
Abstract
The western tarnished plant bug, Lygus hesperus, is a key hemipteran pest of numerous agricultural, horticultural, and industrial crops in the western United States and Mexico. A lack of genetic tools in L. hesperus hinders progress in functional genomics and in developing innovative pest control methods such as gene drive. Here, using RNA interference (RNAi) against cardinal (LhCd), cinnabar (LhCn), and white (LhW), we showed that knockdown of LhW was lethal to developing embryos, while knockdown of LhCd or LhCn produced bright red eye phenotypes, in contrast to wild-type brown eyes. We further used CRISPR/Cas9 (clustered regularly interspaced palindromic repeats/CRISPR-associated) genome editing to generate germline knockouts of both LhCd (Card) and LhCn (Cinn), producing separate strains of L. hesperus characterized by mutant eye phenotypes. Although the cardinal knockout strain Card exhibited a gradual darkening of the eyes to brown typical of the wild-type line later in nymphal development, we observed bright red eyes throughout all life stages in the cinnabar knockout strain Cinn, making it a viable marker for tracking gene editing in L. hesperus. These results provide evidence that CRISPR/Cas9 gene editing functions in L. hesperus and that eye pigmentation genes are useful for tracking the successful genetic manipulation of this insect.
Similar content being viewed by others


Introduction
The western tarnished plant bug, Lygus hesperus Knight (Hemiptera: Miridae) is a major pest of cotton and other crops throughout the western United States and other parts of North America1,2,3. Although an integrated pest management program has been implemented against L. hesperus in Arizona3, its success is dependent on the continued effectiveness of only a few insecticides which have been widely used for many years4. With the rapid evolution of insecticide resistance observed in the closely related Lygus lineolaris5,6,7, new tactics are needed to maintain control over members of this genus.
Among contemporary strategies for controlling arthropod pest species, homing-based gene drives are currently being developed for management of agricultural pests as well as those that vector human disease8,9,10,11,12,13. Clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9 (CRISPR/Cas9) gene drive systems have been developed and shown to effectively drive deleterious genes into laboratory insects, resulting in population crashes11,13,14. CRISPR-based gene drive systems developed in the laboratory include homing, split homing, translocation, X-shredder, killer-rescue, cleave-and-rescue, and TARE (reviewed in15).
CRISPR/Cas9 gene editing is also widely used to ascertain gene function due to its efficiency and specificity in inducing mutations by cleavage and impairment of the genomic target sequences in model and non-model organisms16,17. In insects, genes that control eye pigmentation are frequently targeted because many of the induced mutations produce striking visible changes that facilitate screening for knockout efficacy. A frequent target is White, an ABC transporter that functions in transporting pigment precursor into pigment granules18. For example, CRISPR editing of white produced white-eyed adults in both Helicoverpa armigera19 and Bactrocera dorsalis20, although homozygous mutations were lethal in the former and unexpectedly resulted in the loss of black head spots in the latter. In hemipterans, CRISPR-mediated null mutation of white results in white eyes in nymphs and bright red eyes in adults of Bemisia tabaci21 and lighter red eyes and white ocelli in Nilaparvata lugens22.
Insect eye pigments are primarily from the guanine-derived pteridines and/or the tryptophan-derived ommochromes. Genes involved in the ommochrome pathway of pigment transport and formation are well studied in several model species, including Drosophila melanogaster, Bombyx mori and Tribolium castaneum23,24,25 and typically follow the schematic shown in Fig. 1. Null mutations in the enzymes involved in the step-by-step process of converting tryptophan into ommochromes (Vermilion, Cinnabar, and Cardinal) within this pathway often produce distinct eye color phenotypes and serve as visible markers for detecting successful gene manipulation. In Helicoverpa zea, mutant yellow eyes were observed after CRISPR-mediated knockout of vermilion, tryptophan 2,3-dioxygenase26. Knockouts of kynurenine monooxygenase (cinnabar) in Plutella xylostella manifested in yellow or red eyes depending on the mutation, while knockout of the haem peroxidase gene, cardinal, produced yellow eyes that gradually changed to red27. In N. lugens, knockout of cinnabar generated a red eye phenotype22. Such studies demonstrate that genes in the ommochrome pathway like vermilion, cinnabar, and cardinal can serve as targets that give discernable phenotypes for tracking stable germ line gene edits across multiple generations.

The insect ommochrome eye pigmentation pathway. Genes involved are karmoisin, vermilion, cinnabar, scarlet, white, and cardinal. Genes targeted for gene function in L. hesperus are italicized.
Current knowledge of eye pigmentation in Hemiptera is limited to just a few species and has been produced primarily through genetic manipulation of white, cinnabar, and cardinal20,24,28,29,30. In L. lineolaris, a mutant red eye phenotype occurs naturally under field and laboratory conditions6,31,32, although the genetic basis of this phenotype is unknown. To reproduce such mutants in L. hesperus, we previously used RNAi to knockdown genes in the ommochrome pathway. In that study, late 5th instar nymphs were injected with dsRNA and eye color development was tracked through adult maturation seven days post-eclosion28. However, RNAi only produced red pigment along the margins of the eyes which otherwise looked wild-type. We concluded that the incompleteness of the transformation to a fully red eye was the result of the transient knockdown of the genes and the accrual of pigments during nymphal development28. Thus, an induced null mutation in genes involved in either the synthesis or transport of ommochromes may lead to a more pronounced adult phenotype.
