eluent9.
Protein interaction analysis
Interaction screening of the recombinant aggrecan variants were done by surface plasmon resonance, as previously described9. Briefly, recombinant fibulin-1, tenascin-C (fibronectin type III repeats 3–5) or tenascin-R (fibronectin type III repeats 3–5) were immobilized in CM5 flow cells to 811, 662 and 233 Resonance Units (RU), respectively. Recombinant aggrecan variant G3 domains (E1E2LCt), at 50, 100 or 200 nM in Biacore running buffer (see above), were flowed at 50 µl/min over these surfaces, and binding and dissociation followed in a BIAcore 2000 instrument (BIAcore, Uppsala, Sweden), and data analyzed with BIAevaluation 4.1 software.
References
Roughley, P. J. & Mort, J. S. The role of aggrecan in normal and osteoarthritic cartilage. J. Exp. Orthop. 1(1), 8 (2014).
Aspberg, A. The different roles of aggrecan interaction domains. J. Histochem. Cytochem. 60(12), 987–996 (2012).
Watanabe, H. et al. Mouse cartilage matrix deficiency (cmd) caused by a 7 bp deletion in the aggrecan gene. Nat. Genet. 7(2), 154–157 (1994).
Cavanagh, J. A. et al. Bulldog dwarfism in Dexter cattle is caused by mutations in ACAN. Mamm. Genome 18(11), 808–814 (2007).
Li, H., Schwartz, N. B. & Vertel, B. M. cDNA cloning of chick cartilage chondroitin sulfate (aggrecan) core protein and identification of a stop codon in the aggrecan gene associated with the chondrodystrophy, nanomelia. J. Biol. Chem. 268(31), 23504–23511 (1993).
Gleghorn, L. et al. A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am. J. Hum. Genet. 77(3), 484–490 (2005).
Tompson, S. W. et al. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am. J. Hum. Genet. 84(1), 72–79 (2009).
Gkourogianni, A. et al. Clinical characterization of patients with autosomal dominant short stature due to aggrecan mutations. J. Clin. Endocrinol. Metab. 102(2), 460–469 (2017).
Stattin, E. L. et al. A missense mutation in the aggrecan C-type lectin domain disrupts extracellular matrix interactions and causes dominant familial osteochondritis dissecans. Am. J. Hum. Genet. 86(2), 126–137 (2010).
Stattin, E. L. et al. Familial osteochondritis dissecans associated with early osteoarthritis and disproportionate short stature. Osteoarthr. Cartil. 16(8), 890–896 (2008).
Linden, B. The incidence of osteochondritis dissecans in the condyles of the femur. Acta Orthop. Scand. 47(6), 664–667 (1976).
Bruns, J., Werner, M. & Habermann, C. Osteochondritis dissecans: Etiology, pathology, and imaging with a special focus on the knee joint. Cartilage 9(4), 346–362 (2018).
Sentchordi-Montane, L. et al. Heterozygous aggrecan variants are associated with short stature and brachydactyly: Description of 16 probands and a review of the literature. Clin. Endocrinol. 88(6), 820–829 (2018).
Mancioppi, V. et al. Retrospective diagnosis of a novel ACAN pathogenic variant in a family with short stature: A case report and review of the literature. Front. Genet. 12, 708864 (2021).
Florio, A. et al. A child with a novel ACAN missense variant mimicking a septic arthritis. Ital. J. Pediatr. 45(1), 148 (2019).
Hauer, N. N. et al. Genetic screening confirms heterozygous mutations in ACAN as a major cause of idiopathic short stature. Sci. Rep. 7(1), 12225 (2017).
Nilsson, O. et al. Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. J. Clin. Endocrinol. Metab. 99(8), E1510–E1518 (2014).
Stavber, L. et al. High frequency of pathogenic ACAN variants including an intragenic deletion in selected individuals with short stature. Eur. J. Endocrinol. 182(3), 243–253 (2020).
van der Steen, M. et al. ACAN gene mutations in short children born SGA and response to growth hormone treatment. J. Clin. Endocrinol. Metab. 102(5), 1458–1467 (2017).
Denis, A. et al. Variable expressivity in a family with an aggrecanopathy. Mol Genet. Genomic Med. 10, e1773 (2022).
Ozere, S. et al. The link between aggrecan and familial osteochondritis dissecans. Surgeries 2(2), 128–138 (2021).
Lundell, A. et al. Structural basis for interactions between tenascins and lectican C-type lectin domains; evidence for a crosslinking role for tenascins. Structure 12(8), 1495–1506 (2004).
Barksby, H. E. et al. Interleukin-1 in combination with oncostatin M up-regulates multiple genes in chondrocytes: Implications for cartilage destruction and repair. Arthritis Rheum. 54(2), 540–550 (2006).
