
Abstract
Quality and quantity of food items consumed has a crucial effect on phenotypes. In addition to direct effects mediated by nutrient resources, an individual’s diet can also affect the phenotype indirectly by altering its gut microbiota, a potent modulator of physiological, immunity and cognitive functions. However, most of our knowledge of diet-microbiota interactions is based on mammalian species, whereas little is still known about these effects in other vertebrates. We developed a metabarcoding procedure based on cytochrome c oxidase I high-throughput amplicon sequencing and applied it to describe diet composition in breeding colonies of an insectivorous bird, the barn swallow (Hirundo rustica). To identify putative diet-microbiota associations, we integrated the resulting diet profiles with an existing dataset for faecal microbiota in the same individual. Consistent with previous studies based on macroscopic analysis of diet composition, we found that Diptera, Hemiptera, Coleoptera and Hymenoptera were the dominant dietary components in our population. We revealed pronounced variation in diet consumed during the breeding season, along with significant differences between nearby breeding colonies. In addition, we found no difference in diet composition between adults and juveniles. Finally, our data revealed a correlation between diet and faecal microbiota composition, even after statistical control for environmental factors affecting both diet and microbiota variation. Our study suggests that variation in diet induce slight but significant microbiota changes in a non-mammalian host relying on a narrow spectrum of items consumed.
Similar content being viewed by others
Introduction
Diet has a decisive effect on traits tightly linked with fitness, including growth1, reproduction2, immunity3 and various aspects of physiology4. At the same time, an animal’s condition and/or health can have a causal effect on the composition of the diet consumed5. As an example, the preferred diet may vary with age due to differing nutritional demands related to specific ontogenetic stages6.
In addition to the direct consequences of nutrient compounds on fitness-related traits, diet variation may also impose indirect effects by modulating populations of microbial symbionts hosted in the digestive tract of a given individual. This gut microbiota represents the largest fraction of microbial symbionts associated with animal hosts in terms of both cell count and encoded genes7,8. Notably, the gut microbiota is a potent modulator of host physiology and health status, with strong effects on the immune system, digestive tract morphology and digestion efficiency. At the same time, disruptions to the normal gut microbiota have been associated with a number of adverse consequences to host health9,10.
Diet composition has been identified as one of the main drivers of gut microbiota variation in mammals. At larger phylogenetic scales, for example, repeated transitions between carnivory and herbivory were followed by consistent changes in gut microbiota content11,12,13. In omnivorous species, including humans, both long-term and short-term dietary habits impose gut microbiota changes that partly recapitulate transitions observed at the herbivore-carnivore continuum14,15,16. On the other hand, the effect of diet on gut microbiota has also been observed in species relying on a relatively narrow diet spectrum. For example, significant gut microbiota differences have been detected between folivore and frugivore lemurs17, in bison fed on pasture or a grain diet18 and even between strictly insectivorous bat species differing in consumed prey19.
In comparison with mammals, bird diet appears to be a less important factor modulating gut microbiota20. Most studies undertaken on birds have found some support for the effect of diet on interspecific variation, particularly in passerines21,22,23,24. However, many of these studies had limitations related to the use of indirect dietary data and dietary data based on literature searches, but see25,26,27. A rather high within-species variation in dietary items consumed in some cases could mean that an important source of variation was omitted from these analyses. Consequently, further research of within-species diet variation may improve our understanding of dietary-induced gut microbiota changes in free-living populations.
Analyses of diet in free-living populations are usually based on macroscopic examination of faecal samples or undigested food remains28,29. However, this approach is time consuming and demanding in terms of a researcher's expertise. Moreover, there may be a non-negligible risk of limited taxonomic resolution or other specific biases30,31. Stable isotopes analysis represents complementary macroscopic method that has provided important insights into the foraging ecology of free-living populations. This technique has also been successfully used to study the effects of diet on the gut microbiota26,32,33. On the other hand, information on the taxonomic composition of the ingested diet based on the stable isotopes approach is limited. DNA-based methodologies such as metabarcoding, which rely on deep sequencing of DNA markers bearing taxonomic information, represent a promising alternative that could partly overcome such challenges34,35,36. On the other hand, there are some concerns with metabarcoding, such as the poor amplification of certain taxa and/or the weak correlation between their biomass and the corresponding proportions of the sequences37,38.