Here, we aim to demonstrate that CRISPR/Cas9 can be applied to L. hesperus by targeting genes within the ommochrome pathway. We show that CRISPR/Cas9-induced mutations in cardinal (LhCd) and cinnabar (LhCn) are heritable and that stable lines (named Card and Cinn, respectively) with obvious mutant eye color phenotypes can be established for both genes. Although the Card strain showed pronounced red eyes early in development, coloration eventually reverted to wild-type in late nymphs and adults, suggesting that other genes are involved in driving eye pigmentation in the later stages of L. hesperus development. In contrast, we observed stable red eyes in the Cinn strain, suggesting that LhCn is critical for eye pigmentation throughout development and can potentially be used to track transformation and gene drive experiments that could ultimately lead to alternative L. hesperus control measures.
Results
Knockdown of cardinal, cinnabar, and white in embryos
Using embryonic RNAi, we were able to pre-determine the effects of CRISPR/Cas9-mediated mutations on eye color. In our previous work, RNAi knockdown of LhCd or LhCn in late 5th instars resulted in mature adult eyes that had a bright red band along the medial margins, whereas white (LhW) knockdown led to a high proportion of incomplete adult molts and a commensurate increase in mortality28. Here, injecting dsRNAs targeting either LhCd or LhCn into ~ 1 h old L. hesperus eggs produced in embryos medium red or bright red eyes, respectively (Fig. 2a). These phenotypes were readily apparent 5 days post-injection and differed from brown wild-type eyes. In total, the observed phenotypes were present in 21/120 eggs injected with LhCd dsRNA and 28/120 eggs for the LhCn dsRNA treated group. Among the remaining eggs injected with either LhCd or LhCn dsRNA, 30–50% were either dead or had wild-type eyes.

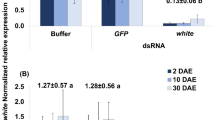
RNA interference knockdown of embryonic eye pigment genes. (a) Lygus hesperus embryos at five days post-injection (dpi) with dsRNA for venus, LhCd, LhCn, or LhW. Embryos injected with dsRNA corresponding to LhW were lethal and resulted in no accumulation of eye pigmentation. Inset shows 3 × magnification of the eyes. Arrowhead and arrow indicate the undeveloped and unorganized but developed parts of the LhW dsRNA injected egg, respectively. (b) Semi-quantification of knockdown in LhCd, LhCn, and LhW transcripts. Gel images of transcript knockdown are representative of four technical replicates and three biological replicates. No template (NT).
Like the post-eclosion lethality previously observed following LhW knockdown in 5th instar nymphs28, embryonic injection of LhW dsRNA resulted in nonviable eggs characterized by incomplete and/or unorganized embryonic development at 5 days post-injection (Fig. 2a). This phenotype, which is distinguishable from the complete lack of development observed when physical trauma (i.e., injection) induces egg mortality, was observed in 53/120 eggs (Fig. 2a). Knockdown of LhCd, LhCn, and LhW transcripts was confirmed by semi-quantitative PCR (Fig. 2b, Supplementary Fig. S1).
CRISPR/Cas9-mediated knockout of LhCd or LhCn
Given the phenotypes generated in the embryonic RNAi experiments, we next sought to assess the viability of embryonic CRISPR/Cas9 knockouts singly targeting LhCd and LhCn. For both transcripts, the sequences corresponding to the 20-nucleotide guide and the protospacer adjacent motif were searched via BLASTn against L. hesperus publicly available data (NCBI organism limit—L. hesperus; taxid 30085). No matches were found that would suggest potential off-target effects (Supplementary Table S1).
The CRISPR/Cas9 knockouts were conducted via injections in two independent experiments. Overall, we achieved a hatch rate between 8.8 and 27.5% for injected individuals compared to 86.3–100% for the noninjected control group (Supplementary Table S2). The survival of the instar nymphs to adults ranged from 22.7 to 71.4% for all Cas9-ribonucleoprotein complex (RNP) injected groups (Supplementary Table S2). Of these RNP-injected adults, 40–100% showed mutant eye phenotypes (Supplementary Table S2).
To generate L. hesperus strains with the mutant eye phenotypes, we crossed surviving G0 adults from both the LhCd- and LhCn-injected lines from experiment 1 according to the scheme depicted in Fig. 3. We estimate that the efficiency of CRISPR/Cas9 gene-editing of the germline was 86% (± 9.6%), as determined by averaging the efficiency of all samples from the Card and Cinn strains (Supplementary Methods and Table S3). Individuals from the Card strain showed a gradual darkening of the eye beginning at the 3rd instar that continued with subsequent molts and during adult maturation (Fig. 5a). Eye coloration in fully mature adult Card females closely resembled those from wild-type, while Card males displayed red eye phenotypes that were brighter than those found in the wild-type strain (Fig. 5b). In contrast, the eye phenotype across all stages of Cinn strain development was characterized by complete bright red pigmentation regardless of sex and stage (Fig. 5).

Crossing schemes used to generate CRISPR/Cas9 Lygus hesperus mutant eye pigmentation strains. (a) The Card strain was initiated from six G0 survivors (two females and four males). Both mutant G0 females were crossed with a mutant male (Card × 1 and × 2) and the remaining mutant males were crossed with four wild-type females (Card × 3–5). G1 offspring of Card × 1 and × 2 were inbred to generate G2. G1 heterozygous offspring from Card × 3–5 were inbred to generate a mix of wild-type and red-eye G2 progeny. The selected G2 (boxed) with red eyes from all Card crosses were pooled to establish the Card colony. Cross numbers are indicated by an “x” and the respective number. (b) Two G0 female from Cinn were crossed with wild-type males generating heterozygous G1 progeny that were subsequently inbred to produce G2. Red-eye mutants (boxed) in G2 were crossed and perpetuated to establish the Cinn colony. L. hesperus models with black or red eyes represent wild type or mutant, respectively, in (a) and (b).
