
Abstract
Growing evidence has demonstrated the functional relevance of long intergenic noncoding RNAs (lincRNAs) to tumorigenesis and immune response. However, immune-related lincRNAs and their value in predicting the clinical outcomes of patients with liver cancer remain largely unexplored. Herein, we utilized the strategy of iterative gene pairing to construct a tumor-specific immune-related lincRNA pairs signature (IRLPS), which did not require specific expression levels, as an indicator of patient outcomes. The 18-IRLPS we developed was associated with overall survival, tumor progression, and recurrence in liver cancer patients. Multivariate analysis revealed that the risk model was an independent predictive factor. A high IRLPS risk was correlated suppressive immune microenvironment, and IRLPS-high patients might benefit more from CD276 blockade or TMIGD2 agonist. Patients in the high-risk group were associated with elevated tumor mutation, increased sensitivity to dopamine receptor antagonists, cisplatin, doxorubicin, and mitomycin but more resistance to vinblastine. Mechanistically, IRLPS high scores might lead to poor prognosis by promoting cell proliferation and metabolic reprogramming. The prognostic significance of the 18-IRLPS was confirmed in independent cancer datasets. These findings highlighted the robust predictive performances of the 18-IRLPS for prognosis and personalized treatment.
Similar content being viewed by others


Introduction
Liver cancer is the sixth most commonly diagnosed cancer and the third leading cause of cancer death worldwide in 2020, while hepatocellular carcinoma (HCC) accounts for 70–85% of the total burden1. Surgical resection and liver transplantation are potentially curative treatment options available for early-stage HCC. However, 5-year survival rates following surgical resection remain relatively low for early-stage disease (17–53%), with recurrence rates as high as 70%2,3. At present, sorafenib and lenvantinib, the multi-targeted kinase inhibitors, are used as first-line therapy for advanced HCC, remaining unsatisfactory in the clinical practice4. Moreover, HCC is a highly heterogeneous tumor, which considerably cuts down the efficacy of clinical treatments and makes the survival prediction quite complicated5. The efficacy and safety of nivolumab, an immunotherapy targeting PD-1, have been explored in patients with HCC. However, only ~ 20% of participants respond to the treatment6,7. Several biomarkers, including PD-L1 expression, microsatellite instability, and tumor mutational burden (TMB), have been approved for selecting patients with other malignancies who will benefit from the immune-checkpoint blockade therapy8. However, there are few robust predictive biomarkers available in HCC, with the use of PD-L1 expression being of limited value6,7. Thus, the perusal of the tumor microenvironment and identifying novel and promising biomarkers have become imperative for improved treatment in HCC patients.
Long noncoding RNAs (lncRNAs), which are RNA transcripts longer than 200 bp and unable to code proteins, could regulate gene expression by interacting with DNA, RNA, and proteins to exhibit either enhancement or inhibition9,10. Increasing evidence support that lncRNAs play critical roles in tumorigenesis and progression of HCC11,12. Moreover, lncRNAs are reported as crucial regulators of cancer immunity, such as antigen release and immune activation, which contribute to the malignant phenotypes of cancer13,14,15. There is growing evidence that immune-related lncRNAs may be novel disease biomolecules for clinical cancer treatment and possess valuable prognostic significance for survival16,17. However, batch effects on the detected gene expression profiles due to different platforms and testing time may lead to inaccuracy of prognostic prediction using gene signatures according to their exact expression levels18.
Recently, a novel algorithm for normalizing and scaling the expression matrix based on the relative ranking of gene expression levels has been proposed to eliminate the potential defects mentioned above19,20. In this study, we were inspired by the strategy of gene pairing and aimed to discover an immune-related intergenic lncRNAs (lincRNAs)-based risk model for predicting clinically relevant outcomes in patients with HCC. By iteratively comparing the relative expression of tumor-specific immune-associated lincRNA pairs in each sample, we developed a valid signature with no requirement of specific expression levels. We estimated its predictive value among patients with HCC for prognostic effectiveness, tumor immune infiltration, and therapeutic liability in immunotherapy, targeted therapy, and chemotherapy. We also found that the signature of lincRNA pairs was associated with enhanced cell cycle and altered metabolism. Lastly, we validated its predictive efficacy in multiple cancer types.
Results
Establishment of immune-related lincRNA pairs signature with prognostic significance
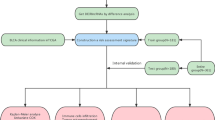
The strategy for identifying tumor-specific immune-related lincRNA pairs signature (IRLPS) in this study is shown in Fig. 1A. First, we selected 454 lincRNAs, which were potential tumor-intrinsic (highly expressed in tumor tissues compared with adjacent normal tissues but not expressed in immune tissues) immune regulators from a previous study21. Next, we retrieved the transcriptome profiles of The Cancer Genome Atlas Liver Hepatocellular Carcinoma (TCGA-LIHC) cohort and separated the data of lncRNA and mRNA using the GENCODE annotation file. Among the 454 tumor-specific immune-associated lincRNAs, 429 lincRNAs were detected in the TCGA-LIHC dataset and selected for subsequent analysis (Supplementary Table 1). Using an iterative 0-or-1 matrix screening, we identified 35,604 valid lincRNA pairs. After a univariate analysis followed by a modified Lasso penalized regression, 39 pairs were extracted. At last, 18 lincRNA pairs were included in the IRLPS using a stepwise method with a multivariate Cox proportional hazards model for overall survival (Fig. 1B). To assess the prognostic performance of the risk model, the areas under curve (AUC) for each ROC curve of the 18-IRLPS was calculated. The AUC values ranged from 0.886 to 0.914 at the 1-, 3-, and 5-year ROC curves for overall survival, confirming the optimality of the IRLPS (Fig. 1C). Next, we determined the maximum inflection point (1.471) as the optimal cutoff value for an ideal IRLPS on the 5-year receiver operating characteristic (ROC) curve using the Akaike information criterion (Fig. 1D). We also compared the 5-year ROC curve of the IRLPS with common clinical characteristics, showing the superiority of the risk model (Fig. 1E). Based on the cutoff value determined above, 365 acceptable patients from the TCGA-LIHC cohort were stratified into the high- and low-risk groups. The distribution of the IRLPS risk score, survival status, and IRLPs expression pattern was revealed in Fig. 1F. Patients in the high-risk group exhibited an inferior clinical outcome compared with those in the low-risk group. Kaplan–Meier analysis showed that patients in the high-risk group had significantly shorter overall survival time than those in the low-risk group (Fig. 1G). Moreover, multivariate Cox regression analysis indicated that the prognostic performance of the risk score was independent of other clinical factors for overall survival prediction after being adjusted by other clinical characteristics, including age, gender, grade, and stage (Fig. 1H). We also investigated the possible correlations between the risk score and clinicopathological features. The alluvial diagram, pie chart, and strip chart showed that the high-risk group was significantly corresponded to more patients being dead and with advanced clinical stage, M stage, and T stage (Supplementary Fig. 1A–C). On the other hand, patients with higher T stage, clinical stage, and tumor grade were significantly associated with increased risk scores (Supplementary Fig. 1D–F).