Aspberg, A. et al. The C-type lectin domains of lecticans, a family of aggregating chondroitin sulfate proteoglycans, bind tenascin-R by protein-protein interactions independent of carbohydrate moiety. Proc. Natl. Acad. Sci. USA 94(19), 10116–10121 (1997).
Aspberg, A. et al. Fibulin-1 is a ligand for the C-type lectin domains of aggrecan and versican. J. Biol. Chem. 274(29), 20444–20449 (1999).
Olin, A. I. et al. The proteoglycans aggrecan and Versican form networks with fibulin-2 through their lectin domain binding. J. Biol. Chem. 276(2), 1253–1261 (2001).
Day, J. M. et al. Alternative splicing in the aggrecan G3 domain influences binding interactions with tenascin-C and other extracellular matrix proteins. J. Biol. Chem. 279(13), 12511–12518 (2004).
Fukuhara, Y. et al. The second report on spondyloepimetaphyseal dysplasia, aggrecan type: A milder phenotype than originally reported. Clin. Dysmorphol. 28(1), 26–29 (2019).
Xu, M. et al. Chondrocytes derived from mesenchymal stromal cells and induced pluripotent cells of patients with familial osteochondritis dissecans exhibit an ER stress response and defective matrix assembly. Stem Cells Transl. Med. 5(9), 1171–1181 (2016).
Benonisdottir, S. et al. Epigenetic and genetic components of height regulation. Nat. Commun. 7, 13490 (2016).
Liang, H. et al. Growth-promoting therapies may be useful in short stature patients with nonspecific skeletal abnormalities caused by acan heterozygous mutations: Six Chinese cases and literature review. Endocr. Pract. 26(11), 1255–1268 (2020).
Lin, L. et al. A high proportion of novel ACAN mutations and their prevalence in a large cohort of chinese short stature children. J. Clin. Endocrinol. Metab. 106(7), e2711–e2719 (2021).
Xu, D. et al. Novel aggrecan variant, p. Gln2364Pro, causes severe familial nonsyndromic adult short stature and poor growth hormone response in Chinese children. BMC Med. Genet. 19(1), 79 (2018).
Bonafe, L. et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am. J. Med. Genet. A 167a(12), 2869–2892 (2015).
Warman, M. L. et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A 155A(5), 943–968 (2011).
Mortier, G. R. et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. A 179(12), 2393–2419 (2019).
Geng, H. et al. Cartilage oligomeric matrix protein specific antibodies are pathogenic. Arthritis Res. Ther. 14(4), R191 (2012).
Elliott, S. et al. Esculetin inhibits cartilage resorption induced by interleukin 1alpha in combination with oncostatin M. Ann. Rheum. Dis. 60(2), 158–165 (2001).
Wang, Y. et al. Quantitative proteomics analysis of cartilage response to mechanical injury and cytokine treatment. Matrix Biol. 63, 11–22 (2017).
Müller, C. et al. Quantitative proteomics at different depths in human articular cartilage reveals unique patterns of protein distribution. Matrix Biol. 40, 34–45 (2014).
Longo, P. A. et al. Transient mammalian cell transfection with polyethylenimine (PEI). Methods Enzymol. 529, 227–240 (2013).
Acknowledgements
We sincerely thank all the patients and family members involved in this study for their participation. We also thank Susann Haraldsson, Department of Medical Biosciences, Umeå University, Umeå, for expert technical assistance.
This study was supported by the Olle Engkvist Foundation, the Crafoord, Anna-Greta Crafoord, Alfred Österlund, Greta and Johan Kock, Gyllenstiernska Krapperups and Swedish Rheumatism Association Foundations, Uppsala University Hospital, and Uppsala University. Mass spectrometers were generously funded by grants from the IngaBritt and Arne Lundberg Foundation.
Funding
Open access funding provided by Lund University.
Author information
Authors and Affiliations
Contributions
A.A. and E.L.S. designed the study, with the help of J.G., A.D., J.D. and E.C. J.G., A.D., J.D. and E.C. recruited patients, performed clinical examinations and collected samples for sequencing with the help of A.Sa., T.R., M.S., C.R. E.L.S. conducted DNA sequencing and performed genetic analysis with the help of E.C. K.L., P.Ö. and A.St. performed experiments with the help of A.A. A.A. and E.L.S. drafted the manuscript with critical revision from P.Ö., A.St., J.G., A.D., J.D. and E.C. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stattin, EL., Lindblom, K., Struglics, A. et al. Novel missense ACAN gene variants linked to familial osteochondritis dissecans cluster in the C-terminal globular domain of aggrecan. Sci Rep 12, 5215 (2022). https://doi.org/10.1038/s41598-022-09211-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-09211-y
This article is cited by
-
Bilateral juvenile osteochondrosis dissecans in monozygotic twins: a case report
Journal of Orthopaedic Surgery and Research (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