Target site mutations in LhCd and LhCn mutant strains
Both gDNA and cDNA corresponding to the gRNA target sites in G0-G3 individuals from the Card and Cinn strains showed mutations (Table 1). For LhCd, we found a total of 43 mutations with 20 corresponding to LhCd1 sgRNA1 and 23 for LhCd2 sgRNA2 (Table 1, Fig. 4). For LhCn, we found 12 and 13 mutations corresponding to LhCn1 sgRNA1 and LhCn2 sgRNA2, respectively (Table 1, Fig. 4). Of these mutations, 25 unique combinations were found for LhCd and 14 for LhCn. Names of the mutations are based on the unique combination of mutations that were found within each individual (Table 1). With the only exception of mutation Cd1.7, in which LhCd showed a wild-type allele at LhCd1 target site, all targeted sites displayed more than one mutation. Four LhCd mutations (Cd1.1-Cd1.4) and two LhCn mutations (Cn1.1 and Cn1.2) were found to occur in gDNA and/or cDNA across generations (Table 1). Cd1.1 was the most common LhCd mutation, as it was found in a single G0 individual and in two G3 individuals from the Card strain (Table 1, Fig. 4). For the Cinn strain, two individuals, including one G0 and one G2, both harbored the Cn1.1 mutation (Table 1, Fig. 4). There were 10 mutations in Card and 5 mutations in Cinn that were in-frame, whereas 14 and 9 mutations in Card and Cinn, respectively generated premature stop codons. Overall, the combination of mutations at the sgRNA1 and 2 target sites resulted in mutant eye color phenotypes regardless of the in-frame effect.

Intergenerational LhCd and LhCn mutations in Lygus hesperus CRISPR/Cas9 mutant strains. Multiple sequence DNA alignments of intergenerational LhCd (a) and LhCn (b) mutations with the corresponding wild-type (Wt) sequences. Aligned regions of LhCd and LhCn only include sequences near the sgRNA target sites (LhCd1-2 and LhCn1-2). A variant allele within the wild-type sequences (Wtv) is shown. Horizontal lines indicate the sgRNA sequences, boxes indicate the PAM sequence trinucleotides, arrowheads indicate the predicted Cas9 cut sites, blue text indicates allele variants, red text denotes substitutions or insertions, and (–) corresponds to a sequence deletion.
Discussion
Successful use of CRISPR/Cas9 in L. hesperus provides a first step towards the use of contemporary molecular control strategies against this pest species, as well as in other related species. The results here show that CRISPR editing of two L. hesperus genes (LhCd and LhCn) resulted in heritable mutations that affected eye pigmentation across nymphal and adult development. RNAi knockdown of L. hesperus genes involved in eye pigment transport in late 5th instars produced adults exhibiting primarily wild-type eyes apart from a red line extending from the rostrum to the antenna along the medial margins28. The extent of the change varied by individual and was often difficult to distinguish without close examination. It is evident that eye pigmentation in L. hesperus is a continuous process throughout the entire course of development (Fig. 5), which can only be marginally impacted by transient knockdown from RNAi. This attribute led us to attempt CRISPR/Cas9 knockout of the same genes previously targeted to produce a more pronounced and lasting change in phenotype. Our strongest result was with LhCn; CRISPR/Cas9-mediated knockout of this gene yielded persistent bright red eyes that were strikingly different from the typical brown coloration in wild-type (Fig. 5). Sequencing of the sgRNA target sites showed multiallelic mutations that confirmed LhCn was indeed knocked out. Although the final eye color appears to be species dependent, the bright red eye phenotype in the Cinn strain is consistent with Cinnabar functioning in ommochrome transport/biosynthesis, as has been found in knockdown or knockout studies in N. lugens22, Nasonia vitripennis33,34, Aedes aegypti35, T. castaneum36, and B. mori37. It is, however, possible that LhCn has other biological roles in L. hesperus. The homologous Cn gene in D. melanogaster modulates post-translational regulation of the mitochondrial fission gene Drp1, such that disruption of Cn activity negatively impacts mitochondrial morphology and function38. However, we have not observed any changes in L. hesperus beyond eye color in this or our previous study of LhCn28.

Eye pigmentation phenotypes for the CRISPR/Cas9 Lygus hesperus mutant strains. (a) Egg (E) and nymphal instars (1st, 2nd, 3rd, 4th, and 5th) corresponding to representative wild-type, Card, and Cinn insects. Insets show 2.5 × magnification of the 3rd instar eye, which underscores gradual reversion to wild-type coloration in the Card line. (b) Ventral and lateral views of female and male eyes at 1-day and 7-days post-emergence.
Knocking down cardinal, another gene in the ommochrome pathway, also impacts eye color in L. hesperus. Manipulation of LhCd expression in late 5th instars had a moderate impact on eye color28, similar to that observed for N. lugens in which RNAi yielded eyes with a mixture of red and brown pigment29. Unlike the LhCn results, LhCd knockout mutant eyes appeared to gradually accumulate brown pigment with each successive developmental stage after the 3rd instar (Fig. 5). Although adult eyes in the LhCd mutants were redder than that of wild-type individuals, they were substantially darker than LhCn mutant eyes. Gradual changes to the eye color have also been found in adults with crimson mutations in Culex pipiens39 and cardinal mutants in Plutella xylostella27. Cardinal functions as the final step in ommochrome biosynthesis to produce xanthommatin. In the absence of a functional cardinal, the oxidative 3-hydroxy kynurenine can auto-dimerize to xanthommatin over time33,40,41. This may explain the gradual darkening in eye pigmentation that was reminiscent of the wild-type phenotype that we observed in L. hesperus.