Construction of immune-related lincRNA pairs signature with prognostic significance. (A) Flow Chart of identifying tumor-specific immune-related lincRNA pairs signature (IRLPS). (B) Forest map showing the 18 IRLPS identified by multivariate Cox proportional hazard regression analysis of overall survival. (C) The 1-, 3-, and 5-year ROC curves of the optimal model for overall survival. (D) The optimal cutoff point for the IRLPS on the 5-year ROC curve. (E) Comparison of 5-year ROC curves of the IRLPS with that of common clinical characteristics. (F) The distribution of the risk score, patients’ survival status, and IRLPs expression pattern. (G) Kaplan–Meier survival curves of overall survival between patients with higher and lower risk scores of the IRLPS. (H) Univariate (left) and multivariate (right) Cox hazard ratio analysis of overall survival.
To further examine whether the IRLPS is related to tumor progression and recurrence, we used progression-free survival and disease-free interval to investigate the effectiveness of the IRLPS for risk prediction. The AUC values for predicting patients' progression status by the IRLPS were 0.727, 0.704, and 0.770 at the 1-, 3-, and 5-year ROC curves and achieved better predictive performance than other clinical characteristics (Supplementary Fig. 2A,B). Kaplan–Meier analysis demonstrated that patients in the high-risk group significantly correlated with unfavorable progression-free survival (Supplementary Fig. 2C). We also performed multivariate Cox regression analysis of progression-free survival and found that the IRLPS remained an independent predictor for progression-free survival (HR = 1.050, 95% CI 1.027–1.074; Supplementary Fig. 2C). The IRLPS also exhibited the superiority for predicting tumor recurrence, with the AUC values being 0.757, 0.694, and 0.797 at the 1-, 3-, and 5-year ROC curves (Supplementary Fig. 2E,F). Patients in the high-risk group experienced significantly reduced disease-free interval than those in the low-risk group (Supplementary Fig. 2G). Multivariate Cox regression analysis of disease-free interval indicated that increasing IRLPS risk score was independently associated with a greater probability of disease recurrence (HR = 1.056, 95% CI 1.025–1.088; Supplementary Fig. 2H).
Correlation between the risk model and tumor immune microenvironment
We subsequently investigated whether the IRLPS was related to the tumor immune microenvironment. Kaplan–Meier survival curves revealed that patients with low immune scores (representing the infiltration of immune cells in tumor tissue) or ESTIMATE scores (inferring tumor purity) from the TCGA-LIHC cohort had a worse progression-free survival and disease-free interval (Fig. 2A,B). Intriguingly, the immune scores of samples within the IRLPS-high group showed a declining trend, whereas the ESTIMATE scores were significantly lower in tumors with higher risk scores (Fig. 2C,D). We applied Spearman’s correlation analysis to the IRLPS risk score and the enrichment scores of 68 immune signatures calculated using the single-sample gene set enrichment analysis (ssGSEA)21. Results showed that the risk score was positively associated with neutrophil cells but inversely correlated with cytotoxic T cell, DC cells, and IFN signaling (Fig. 2E). We also quantified the enrichment of a set of marker genes for tumor-infiltrating immune cells to assess immune functions22. Consistently, the enrichment scores of cytolytic activity, T cell co-stimulation, HLA, type I IFN response, type II IFN response, and inflammation-promoting were significantly lower in the high-risk group (Supplementary Fig. 3A). We further explored the potential correlation between the risk score of the IRLPS and immune cell infiltration abundance calculated by seven commonly acknowledged deconvolution methods, including TIMER, XCELL, QUANTISEQ, MCPcounter, EPIC, CIBERSORT, and CIBERSORT-ABS23. Results showed a higher risk score was negatively associated with central memory CD8+ T cells while positively related to Th2 CD4+ T cells and M0 macrophages (Fig. 2F, Supplementary Fig. 3B,C). Notably, patients with a low infiltration level of central memory CD8+ T cells or high abundance of Th2 CD4+ T cells and M0 macrophages exhibited significantly reduced overall survival probability (Fig. 2G, Supplementary Fig. 3D). Additionally, we investigated the relationship between the risk score of IRLPS and chemokine expression levels using the TISIDB database24. Elevated expression of CXCL1, CXCL3, CXCL5, CXCL6, CXCL8, CCL20, and CCL26 was observed in the high-risk group while CCL14, CCL18, CCL21, CCL23, CXCL2, CXCL12, and CXCL23 were declined (Fig. 2H). Immune subtypes spanning multiple cancer types have been proposed to define immune response patterns affecting patient prognosis. Subtype C1 (wound healing) had a high proliferation rate and a Th2 cell bias. In contrast, the C3 (inflammatory) subtype is characterized by low to moderate tumor cell proliferation and lower levels of somatic copy number alterations than the other subtypes25. For liver cancer, patients within the C3 subtype displayed the most favorable prognosis while C1 the worst. Moreover, there were more C1 subtypes and fewer C3 subtypes in the IRLPS-high group than the low-risk group (Fig. 2I). On the other hand, C3 subtype constituent tumors showed remarkably lower risk scores of the IRLPS than C1 (Supplementary Fig. 3E).

Correlation between the prognostic risk score with tumor immune microenvironment. (A, B) Low immune score (A) or ESTIMATE score (B) was associated with shortened progression-free survival (left) and disease-free interval (right) in TCGA-LIHC patients. (C, D) Box plot comparing the immune score (C) or ESTIMATE score (D) between the high- and low-risk groups. (E) Correlation heatmap of the IRLPS risk score and enrichment scores of representative immune-related signatures. (F) Association between the risk score of IRLPS and tumor-infiltrating immune cells including central memory CD8+ T cells, Th2 CD4+ T cells, and M0 macrophages. (G) Kaplan–Meier survival curves of overall survival between patients with high- and low-abundance of tumor-infiltrating central memory CD8+ T cells, Th2 CD4+ T cells, and M0 macrophages. (H) Box plot comparing gene expression of chemokines between the high- and low-risk groups by Wilcoxon signed-rank test. (I) Heatmap and table showing the distribution of immune subtypes between the IRLPS risk groups, compared using the chi-square test (top). Kaplan–Meier survival curves of overall survival between patients within the indicated immune subtypes (bottom).
Correlation of the IRLPS to immune checkpoint-related genes and somatic alteration
To further explore the relationship between the IRLPS and tumor immunity, we assessed the risk score and immunotherapy-relevant biomarkers correlation. Immune checkpoint-related genes could trigger the immunosuppressive tumor environment and are reported as predictive biomarkers for immunotherapy in multiple malignancies. Gene expression levels of TNFSF4, TNFRSF18, CD276, CD80, HHLA2, LGALS9, VTCN1, and TNFRSF14, but not HAVCR2, LAG3, CTLA4, and PDCD1, were significantly upregulated in IRLPS-high patients (Fig. 3A). Moreover, we found that the risk score was positively correlated with HHLA2 (r = 0.292), CD276 (r = 0.259), TNFSF4 (r = 0.215) and inversely related to TMIGD2 (r = − 0.221; Fig. 3B). Notably, Kaplan–Meier analysis showed that the higher expression of HHLA2, CD276, and TNFSF4 was significantly associated with inferior prognosis in overall survival while patients with lower TMIGD2 levels had worse progression-free survival (Fig. 3C). We further analyzed their correlation with the IRLPS-associated tumor-infiltrating immune cells and immune signatures (Fig. 2E,F). Results indicated that CD276 was positively correlated with the abundance of Th2 CD4+ T cells (r = 0.301) and M0 macrophages (r = 0.236) while TMIGD2 was related to enriched central memory CD8+ T cells (r = 0.487; Fig. 3D). We also observed an association of CD276 with neutrophil cells (TREM1_data; r = 0.269) and TMIGD2 with DC cells (CD103pos_mean, r = 0.569; CD103pos_CD103neg_ratio, r = 0.332), cytotoxic T cell (CD8_PCA, r = 0.479; MCD3_CD8, r = 0.287), and IFN Signaling (IR7_scorer, r = 0.423; Fig. 3E). These results indicated that patients in the IRLPS-high group might benefit more from CD276 blockade or TMIGD2 agonist.

Correlation of the IRLPS to immune checkpoint-related genes and somatic alteration. (A) Expression of 20 immune checkpoint-related genes in high- and low-risk groups. (B) Lollipop diagram showing the Spearman's correlation between the indicated immune genes with the risk score of IRLPS. (C) Kaplan–Meier survival curves between patients with the high and low expression level of HHLA2, CD276, TNFSF4, and TMIGD2. (D) Correlation matrix of the indicated immune gene expression and tumor-infiltrating M0 macrophages, Th2 CD4+ T cells, and central memory CD8+ T cells. (E) Correlation matrix of the indicated immune gene expression and enrichment scores of representative immune-related signatures. (F) Comparison of tumor mutational burden (TMB) between the low- and high-risk groups in the TCGA-LIHC cohort. (G) Scatterplot depicting a positive correlation between risk score and mutation load. (H) Kaplan–Meier survival curves for patients with high and low TMB, dividing with the median TMB as a cutoff. (I) Kaplan–Meier survival curves of overall survival among four patient groups stratified by TMB and the risk score of IRLPS. (J) The oncoPrint of tumors with high (left) and low (right) risk scores. Individual patients are represented in each column.
Given the clinical implications of TMB, it is worth exploring its correlation with the risk score of IRLPS. TMB was significantly higher in patients from the high-risk group than those from the low-risk group and positively correlated with the risk score (Fig. 3F,G). However, no significant difference in overall survival was observed between patients with high and low TMB (Fig. 3H). We next evaluated the synergistic effect of the IRLPS and TMB in prognostic prediction. Stratified survival analysis of the four patient groups revealed that the TMB status did not interfere with IRLPS and the IRLPS subgroups remained significant survival differences in both high and low TMB subgroups (Fig. 3I), suggesting that the IRLPS could serve as a predictive indicator independent of TMB. Furthermore, we compared the distribution of somatic varients in the top 25 driver genes with the highest alteration frequency between the high- and low-risk groups (Fig. 3J). Results showed a significantly greater alteration frequency of TP53, DOCK2, DNAH7, HMCN1, and LRP1B in the high-risk groups (Supplementary Table 2), which provided potential directions for interpreting the underlying mechanisms of the IRLPS.
Candidate compounds/chemotherapies targeting the IRLPS
To understand the potential therapeutic value of the IRLPS in drug sensitivity prediction, we first employed the Connectivity Map (CMap) to discover candidate small molecular compounds that might target the IRLPS-associated pathways. A total of 43 compounds were significantly enriched (Fig. 4A). CMap mode-of-action (MoA) analysis revealed 34 mechanisms of action shared by these compounds (Fig. 4B). Five compounds (chlorpromazine, fluspirilene, prochlorperazine, thioridazine, and trifluoperazine) shared the MoA of dopamine receptor antagonist, which has been reported to inhibit stemness-related tumorigenicity26,27. We also identified shared MoA of adrenergic receptor antagonist (doxazosin and phenoxybenzamine), estrogen receptor agonist (alpha-estradiol and estriol), GABA receptor modulator (etomidate and tracazolate), glucocorticoid receptor agonist (medrysone and rimexolone), and the enrichment of the cell proliferation inhibitor apigenin.