The white gene functions in both pteridine and ommochrome transport18, where White dimerizes with either Brown or Scarlet to transport pigments into pigment granules. It is frequently used as a marker of insect genetic manipulation20,30,42,43,44. Although knockdown of LhW was frequently fatal in dsRNA-injected 5th instar L. hesperus nymphs, it did disrupt pigment accumulation in survivors28. Here, knockdown of LhW in embryos completely disrupted embryonic development prior to visible eye formation, we were thus unable to confirm if LhW is involved in L. hesperus eye pigmentation. Similar mortality induced by white knockout was observed in H. armigera19 and Oncopeltus fasciatus30. Given the pronounced effect of silencing white, LhW may be essential for embryogenesis in L. hesperus, and likely contributes to other key processes. Knockout of white in D. melanogaster produces retinal degeneration, shortened life span, and progressive loss of ability to climb45. The multitude of biological processes affected by white indicate that its substrate specificity is not limited to pigment precursors. White has been shown to be expressed in the nervous and excretory system of Drosophila and functions in the transport of important substrates like biogenic amines46 and cyclic guanosine monophosphate47, respectively. The exact functions of LhW remain to be elucidated, but are clearly crucial for normal development.
As is evident from the changes to L. hesperus eye color, our methods produced efficient and on-target gene editing that was inherited among increasing proportions of progeny for each new generation. Both Card and Cinn colonies have been reared beyond 10 generations with no changes in the mutant eye phenotypes, indicating that respective mutant lines can be stably generated under established laboratory rearing conditions without any observable negative impacts on fitness. Functional genomic studies can now be conducted on key genes, such as those regulating development, reproduction, and insecticide resistance. Use of relatively new molecular approaches, such as Receptor Mediated Ovarian Transduction of Cargo (ReMOT Control) and/or branched amphiphilic peptide capsules (BAPC), can also be applied to facilitate and accelerate functional genomic studies in L. hesperus. ReMOT Control uses an ovary specific-ligand to target the RNP cargo to developing oocytes by injecting the abdomens of mature adult females21,34,48,49,50,51. This technology has potential for generating mutant insects at a higher rate and reduced cost. In contrast, BAPC assisted-delivery of CRISPR/Cas9 into the developing oocyte by injecting the BAPC-RNP mixture near the ovaries may enable enhanced uptake and improve efficiency of editing48. The immediate next step in optimizing genetic engineering of L. hesperus is to use CRISPR/Cas9 to target genes that reduce fitness, alter sex differentiation, or induce mortality. The generation of conditional gene-drive systems in other model insects exemplifies the feasibility of driving traits with associated negative phenotypes into populations8,11,52. Future work in L. hesperus will continue to focus on expanding our understanding of basic functional genomics as it relates to pest biology, but also begin to develop new transgenic and/or gene-drive practical approaches for potential pest management purposes.
Methods
Lygus hesperus rearing
A laboratory colony of L. hesperus, collected in Maricopa, AZ, served as the source of insects. Adults were maintained in 0.03 m3 screened plastic cages containing shredded paper that were housed in an environmental chamber set at 27 ± 1 °C, 40–60% RH, and a 14:10 (L:D) h photoperiod. Fresh green beans (Phaseolus vulgaris L.), an artificial diet pack53, and a bottle of water with a wick were used to nourish and hydrate the colony and were replaced as needed.
Embryonic RNAi
dsRNA targeting LhCd (MH806847), LhCn (MH806848), and LhW (MH806842) was produced as described in Brent and Hull (2019) to a concentration of 1 µg/µl. In brief, ~ 500-bp products were PCR amplified from validated plasmid DNAs harboring LhCd, LhCn, or LhW using T7 promoter containing primers (Supplementary Table S4). PCR products were purified then used as templates for in vitro transcription using a MEGAscript RNAi kit (Thermo Fisher Scientific). dsRNAs corresponding to the fluorescent protein gene venus, which was injected as a negative control, were similarly generated.
Gel packs, made of Parafilm M (Pechiney Plastic Packaging, Chicago, IL) and filled with carrageenan (1.25% w/v), were provided to L. hesperus as an oviposition substrate for one hour. The gel was removed from the packs and the egg-embedded parafilm sheet retained. The parafilm was stretched to release the eggs, which were then transferred to a moistened filter paper using a wet, fine-tip paintbrush. Eggs were aligned in 4 rows of 10 for a total of 40 eggs per treatment per experiment (LhCd, LhCn, LhW, and venus) and then gently covered with a No.1, 24 × 40 mm coverslip coated with permanent linerless double-sided Scotch tape (3 M, Maplewood, MN). The bare side of the coverslip was mounted on a glass slide with double-sided tape that adhered to the corners of the coverslip. The slide was placed on a Leica DMIL scope (Allendale, New Jersey). An IM 300 Microinjector (Narishige International USA, Amityville, NY) with a quartz needle loaded with dsRNA was used to inject the embryos at the posterior pole. Needles were produced by pulling capillary tubes with filament using a P-2000 needle puller (Sutter Instrument, Novato, CA) with the following two-line program: Line 1) Heat = 850, Filament = 5, Velocity = 25, Delay = 128; and line 2) Heat = 700, Filament = 5, Velocity = 50, Delay = 150. Needles were beveled using a Model EG-44 micropipette grinder (Narishige) at a 30° angle and an approximate rotor speed of 1800 rpm or 90% of the maximum speed. Needles were backfilled using a Microloader tip (Eppendorf, Enfield, CT). Following injection, coverslips with eggs were placed in a covered plastic petri dish containing 1% agarose (1 g agarose in 100 ml of distilled water), which was sealed with parafilm and placed in a growth chamber with the same settings as the laboratory colony. Images of eggs were taken 5 days post-injection using a Nikon SMZ18 microscope equipped with a Nikon D5-Ri2 camera (Nikon Instruments Inc., Melville, NY).