Small molecular compounds identification and sensitivity prediction for chemotherapies. (A) Heatmap showing the enrichment score of candidate compounds from the CMap. (B) Heatmap showing each compound from the CMap that shares mechanisms of action (rows), sorted by descending number of compounds that share mechanisms of action. (C) Sensitivity of the indicated drugs in the two risk groups, compared by Wilcoxon signed-rank test.
We further explored the role of IRLPS in the sensitivity prediction for common administrating chemotherapeutic drugs, including doxorubicin, cisplatin, mitomycin, vinblastine, and sorafenib. Patients in the high-risk group showed increased sensitivity to cisplatin, doxorubicin, and mitomycin but more resistance to vinblastine. However, no significant difference for sorafenib was observed between the two risk groups (Fig. 4C). Together, these results imply that the IRLPS may be a potential biomarker for identifying patients who are more likely to benefit from a tailored treatment strategy.
Pathway and gene network features associated with the IRLPS
To identify the potential biological processes related to the IRLPS, GSEA was employed to assess the Hallmark and KEGG pathways. We found that the high-risk group was significantly associated with carcinogenic pathways such as cell cycle (E2F targets, G2M checkpoint, mitotic spindle, MYC targets), DNA repair, and glycolysis (Fig. 5A,B). In contrast, bile acid metabolism and amino acid metabolism were enriched in the low-risk group (Fig. 5C,D). We further identified 481 significantly upregulated and 190 downregulated genes in the high-risk group compared with the low-risk group (Fig. 5E,F). Enrichment analysis was performed to illustrate the functional annotations of these differentially expressed genes. We observed the enrichment of cell division- and cell cycle regulation-related GO terms (Fig. 5G), as well as KEGG cell cycle and carbon metabolism pathways (Fig. 5H). Collectively, these findings suggested that the IRLPS might lead to poor prognosis by promoting cell proliferation and metabolic reprogramming.

Pathway and gene network features associated with the IRLPS. (A, B) Top ten Hallmark (A) and KEGG (B) pathways significantly associated with the high-risk group in TCGA-LIHC. (C, D) The significantly enriched Hallmark (C) and KEGG (D) pathways in the low-risk group. (E) Heatmap of the differentially expressed genes. (F) Volcano plot of significant differentially expressed genes between the two risk groups. Significantly upregulated and downregulated genes in the high-risk group are represented as red and blue dots, respectively. The top ten hub genes are labeled. (G) GO enrichment analyses of the differentially expressed genes. (H) KEGG pathway enrichment analyses of the differentially expressed genes.
To further narrow the scope, a protein–protein interaction network (containing 284 nodes and 1827 edges) was constructed (Fig. 6A). Then the top ten hub genes, including CDK1, CDC20, CCNB1, CCNB2, BUB1, AURKB, PLK1, BUB1B, CDCA8, and CCNA2, were identified (Fig. 6B). Their expression was positively correlated with the IRLPS risk score (r > 0.3; Fig. 6C). Nine of the ten hub genes, except CCNA2, were significantly associated with increased hazard ratios for overall survival, progression-free survival, and disease-free interval based on Cox regression analysis (Fig. 6D–F).

Identification of hub genes involved in the IRLPS. (A) A PPI network of the differentially expressed genes with the STRING confidence score > 0.9. Thicker edges between nodes indicate stronger combined scores. (B) Identification of the top ten hub genes in the PPI network using the multi-network clustering (MNC) algorithm. Red nodes represent genes with high MNC scores, while yellow nodes represent genes with lower MNC scores. (C) Lollipop diagram showing the Spearman's correlation between the ten hub genes with the IRLPS risk score. (D–F) Forest plot of univariate Cox regression analyses for overall survival (D), progression-free survival (E), and disease-free interval (F) in TCGA-LIHC, showing hub genes with significance.
Confirmation of the IRLPS in independent cancer datasets
To assess the robustness of the 18-IRLPS risk model, we test its predictive power on external cancer datasets with lincRNA expression and clinical information. Time-dependent ROC analysis revealed significant performances to predict overall survival for other types of cancer in the TCGA project, among which adrenocortical carcinoma (ACC), cholangiocarcinoma (CHOL), and low grade glioma (LGG) showed the highest AUC values (Fig. 7A–C and data not shown). Kaplan–Meier survival curves demonstrated that the 18-IRLPS could stratify patients from the TCGA-ACC, TCGA-CHOL, and TCGA-LGG datasets into high- and low-risk groups with significantly different overall survival using the same cutoff value of risk score (1.471) obtained from TCGA-LIHC (Fig. 7D–F). A similar extent of effectiveness was observed for progression-free survival of the three TCGA cancer types (Fig. 7G–L), confirming the powerful predictive performances of the IRLPS for overall survival and tumor progression.

Prognostic significance validation of the 18-IRLPS in independent cohorts. (A–C) The time-dependent ROC curves of the IRLPS for predicting overall survival in TCGA-ACC (A), TCGA-CHOL (B), and TCGA-LGG (C). (D–F) Kaplan–Meier survival curves of overall survival between patients with higher and lower risk scores of the IRLPS in TCGA-ACC (D; n = 79), TCGA-CHOL (E; n = 36), and TCGA-LGG (F; n = 506). (G–I) The 1-, 3-, and 5-year ROC curves of the IRLPS for predicting progression-free survival in TCGA-ACC (G), TCGA-CHOL (H), and TCGA-LGG (I). (J–L) Kaplan–Meier survival curves of progression-free survival between patients with higher and lower risk scores of the IRLPS in TCGA-ACC (J), TCGA-CHOL (K), and TCGA-LGG (L).
Additionally, the role of the ten hub genes identified in TCGA-LIHC was explored in the three TCGA cancer types using Spearman's correlation and Cox regression analysis. The hub genes identified above were also significantly correlated with the IRLPS risk score in TCGA-ACC, TCGA-CHOL, and TCGA-LGG (r > 0.3; Supplementary Fig. 4A). They were also significantly associated with overall survival and progression-free survival (Supplementary Fig. 4B,C) in TCGA-ACC and TCGA-LGG. These findings demonstrate that the poor prognosis of IRLPS-high patients may be mediated by common underlying mechanisms in multiple cancer types.