To confirm knockdown of targeted transcripts, expression of LhCd, LhCn, and LhW was measured by semi-quantitative RT-PCR, using actin (GDHC01004191) as a loading control. Total RNA was isolated from four replicated groups of three eggs using a Quick-RNA Microprep kit (Zymo Research, Irvine, CA). RNA quality and quantity were assessed using the Take3 module on a Synergy H4 Hybrid Multi-Mode Microplate Reader (Biotek Instruments, Winooski, VT). Total RNA (250 ng) was treated with DNase I (New England Biolabs, Ipswich, MA). cDNAs were generated from 250 ng RNA using a SuperScript III First-Strand Synthesis System (Life Technologies) and custom-made random pentadecamers (Integrated DNA Technologies, San Diego, CA). Fragments (~ 500 bp) of the genes of interest were amplified in a 20 μl reaction volume using SapphireAmp Fast PCR Master Mix (Clontech Laboratories Inc., Mountain View, CA) and primers listed in Supplementary Table S4. PCR conditions consisted of an initial denaturation at 95 °C for 2 min followed by 35 cycles of 95 °C for 20 s, 56 °C for 20 s, 72 °C for 30 s, and a final extension at 72 °C for 5 min. Gel images were obtained using an Azure 200 Gel Imaging Workstation (Azure Biosystems, Dublin, CA) and processed in Adobe Photoshop v21.2.12 (Adobe Systems Inc., San Jose, CA). Independent RNAi experiments were repeated three times.
Design and synthesis of sgRNAs
sgRNAs were designed using LhCd and LhCn with a focus on identifying guide sites near the 5′-end of the gene using CRISPOR54. Both sets of gene-specific sgRNAs were designed in proximity to one another; LhCd1 and 2 are separated by 8 nucleotides while LhCn1 and 2 are separated by 5 nucleotides. sgRNAs were screened for potential off-target sites by BLASTn of the 20-bp target sequence and the PAM sequence against the L. hesperus taxid 30085 database. Potential off-target sites were determined by comparing the BLASTn hit sequences that exactly matched the 3′ end of each sgRNA and the PAM sequence.
Double-stranded gBlock DNA fragments were synthesized by Integrated DNA Technologies (Coralville, Iowa), with each containing a T7 RNA polymerase binding site (5′-TAATACGACTCACTATA-3′), the 20-bp L. hesperus-specific target region (Supplementary Table S5), and the 80-bp common stem-loop tracrRNA sequence (5′-GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTT-3′). Each gBlock was used as a template for sgRNA synthesis using the HiScribe T7 High Yield RNA synthesis Kit (New England Biolabs). Transcribed sgRNAs were purified using RNAClean XP (Thermo Fisher Scientific) following the manufacturer’s protocol.
Creation of CRISPR/Cas9 eye pigmentation mutant strains
The experimental design included two independent injection groups; the first injections used a Cas9 protein with a nuclear localization signal (PNA Bio, Newbury Park, CA), whereas the second set used the Alt-R Streptococcus pyogenes HiFi Cas9 nuclease V3 (Integrated DNA Technology, Coralville, Iowa). The injection mixture consisted of the RNP complex of Cas9 (300 ng/µl) with two sgRNAs each at 150 ng/µl or a total of 300 ng/µl. Each sgRNA was preincubated with Cas9 at room temperature for 15 min and both solutions of RNP were combined to make the injection mixture. Negative controls include “no injection” and water only. Embryos were prepared and injected as previously described for the RNAi experiments. A total of 80 eggs per treatment (LhCd1 + 2, LhCn1 + 2, non-inject) were injected in the first experiment and the second experiment included 160, 80, 80, and 20 eggs for LhCd1 + 2, LhCn1 + 2, non-inject, and water, respectively.
Six to nine days post-injection, 1st instar nymphs that hatched were collected into a 355 ml mesh lidded paper cup and reared to adulthood under rearing conditions identical to those outlined above. Pairs from each subsequent generation were mated using the crossing schemes shown in Fig. 3. Card cross 1 (× 1) between a mutant male and a mutant female produced 19 G1 males and 15 G1 females. Card × 2, which also crossed mutants of both sexes, generated 9 G1 males and 12 G1 females. All G1 males from Card × 1 were group-mated with females from Card × 2 and vice versa to generate G2. To perpetuate the mutant line, G2 with red eyes were selected and transferred to new cages. In addition, Card × 3–5 each consisted of one mutant male and four wild type females. The G1 progeny with wild-type eye color from Card × 3–5 were combined and group-mated to generate a mix of wild-type and mutant eye color G2 progeny. From the resulting G2 progeny, 19 males with the mutant eye phenotype were crossed with 38 G2 mutant females from the Card × 1 and × 2 lines. Mutant progeny arising from this group-mating, as well as individuals from the ongoing mutant lines of Card × 1 and × 2 formed the Card colony.
To generate the Cinn colony, two mutant G0 females were crossed with two wild-type males to produce G1 progeny with wild-type eyes. Of these, three females and four males from Cinn × 1 were backcrossed with three males and 13 females from Cinn × 2, respectively. G2 progeny with the mutant eye phenotype were selected to perpetuate the Cinn colony. Mutant colonies were reared in 355 ml paper cups covered with mesh lids with up to 50 individuals. To prevent overcrowding in larger groups of > 50 individuals, 1.89 L paper cups were used55. Fresh green beans and sunflower seeds were provided twice a week. Diet and oviposition carrageenan packs were placed into rearing cups one week post-adult emergence.
Sequencing LhCd and LhCn from mutant eye pigment strains
Representative insects from CRISPR strains that displayed altered eye pigmentation were collected at G0, G1, G2, and/or G3 and stored at -80 °C in RNALater (Invitrogen, Carlsbad, CA). gDNA was extracted using a DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany). Total RNA was extracted using TRI Reagent following the manufacturer’s protocol. Total RNA was treated with DNase I (Thermo Fisher Scientific) and cDNA was synthesized using a SuperScript IV First-Strand Synthesis kit (Invitrogen). LhCd and LhCn were PCR amplified from gDNA and cDNA using a Phusion High-Fidelity PCR kit (Thermo Fisher Scientific) with gene-specific primer pairs (Supplementary Table S4) and thermocycler conditions of 1 cycle at 98 °C for 30 s; 35 cycles at 98 °C for 5 s, 60 °C for 10 s, 72 °C for 5 s; and 1 cycle at 72 °C for 5 min. PCR products were cloned into pJET1.32/blunt vector (Thermo Fisher Scientific) and transformed into One Shot OmniMAX 2 T1 Chemically Competent E. coli (Thermo Fisher Scientific). Multiple clones (n = 4–10) from each transformation reaction were Sanger sequenced (Retrogen Inc., San Diego, CA).
References
Scott, D. R. An annotated listing of host plants of Lygus hesperus Knight. Bull. Entomol. Soc. Am. 23, 19–22. https://doi.org/10.1093/besa/23.1.19 (1977).
Wheeler, A. G. Biology of the Plant Bugs (Hemiptera: Miridae): Pests, Predators, Opportunists (Cornell University Press, 2001).
Naranjo, S. E. & Ellsworth, P. C. Fifty years of the integrated control concept: Moving the model and implementation forward in Arizona. Pest Manag. Sci. 65, 1267–1286. https://doi.org/10.1002/ps.1861 (2009).
Palumbo, J. C., Horowitz, A. R. & Prabhaker, N. Insecticidal control and resistance management for Bemisia tabaci. Crop Prot. 20, 739–765. https://doi.org/10.1016/S0261-2194(01)00117-X (2001).
Snodgrass, G. L. Insecticide resistance in field populations of the tarnished plant bug (Heteroptera: Miridae) in cotton in the Mississippi delta. J. Econ. Entomol. 89, 783–790. https://doi.org/10.1093/jee/89.4.783 (1996).
Snodgrass, G. L. & Scott, W. P. Tolerance to acephate in tarnished plant bug (Heteroptera: Miridae) populations in the Mississippi River Delta. Southwest. Entomol. 27, 191–199 (2002).
Snodgrass, G. L., Gore, J., Abel, C. A. & Jackson, R. Acephate resistance in populations of the tarnished plant bug (Heteroptera: Miridae) from the Mississippi River Delta. J. Econ. Entomol. 102, 699–707. https://doi.org/10.1603/029.102.0231 (2009).
Gantz, V. M. et al. Highly efficient Cas9-mediated gene drive for population modification of the malaria vector mosquito Anopheles stephensi. Proc. Natl. Acad. Sci. 112, E6736–E6743. https://doi.org/10.1073/pnas.1521077112 (2015).
Hammond, A. et al. A CRISPR–Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat. Biotechnol. 34, 79–83. https://doi.org/10.1038/nbt.3439 (2016).
Galizi, R. et al. A CRISPR–Cas9 sex-ratio distortion system for genetic control. Sci. Rep. 6, 31139. https://doi.org/10.1038/srep31139 (2016).
Kyrou, K. et al. CRISPR–Cas9 gene drive targeting doublesex causes complete population suppression in caged Anopheles gambiae mosquitoes. Nat. Biotechnol. 36, 1062–1066. https://doi.org/10.1038/nbt.4245 (2018).
Scott, M. J. et al. Agricultural production: Assessment of the potential use of Cas9-mediated gene drive systems for agricultural pest control. J. Responsib. Innov. 5, S98–S120. https://doi.org/10.1080/23299460.2017.1410343 (2017).
Simoni, A. et al. A male-biased sex-distorter gene drive for the human malaria vector Anopheles gambiae. Nat. Biotechnol. 38, 1054–1060. https://doi.org/10.1038/s41587-020-0508-1 (2020).
Hammond, A. et al. Gene-drive suppression of mosquito populations in large cages as a bridge between lab and field. Nat. Commun. 12, 4589. https://doi.org/10.1038/s41467-021-24790-6 (2021).
Raban, R. R., Marshall, J. M. & Akbari, O. S. Progress towards engineering gene drives for population control. J. Exp. Biol. 223, jeb208181. https://doi.org/10.1242/jeb.208181 (2020).
Cui, Y., Sun, J.-L. & Yu, L. Application of the CRISPR gene-editing technique in insect functional genome studies—A review. Entomol. Exp. Appl. 162, 124–132. https://doi.org/10.1111/eea.12530 (2017).
Sun, D., Guo, Z., Liu, Y. & Zhang, Y. Progress and prospects of CRISPR/Cas systems in insects and other arthropods. Front. Physiol. 8, 608. https://doi.org/10.3389/fphys.2017.00608 (2017).
Mackenzie, S. M. et al. Mutations in the white gene of Drosophila melanogaster affecting ABC transporters that determine eye colouration. Biochim. Biophys. Acta. 1419, 173–185. https://doi.org/10.1016/S0005-2736(99)00064-4 (1999).
Khan, S. A., Reichelt, M. & Heckel, D. G. Functional analysis of the ABCs of eye color in Helicoverpa armigera with CRISPR/Cas9-induced mutations. Sci. Rep. 7, 40025. https://doi.org/10.1038/srep40025 (2017).