Discussion
HCC is highly heterogeneous both from molecular and clinical standpoints28,29, making insufficient responses to monotherapy in many clinical cases and the survival prediction quite complicated. Due to characteristics of the immune contexture substantially impacting immune therapy outcome30, the IRLPS was established from the tumor-specific immune-related lincRNAs as a prognostic indicator. Several recent reports screened lncRNAs associated with prognosis or immune response of HCC, using two to ten lncRNAs to construct a risk score formula31,32,33,34,35. Unlike traditional prognostic models, the pairwise comparison and score calculation of each IRLPs are based entirely on the lincRNA expression in the same patient, overcoming batch effects. Previous studies utilized a similar modeling strategy, of which 12 or 30 differentially expressed immune-related lncRNA pairs were included36,37. In contrast, we focused on only the intergenic subtype of lncRNA (lincRNA) and did not use arbitrary cutoffs for differential expression significance.
The 18-IRLPS consisted of 34 lincRNAs, some of which have been reported to be involved in the occurrence and development of liver cancer. For instance, the expression of DIO3OS is lower in HCC, and upregulation of DIO3OS represses malignant behaviors38. LINC01352 downregulation mediated by the HBx/ERα complex promotes HCC cell growth and metastasis39. Correspondingly, pairs of DIO3OS|RP11-672L10.6 and LINC01352|RP11-43F13.4 have an HR lower than 1 (Fig. 1B), indicating that higher expression of DIO3OS compared with RP11-672L10.6 or higher expression of LINC01352 compared with RP11-43F13.4 in a specific sample is associated with a better prognosis of patients with liver cancer.
The assessment of the immune status in the tumor microenvironment is essential for a comprehensive understanding of the tumor. A large number of studies have shown that dense infiltration of T cells, especially CD8+ T cells, predicts a good prognosis22,40. In most cancers, Th2 CD4+ T cells have been shown to support tumor growth and progression, which could form an immunosuppressive milieu to hamper the activation of CD8+ T cells for eradicating the tumor cells41. By integrating analysis, we revealed that the suppressive Th2 CD4+ T cells and M0 macrophages are more abundant in the IRLPS-high group, while the fraction of central memory CD8+ T cells decreased (Fig. 2F). Correlation analysis with immune-related gene sets also indicates that patients in the high-risk group were inversely associated with cytotoxic T cell, antigen presentation, and IFN signaling (Fig. 2E and Supplementary Fig. 3A). The infiltration of diverse types of immune cells is tightly regulated by various chemokines, which modulate tumor immunity, the biological phenotype of the tumors, and the prognosis for patients42. We found that the IRLPS-high group had decreased expression of CCL23 (Fig. 2H), in line with our previous study that CCL23 could serve as a tumor suppressor through recruiting CD8+ T cell infiltration in liver cancer43. Thus, the IRLPS may work through regulating immune cells infiltration and has a critical role in the immunosuppressive tumor microenvironment.
CD276, also known as B7-H3, is a newly identified target for cancer immunotherapy because it is overexpressed in tumor tissues while showing limited expression in most normal tissues44. To date, several CD276-based immunotherapeutic strategies have demonstrated potent antitumor activity and acceptable safety profiles in preclinical models45. In HCC, aberrantly expressed CD276 could promote tumor progression and inhibit the proliferation of CD8+ T cells46. Consistently, the expression level of CD276 was positively correlated with the IRLPS risk score and significantly associated with poor prognosis in overall survival. CD276 also had a positive correlation with Th2 CD4+ T cells and M0 macrophages (Fig. 3A–D), supporting the potential value of CD276 blockade in the IRLPS-based immunotherapy.
Several candidate compounds were identified to be potential treatments targeting the IRLPS in HCC patients. The dopamine receptor antagonists thioridazine and prochlorperazine are potential compounds targeting undifferentiated tumors47. A recent study shows that thioridazine is a potential drug against HCC through inhibition of the PI3K/AKT pathway, ROS induction, and angiogenesis48. Prochlorperazine possesses anticancer activity by affecting cell cycle stages, stimulating apoptosis, and inhibiting migration and invasiveness49. Cell proliferation inhibitor apigenin was also enriched in the high-risk group. Apigenin, a natural flavonoid, has low intrinsic toxicity and possesses preventive and therapeutic potential against cancers50. In HCC, apigenin could inhibit the growth and epithelial-mesenchymal transition of cancer cells51,52.
Aberrant expression of the identified hub genes may contribute to HCC occurrence and development. For instance, a recent study has demonstrated that CDCA8 promotes HCC growth and stemness through the AKT/β-catenin signaling53. BUB1B exerts an oncogenic effect in HCC cell proliferation, migration, and invasion partially by affecting mitochondrial function54. CDC20 could enhance cell proliferation and is associated with the development and progression of HCC55.
In summary, we established an 18-IRLPS, regardless of specific expression levels, as an independent prognostic indicator for patients with liver cancer. The risk model may also help distinguish immune and molecular characteristics and predict therapeutic sensitivity, highlighting the promising clinical significance for cancer patients’ personalized treatment and prognosis (Supplementary Fig. 5).
Materials and methods
Data collection and preprocessing
Transcriptome profiling (RNA-seq), clinical, and mutation data of the TCGA-LIHC project consisting of 50 normal and 374 tumor samples were obtained from the GDC Data Portal (https://portal.gdc.cancer.gov/). Patients with a follow-up time of 0 days were excluded. The GENCODE v22 GTF file for annotation and distinguish the mRNAs and lncRNAs was downloaded from https://gdc.cancer.gov/about-data/gdc-data-processing/gdc-reference-files. To determine tumor mutational burden (TMB), the total number of non-synonymous mutations was counted. The number of mutation events per million bases was calculated as the TMB for 361 samples. Tumor driver genes were identified by applying the maftool R package56.
Immune scores and Estimate scores, calculated by the ESTIMATE algorithm57, for samples within the TCGA-LIHC dataset were downloaded from https://bioinformatics.mdanderson.org/estimate/disease.html?liver%20hepatocellular%20carcinoma_RNAseqV2. Data of immune subtypes dominated in liver cancer, including C1 (wound healing), C2 (IFN-γ dominant), C3 (inflammatory), and C4 (lymphocyte depleted), were extracted from the supplementary table of a previous immunogenomic analysis 25.