Bai, X. et al. CRISPR/Cas9-mediated knockout of the eye pigmentation gene white leads to alterations in colour of head spots in the oriental fruit fly, Bactrocera dorsalis. Insect Mol. Biol. 28, 837–849. https://doi.org/10.1111/imb.12592 (2019).
Heu, C. C., McCullough, F. M., Luan, J. & Rasgon, J. L. CRISPR–Cas9-based genome editing in the silverleaf whitefly (Bemisia tabaci). CRISPR J. 3, 89–96. https://doi.org/10.1089/crispr.2019.0067 (2020).
Xue, W.-H. et al. CRISPR/Cas9-mediated knockout of two eye pigmentation genes in the brown planthopper, Nilaparvata lugens (Hemiptera: Delphacidae). Insect Biochem. Mol. Biol. 93, 19–26. https://doi.org/10.1016/j.ibmb.2017.12.003 (2018).
Osanai-Futahashi, M. et al. Identification of the Bombyx red egg gene reveals involvement of a novel transporter family gene in late steps of the insect ommochrome biosynthesis pathway. J. Biol. Chem. 287, 17706–17714. https://doi.org/10.1074/jbc.M111.321331 (2012).
Shamin, G., Ranjan, S. K., Pandey, D. M. & Ramani, R. Biochemistry and biosynthesis of insect pigments. Eur. J. Entomol. 111, 149–164. https://doi.org/10.14411/eje.2014.021 (2014).
Ryall, R. L. & Howells, A. J. Ommochrome biosynthetic pathway of Drosophila melanogaster: Variations in levels of enzyme activities and intermediates during adult development. Insect Biochem. 4, 47–61. https://doi.org/10.1016/0020-1790(74)90041-9 (1974).
Perera, O. P., Little, N. S. & Pierce, C. A. III. CRISPR/Cas9 mediated high efficiency knockout of the eye color gene Vermillion in Helicoverpa zea (Boddie). PLoS ONE 13, e0197567. https://doi.org/10.1371/journal.pone.0197567 (2018).
Xu, X., Harvey-Samuel, T., Yang, J., Alphey, L. & You, M. Ommochrome pathway genes kynurenine 3-hydroxylase and cardinal participate in eye pigmentation in Plutella xylostella. BMC Mol. Cell Biol. 21, 63. https://doi.org/10.1186/s12860-020-00308-8 (2020).
Brent, C. S. & Hull, J. J. RNA interference-mediated knockdown of eye coloration genes in the western tarnished plant bug (Lygus hesperus Knight). Arch. Insect Biochem. Physiol. 100, e21527. https://doi.org/10.1002/arch.21527 (2019).
Liu, S.-H., Luo, J., Yang, B.-J., Wang, A.-Y. & Tang, J. karmoisin and cardinal ortholog genes participate in the ommochrome synthesis of Nilaparvata lugens (Hemiptera: Delphacidae). Insect Sci. 26, 35–43. https://doi.org/10.1111/1744-7917.12501 (2017).
Reding, K. & Pick, L. High-efficiency CRISPR/Cas9 mutagenesis of the white gene in the milkweed bug Oncopeltus fasciatus. Genetics 215, 1027–1037. https://doi.org/10.1534/genetics.120.303269 (2020).
Allen, M. L. Genetics of a sex-linked recessive red eye color mutant of the tarnished plant bug, Lygus lineolaris. Open J. Anim. Sci. 3, 1–9. https://doi.org/10.4236/ojas.2013.32A001 (2013).
Slaymaker, P. H. & Tugwell, N. P. Inheritance of red eye color in Lygus lineolaris (Palisot de Beauvois) (Hemiptera: Miridae) an abnormal trait. J. Kans. Entomol. 57, 343–344 (1984).
Li, M. et al. Generation of heritable germline mutations in the jewel wasp Nasonia vitripennis using CRISPR/Cas9. Sci. Rep. 7, 901. https://doi.org/10.1038/s41598-017-00990-3 (2017).
Chaverra-Rodriguez, D. et al. Germline mutagenesis of Nasonia vitripennis through ovarian delivery of CRISPR–Cas9 ribonucleoprotein. Insect Mol. Biol. 29, 569–577. https://doi.org/10.1111/imb.12663 (2020).
Cornel, A. J., Benedict, M. Q., Rafferty, C. S., Howells, A. J. & Collins, F. H. Transient expression of the Drosophila melanogaster cinnabar gene rescues eye color in the white eye (WE) strain of Aedes aegypti. Insect Biochem. Mol. Biol. 27, 993–997. https://doi.org/10.1016/S0965-1748(97)00084-2 (1997).
Lorenzen, M. D., Brown, S. J., Denell, R. E. & Beeman, R. W. Cloning and characterization of the Tribolium castaneum eye-color genes encoding tryptophan oxygenase and kynurenine 3-monooxygenase. Genetics 160, 225–234. https://doi.org/10.1093/genetics/160.1.225 (2002).
Quan, G. X. et al. Characterization of the kynurenine 3-monooxygenase gene corresponding to the white egg 1 mutant in the silkworm Bombyx mori. Mol. Genet. Genom. 267, 1–9. https://doi.org/10.1007/s00438-001-0629-2 (2002).
Maddison, D. C. et al. A novel role for kynurenine 3-monooxygenase in mitochondrial dynamics. PLoS Genet. 16, e1009129. https://doi.org/10.1371/journal.pgen.1009129 (2020).
Rasgon, J. L. & Scott, T. W. Crimson: A novel sex-linked eye color mutant of Culex pipiens L. (Diptera: Culicidae). J. Med. Entomol. 41, 385–391. https://doi.org/10.1603/0022-2585-41.3.385 (2004).