Immune infiltration analysis
To investigate the association between the IRLPS risk score and immune infiltrates in liver cancer, infiltration estimation for TCGA-LIHC was obtained from TIMER 2.0 (http://timer.comp-genomics.org/)23, which utilizes multiple immune deconvolution methods including TIMER58, XCELL59, QUANTISEQ60, MCPcounter61, EPIC62, CIBERSORT63, and CIBERSORT-ABS. The correlation (cor) values and P-values were acquired using Spearman's rank correlation test, with P < 0.05 as a significance threshold. The results were visualized in lollipop diagrams and scatter plots by the ggplot2 R package.
Pairing immune-related lincRNAs
The relative expression levels of 429 tumor-specific immune-related lincRNA in each sample were pairwise compared as described previously19,36. Briefly, if the expression value of the first lincRNA is greater than the second in a specific sample, the score of this pair in this sample is 1; otherwise, it is 0. The score of each lincRNA pair was iteratively calculated for all samples. Some lincRNA pairs might be assigned to constant values (0 or 1 in most samples) because of biologically preferential transcription, making them unable to discriminate prognosis from one patient to another and non-informative. Therefore, lincRNA pairs with low variation (the score is 1 or 0 in more than 80% of the samples) were filtered36, and 35,604 valid lincRNA pairs were identified. After combining the survival data from the TCGA database, univariate Cox proportional hazards regression analysis was performed with P < 0.05 as the threshold, and 4288 overall survival-associated lincRNA pairs were extracted. Next, the least absolute shrinkage and selection operator (LASSO) regression was run 1000 times with the glmnet R package64 to prevent overfitting, and 39 lincRNA pairs were selected. Stepwise multivariate Cox regression analysis was conducted to further screen out an optimal combination from these pairs, and the 18 overall survival-associated signatures were obtained. P < 0.05 was set as the inclusion criteria.
The risk score for each patient was calculated based on the following formula: \({\text{risk}}\;{\text{score}} = \sum\nolimits_{i = 1}^{n} {\beta i*\lambda i}\), where n represents the numbers of lincRNA pairs included to construct the signature, β depicts the regression coefficient, and λ represents the 0-or-1 matrix of each lincRNA pair, respectively. We determined the optimal cutoff value based on a time-dependent receiver operating characteristic (ROC) curve to divide patients into high- or low-risk groups. The timeROC R package (version 0.4, weighting = ‘marginal’) was used to plot the time-dependent ROC curves and evaluate the values of area under the ROC curve (AUC)65.
Small molecular compounds identification
To screen out candidate target compounds, differential expression analysis between the high- and low-risk groups was performed using the limma R package66 with the criteria of |log2(foldchange)| ≥ 0.585 and FDR-adjusted P < 0.05. Differentially expressed genes (481 upregulated and 190 downregulated) were uploaded to the Connectivity Map web-server (https://portals.broadinstitute.org/cmap/).
Chemotherapeutic response prediction
Chemosensitivity was predicted using the pRRophetic R package67 based on a public pharmacogenomics database, Genomics of Drug Sensitivity in Cancer. The half-maximal inhibitory concentration (IC50) on each sample was estimated by ridge regression. The prediction accuracy was assessed by tenfold cross-validation based on the training set.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was conducted between patients in different risk groups from the TCGA-LIHC cohort utilizing GSEA software (v4.1.0). We employed h.all.v7.4.symbols.gmt and c2.cp.kegg.v7.4.symbols.gmt as reference gene sets. The screening criteria of items were set as nominal P < 0.05 and FDR-adjusted q < 0.05.
GO and KEGG enrichment analyses
To obtain the potential functions of the differentially expressed genes, GO and KEGG pathway enrichment analyses were implemented using the clusterProfiler and enrichplot R packages (P < 0.05, q < 0.05).
Identification of hub genes
The STRING platform (https://www.string-db.org/) was employed to construct the protein–protein interaction (PPI) network using the differentially expressed genes with a confidence score > 0.9. The network was visualized using Cytoscape (v3.8.0). The cytoHubba plugin in Cytoscape was utilized to perform modular analysis, and the top ten hub genes were identified by the multi-network clustering algorithm.
Statistical analyses
The statistical analyses were performed in R (v.4.1.0)68 with appropriate packages. The association of the 18-IRLPS or hub genes with patients' survival was examined by Cox hazards regression analysis using the survival R package69. The difference in survival between the high-risk and low-risk group of patients was determined using log-rank tests with the survival R package. The chi-square test was used for the categorical data, while the Wilcoxon and Kruskal tests were applied for two or more sets of continuous data.
Ethics declarations
All methods were carried out in accordance with relevant guidelines and regulations.
Data availability
The data sets analysed during this study are available in public, open access repositories listed in this article.
References
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249. https://doi.org/10.3322/caac.21660 (2021).
Zheng, J. et al. Prediction of hepatocellular carcinoma recurrence beyond milan criteria after resection: Validation of a clinical risk score in an international cohort. Ann. Surg. 266, 693–701. https://doi.org/10.1097/sla.0000000000002360 (2017).
Ruan, D.-Y. et al. Nomogram for preoperative estimation of long-term survival of patients who underwent curative resection with hepatocellular carcinoma beyond Barcelona clinic liver cancer stage A1. Oncotarget 7, 61378–61389. https://doi.org/10.18632/oncotarget.11358 (2016).
Kudo, M. et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet (London, England) 391, 1163–1173. https://doi.org/10.1016/s0140-6736(18)30207-1 (2018).
Zhang, Q. et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas. Gut 68, 2019–2031. https://doi.org/10.1136/gutjnl-2019-318912 (2019).
El-Khoueiry, A. B. et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389, 2492–2502. https://doi.org/10.1016/S0140-6736(17)31046-2 (2017).
Zhu, A. X. et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 19, 940–952. https://doi.org/10.1016/s1470-2045(18)30351-6 (2018).
Wang, Y. et al. FDA-approved and emerging next generation predictive biomarkers for immune checkpoint inhibitors in cancer patients. Front. Oncol. 11, 683419. https://doi.org/10.3389/fonc.2021.683419 (2021).
St Laurent, G., Wahlestedt, C. & Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. TIG. 31, 239–251. https://doi.org/10.1016/j.tig.2015.03.007 (2015).
Ulitsky, I. & Bartel, D. P. lincRNAs: Genomics, evolution, and mechanisms. Cell 154, 26–46. https://doi.org/10.1016/j.cell.2013.06.020 (2013).
Zhao, J. & Lawless, M. W. Long noncoding RNAs and their role in the liver cancer axis. Nat. Rev. Gastroenterol. Hepatol. 10, 703. https://doi.org/10.1038/nrgastro.2013.87-c1 (2013).
Wei, L. et al. The emerging role of microRNAs and long noncoding RNAs in drug resistance of hepatocellular carcinoma. Mol. Cancer 18, 147. https://doi.org/10.1186/s12943-019-1086-z (2019).
Atianand, M. K., Caffrey, D. R. & Fitzgerald, K. A. Immunobiology of long noncoding RNAs. Annu. Rev. Immunol. 35, 177–198. https://doi.org/10.1146/annurev-immunol-041015-055459 (2017).
Carpenter, S. & Fitzgerald, K. A. Cytokines and long noncoding RNAs. Cold Spring Harbor Perspect. Biol. https://doi.org/10.1101/cshperspect.a028589 (2018).
Denaro, N., Merlano, M. C. & Lo Nigro, C. Long noncoding RNAs as regulators of cancer immunity. Mol. Oncol. 13, 61–73. https://doi.org/10.1002/1878-0261.12413 (2019).
Xu, J. et al. Long noncoding RNA MIR17HG promotes colorectal cancer progression via miR-17-5p. Can. Res. 79, 4882–4895. https://doi.org/10.1158/0008-5472.Can-18-3880 (2019).
Zhou, Y., Zhu, Y., Xie, Y. & Ma, X. The role of long non-coding RNAs in immunotherapy resistance. Front. Oncol. 9, 1292. https://doi.org/10.3389/fonc.2019.01292 (2019).
Leek, J. T. et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 11, 733–739. https://doi.org/10.1038/nrg2825 (2010).
Heinaniemi, M. et al. Gene-pair expression signatures reveal lineage control. Nat. Methods 10, 577–583. https://doi.org/10.1038/nmeth.2445 (2013).
Wu, J., Zhao, Y., Zhang, J., Wu, Q. & Wang, W. Development and validation of an immune-related gene pairs signature in colorectal cancer. Oncoimmunology 8, 1596715. https://doi.org/10.1080/2162402x.2019.1596715 (2019).
Guo, W. et al. LincRNA-immunity landscape analysis identifies EPIC1 as a regulator of tumor immune evasion and immunotherapy resistance. Sci. Adv. https://doi.org/10.1126/sciadv.abb3555 (2021).
Bindea, G. et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. https://doi.org/10.1016/j.immuni.2013.10.003 (2013).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48, W509–W514. https://doi.org/10.1093/nar/gkaa407 (2020).
Ru, B. et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 35, 4200–4202. https://doi.org/10.1093/bioinformatics/btz210 (2019).
Thorsson, V. et al. The immune landscape of cancer. Immunity 48, 812-830.e814. https://doi.org/10.1016/j.immuni.2018.03.023 (2018).
Cheng, H. W. et al. Identification of thioridazine, an antipsychotic drug, as an antiglioblastoma and anticancer stem cell agent using public gene expression data. Cell Death Dis. 6, e1753. https://doi.org/10.1038/cddis.2015.77 (2015).
Dolma, S. et al. Inhibition of dopamine receptor D4 impedes autophagic flux, proliferation, and survival of glioblastoma stem cells. Cancer Cell 29, 859–873. https://doi.org/10.1016/j.ccell.2016.05.002 (2016).
Cancer Genome Atlas Research Network. Electronic address, w. b. e. & Cancer Genome Atlas Research, N. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 169, 1327-1341.e1323. https://doi.org/10.1016/j.cell.2017.05.046 (2017).
Woo, H. G. & Kim, Y. J. Multiplatform genomic roadmap of hepatocellular carcinoma: A matter of molecular heterogeneity. Hepatology (Baltimore, MD) 68, 2029–2032. https://doi.org/10.1002/hep.29925 (2018).
Zhang, Q. et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell 179, 829-845.e820. https://doi.org/10.1016/j.cell.2019.10.003 (2019).
Zhang, Y. et al. Immune-related long noncoding RNA signature for predicting survival and immune checkpoint blockade in hepatocellular carcinoma. J. Cell Physiol. 235, 9304–9316. https://doi.org/10.1002/jcp.29730 (2020).
Li, M. et al. Four immune-related long non-coding RNAs for prognosis prediction in patients with hepatocellular carcinoma. Front. Mol. Biosci. 7, 566491. https://doi.org/10.3389/fmolb.2020.566491 (2020).
Kong, W. et al. Development and validation of an immune-related lncRNA signature for predicting the prognosis of hepatocellular carcinoma. Front. Genet. 11, 1037. https://doi.org/10.3389/fgene.2020.01037 (2020).
Bai, Y., Lin, H., Chen, J., Wu, Y. & Yu, S. Identification of prognostic glycolysis-related lncRNA signature in tumor immune microenvironment of hepatocellular carcinoma. Front. Mol. Biosci. 8, 645084. https://doi.org/10.3389/fmolb.2021.645084 (2021).
Yuan, M. et al. Identification of a nine immune-related lncRNA signature as a novel diagnostic biomarker for hepatocellular carcinoma. Biomed. Res. Int. 2021, 9798231. https://doi.org/10.1155/2021/9798231 (2021).
Hong, W. et al. Immune-related lncRNA to construct novel signature and predict the immune landscape of human hepatocellular carcinoma. Mol. Therapy. Nucleic Acids 22, 937–947. https://doi.org/10.1016/j.omtn.2020.10.002 (2020).
Deng, M. et al. Construction of a novel immune-related lncRNA signature and its potential to predict the immune status of patients with hepatocellular carcinoma. BMC Cancer 21, 1347. https://doi.org/10.1186/s12885-021-09059-x (2021).
Wang, Z., Song, L., Ye, Y. & Li, W. Long noncoding RNA DIO3OS hinders cell malignant behaviors of hepatocellular carcinoma cells through the microRNA-328/Hhip axis. Cancer Manag. Res. 12, 3903–3914. https://doi.org/10.2147/cmar.S245990 (2020).