Figon, F. & Casas, J. Ommochromes in invertebrates: Biochemistry and cell biology. Biol. Rev. Camb. Philos. Soc. 94, 156–183. https://doi.org/10.1111/brv.12441 (2018).
Zhuravlev, A. V., Vetrovoy, O. V. & Savvateeva-Popova, E. Enzymatic and non-enzymatic pathways of kynurenines’ dimerization: The molecular factors for oxidative stress development. PLoS Comput. Biol. 14, e1006672. https://doi.org/10.1371/journal.pcbi.1006672 (2018).
Meccariello, A. et al. Highly efficient DNA-free gene disruption in the agricultural pest Ceratitis capitata by CRISPR–Cas9 ribonucleoprotein complexes. Sci. Rep. 7, 10061. https://doi.org/10.1038/s41598-017-10347-5 (2017).
Ren, X. et al. Optimized gene editing technology for Drosophila melanogaster using germ line-specific Cas9. Proc. Natl. Acad. Sci. 110, 19012–19017. https://doi.org/10.1073/pnas.1318481110 (2013).
Yan, Y., Ziemek, J. & Schetelig, M. F. CRISPR/Cas9 mediated disruption of the white gene leads to pigmentation deficiency and copulation failure in Drosophila suzukii. J. Insect Physiol. 126, 104091. https://doi.org/10.1016/j.jinsphys.2020.104091 (2020).
Ferreiro, M. J. et al. Drosophila melanogaster white mutant w1118 undergo retinal degeneration. Front. Neurosci. 11, 732. https://doi.org/10.3389/fnins.2017.00732 (2018).
Borycz, J., Borycz, J. A., Kubow, A., Lloyd, V. & Meinertzhagen, A. Drosophila ABC transporter mutants white, brown and scarlet have altered contents and distribution of biogenic amines in the brain. J. Exp. Biol. 211, 3454–3466. https://doi.org/10.1242/jeb.021162 (2008).
Evans, J. M., Day, J. P., Cabrero, P., Dow, J. A. T. & Davies, S.-A. A new role for a classical gene: White transports cyclic GMP. J. Exp. Biol. 211, 890–899. https://doi.org/10.1242/jeb.014837 (2008).
Chaverra-Rodriguez, D. et al. Targeted delivery of CRISPR–Cas9 ribonucleoprotein into arthropod ovaries for heritable germline gene editing. Nat. Commun. 9, 3008. https://doi.org/10.1038/s41467-018-05425-9 (2018).
Macias, V. M. et al. Cas9-mediated gene-editing in the malaria mosquito Anopheles stephensi by ReMOT control. G3 (Bathesda) 10, 1353–1360. https://doi.org/10.1534/g3.120.401133 (2020).
Shirai, Y. & Daimon, T. Mutations in cardinal are responsible for the red-1 and peach eye color mutants of the red flour beetle Tribolium castaneum. Biochem. Biophys. Res. Commun. 529, 372–378. https://doi.org/10.1016/j.bbrc.2020.05.214 (2020).
Sharma, A. et al. Cas9-mediated gene-editing in the black-legged tick, Ixodes scapularis, by embryo injection and ReMOT control. SSRN Electron J. https://doi.org/10.2139/ssrn.3691041 (2020).
Bier, E. Gene drives gaining speed. Nat. Rev. Genet. https://doi.org/10.1038/s41576-021-00386-0 (2021).
Debolt, J. W. Meridic diet for rearing successive generations of Lygus hesperus. Ann. Entomol. Soc. Am. 75, 119–122. https://doi.org/10.1093/aesa/75.2.119 (1982).
Concordet, J.-P. & Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucl. Acids Res. 46, W242–W245. https://doi.org/10.1093/nar/gky354 (2018).
Brent, C. S. Stage-specific effects of population density on the development and fertility of the western tarnished plant bug, Lygus hesperus. J. Insect Sci. 10, 49. https://doi.org/10.1673/031.010.4901 (2010).
Acknowledgements
We thank Daniel Langhorst for technical assistance in this project. This research was supported in part by an appointment to the Agricultural Research Service (ARS) Research Participation Program administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy (DOE) and the U.S. Department of Agriculture (USDA). ORISE is managed by ORAU under DOE contract number DE-SC0014664. All opinions expressed in this paper are the author's and do not necessarily reflect the policies and views of USDA, DOE, or ORAU/ORISE. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Funding
This material is based upon work supported by Cotton Incorporated under the Grant No. 19-218.
Author information
Authors and Affiliations
Contributions
C.C.H., J.J.H., C.S.B., and J.A.F. designed the study. C.C.H., R.J.G., K.P.L., D.M.L., B.F., and J.J.H. performed the experiments. C.C.H., J.J.H., C.S.B., and J.A.F. analyzed the data. C.C.H., J.J.H., C.S.B., and J.A.F. wrote the manuscript. All authors have read and approved the manuscript for publication.
Corresponding author
Ethics declarations
Competing interests
This investigation was partially funded by Cotton Inc. (#19-218) to C.S.B., J.J.H., and J.A.F. C.C.H., R.J.G., K.L., D.M.L., and B.F. declare no potential conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Heu, C.C., Gross, R.J., Le, K.P. et al. CRISPR-mediated knockout of cardinal and cinnabar eye pigmentation genes in the western tarnished plant bug. Sci Rep 12, 4917 (2022). https://doi.org/10.1038/s41598-022-08908-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-08908-4
This article is cited by
-
Genome editing of the vermilion locus generates a visible eye color marker for Oncopeltus fasciatus
Scientific Reports (2023)
-
Inter and intra individual genomic edits contributing to white eye phenotype in the mango fruit fly, Bactrocera dorsalis Hendel (Diptera: Tephritidae) obtained through microinjection of ribo nucleo protein complex
The Nucleus (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