Huang, P. et al. HBx/ERα complex-mediated LINC01352 downregulation promotes HBV-related hepatocellular carcinoma via the miR-135b-APC axis. Oncogene 39, 3774–3789. https://doi.org/10.1038/s41388-020-1254-z (2020).
Fridman, W. H., Zitvogel, L., Sautès-Fridman, C. & Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 14, 717–734. https://doi.org/10.1038/nrclinonc.2017.101 (2017).
Vinogradov, S., Warren, G. & Wei, X. Macrophages associated with tumors as potential targets and therapeutic intermediates. Nanomedicine (London) 9, 695–707. https://doi.org/10.2217/nnm.14.13 (2014).
Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 4, 540–550. https://doi.org/10.1038/nrc1388 (2004).
Meng, J. et al. CCL23 suppresses liver cancer progression through the CCR1/AKT/ESR1 feedback loop. Cancer Sci. 112, 3099–3110. https://doi.org/10.1111/cas.14995 (2021).
Flem-Karlsen, K., Fodstad, Ø., Tan, M. & Nunes-Xavier, C. E. B7–H3 in cancer—Beyond immune regulation. Trends Cancer 4, 401–404. https://doi.org/10.1016/j.trecan.2018.03.010 (2018).
Picarda, E., Ohaegbulam, K. C. & Zang, X. Molecular pathways: Targeting B7–H3 (CD276) for human cancer immunotherapy. Clin. Cancer Res. 22, 3425–3431. https://doi.org/10.1158/1078-0432.Ccr-15-2428 (2016).
Wang, F. et al. B7–H3 was highly expressed in human primary hepatocellular carcinoma and promoted tumor progression. Cancer Invest. 32, 262–271. https://doi.org/10.3109/07357907.2014.909826 (2014).
Malta, T. M. et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 173, 338-354.e315. https://doi.org/10.1016/j.cell.2018.03.034 (2018).
El-SayedIbrahim, N., Morsy, H. & Abdelgwad, M. The comparative effect of nisin and thioridazine as potential anticancer agents on hepatocellular carcinoma. Rep. Biochem. Mol. Biol. 9, 452–462. https://doi.org/10.52547/rbmb.9.4.452 (2021).
Otręba, M. & Kośmider, L. In vitro anticancer activity of fluphenazine, perphenazine and prochlorperazine. A review. J. Appl. Toxicol. JAT. 41, 82–94. https://doi.org/10.1002/jat.4046 (2021).
Madunić, J., Madunić, I. V., Gajski, G., Popić, J. & Garaj-Vrhovac, V. Apigenin: A dietary flavonoid with diverse anticancer properties. Cancer Lett. 413, 11–22. https://doi.org/10.1016/j.canlet.2017.10.041 (2018).
Wang, S. M. et al. Apigenin inhibits the growth of hepatocellular carcinoma cells by affecting the expression of microRNA transcriptome. Front. Oncol. 11, 657665. https://doi.org/10.3389/fonc.2021.657665 (2021).
Qin, Y. et al. Apigenin inhibits NF-κB and snail signaling, EMT and metastasis in human hepatocellular carcinoma. Oncotarget 7, 41421–41431. https://doi.org/10.18632/oncotarget.9404 (2016).
Jeon, T. et al. Silencing CDCA8 suppresses hepatocellular carcinoma growth and stemness via restoration of ATF3 tumor suppressor and inactivation of AKT/β-catenin signaling. Cancers (Basel). https://doi.org/10.3390/cancers13051055 (2021).
Fu, J. et al. Identification of the hub gene BUB1B in hepatocellular carcinoma via bioinformatic analysis and in vitro experiments. PeerJ 9, e10943. https://doi.org/10.7717/peerj.10943 (2021).
Li, J., Gao, J. Z., Du, J. L., Huang, Z. X. & Wei, L. X. Increased CDC20 expression is associated with development and progression of hepatocellular carcinoma. Int. J. Oncol. 45, 1547–1555. https://doi.org/10.3892/ijo.2014.2559 (2014).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756. https://doi.org/10.1101/gr.239244.118 (2018).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612. https://doi.org/10.1038/ncomms3612 (2013).
Li, B. et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 17, 174. https://doi.org/10.1186/s13059-016-1028-7 (2016).
Aran, D., Hu, Z. & Butte, A. J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220. https://doi.org/10.1186/s13059-017-1349-1 (2017).
Finotello, F. et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 11, 34. https://doi.org/10.1186/s13073-019-0638-6 (2019).
Becht, E. et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 17, 218. https://doi.org/10.1186/s13059-016-1070-5 (2016).
Racle, J., de Jonge, K., Baumgaertner, P., Speiser, D. E. & Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife https://doi.org/10.7554/eLife.26476 (2017).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. https://doi.org/10.1038/nmeth.3337 (2015).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010).
Wang, Q. W., Lin, W. W. & Zhu, Y. J. Comprehensive analysis of a TNF family based-signature in diffuse gliomas with regard to prognosis and immune significance. Cell Commun. Signal 20, 6. https://doi.org/10.1186/s12964-021-00814-y (2022).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Geeleher, P., Cox, N. & Huang, R. S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 9, e107468. https://doi.org/10.1371/journal.pone.0107468 (2014).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2021).
Therneau, T. M. A Package for Survival Analysis in R. (2021).
Acknowledgements
We thank Chenhao Jiang (Shihezi University School of Medicine) for his helpful comments. This work was supported by the Science and Technology Cooperation Program of Xinjiang Production and Construction Corps (2021BC002), the Youth Science and Technology Innovation Leading Talents Project of Xinjiang Production and Construction Corps (2020CB015), the Youth Innovation Talents Project of Shihezi University (CXBJ201907), and the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2020-PT330-003).
Author information
Authors and Affiliations
Contributions
Y.Z., J.H., and L.W. conceived the study. Y.Z., X.Y., L.Z., and X.G. searched and analyzed the data. Y.Z. and L.W. wrote the draft of the paper. Y.Z., X.C., X.W., J.H., and L.W. revised the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Y., Yang, X., Zhou, L. et al. Immune-related lincRNA pairs predict prognosis and therapeutic response in hepatocellular carcinoma. Sci Rep 12, 4259 (2022). https://doi.org/10.1038/s41598-022-08225-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-08225-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