Studies on diet-microbiota interactions in wild populations of birds are still rare, probably due to scarcity of data on interindividual variation in diet. To our knowledge, there is only one study exploring the effects of natural within-species diet variation on gut microbiota content in birds39. Furthermore, Teyssier et al.40 demonstrated the effect of diet on intraspecific gut microbiota variation in an omnivorous passerine bird through experimentally induced dietary changes. In this study, we developed procedures for metabarcoding-based diet profiling in insectivorous birds and applied this approach in studying interactions between diet and gut microbiota in breeding population of a migratory passerine bird, the barn swallow (Hirundo rustica). In the first step, we explored potential drivers of diet composition in the barn swallow, e.g. temporal variation during the course of the breeding season, spatial variation between breeding colonies and variation between adults and their nestlings. As a further step, we combined existing data on barn swallow faecal microbiota41 with diet profiles for the same individuals to test whether interindividual variation in diet was a predictor of gut microbiota composition and its predicted functions. To our knowledge, our contribution represents the first attempt to integrate individual-based data on metabarcoding-based diet and faecal microbiota composition in an insectivorous bird.
Material and methods
Sample collection
We used previously extracted metagenomics DNA samples collected for our previous study on the faecal microbiota of barn swallows41. Collection of faecal samples from both adults and nestlings (6–12 days after hatching) was conducted at two colonies (Šaloun farm, Lomnice nad Lužnicí [49° 4′ 7.762″ N, 14° 42′ 36.521″ E]; Hamr farm, Lužnice [49° 3′ 25.288″ N, 14° 46′ 10.82″ E]) in the Třeboňsko Protected Landscape Area (Czech Republic; distance between populations = 4.5 km) during the barn swallow breeding season from May to August 2014. The average Julian date of sampling did not differ between the two sites (Welsh t-test: d.f. = 79.9, t = 1.329, p = 0.1843). Both farms are located in a landscape dominated by intensive agriculture and consisted of a mosaic of agricultural fields, hay meadows, fish ponds, and small secondary coniferous or mixed forest patches. The farms differed in terms of the species farmed, with sheep and goats being more prevalent in Šaloun, while production in Hamr farm was more focused on cows and pigs.
To collect faecal samples, adults were placed in a paper bag and nestlings in a plastic beaker filled with paper towels, where they were kept for approx.30 min. Faeces were harvested using a sterile microbiological swab (Copan, Italy), placed in sterile DNA/RNA free cryotubes (Simport, Canada) and stored in liquid nitrogen or at −80 °C for further laboratory analysis. For details on field procedures and faecal sample collection see Kreisinger et al.41 and Petrželková et al.42. Only a single sample was analysed for each individual. We included 140 individuals (47 adults and 93 juveniles) in this study. However, as explained later, we were able to generate useful sequencing data only for 82 individuals (17 adults and 65juveniles), which were included in the final analyses (Supporting information Table A1).
Laboratory analysis
Metagenomic DNA from faecal samples was extracted using commercial PowerSoil kits (MoBio), with faecal microbiota subsequently profiled through high-throughput sequencing of 16S rRNA amplicons, as described in our previous studies41,43. In brief, the V3-V4 variable regions of 16S rRNA were amplified through a polymerase chain reaction (PCR) using universal primers S-D-Bact-0341-b-S-17 (CCTACGGGNGGCWGCAG) and S-D-Bact-0785-a-A-21 (GACTACHVGGGTATCTAATCC)44. Next, sequencing libraries were prepared using TruSeq nano kits (Illumina) and sequenced on Illumina Miseq using the v3 kit (300 bp paired-end reads) at Montpellier-SupAgro (France).
For the purpose of diet profiling, we used universal Cytochrome c oxidase subunit I (COI) primers (BF2-GCHCCHGAYATRGCHTTYCC and BR2-TCDGGRTGNCCRAARAAYCA) targeting a broad range of invertebrate taxa45. We selected these primers as previous in vitro and in silico tests indicated that these primers exhibit a low level of PCR bias compared to existing alternatives45.
To reduce problems associated with the formation of primer-dimers, sequencing libraries were prepared in three PCR steps:
-
1.
COI pre-amplification by gene-specific primers, using a PCR mixture consisting of 5 µl of PCR mastermix, 0.6 µM of forward and reverse COI-specific primer and 3.8 µl of metagenomic DNA. Our pilot PCR analysis revealed that the primers showed a strong affinity to the host DNA. To avoid amplification of host COI, 6 µM of a custom blocking primer containing C3 spacer modification on the 3' end and exhibiting a perfect match to the host COI (ACCGAAGAACCAGAATAGGTGTTGGTAAAGTAC) was added to the PCR reaction. To evaluate potential biases associated with this technique, a subset of samples (n = 23) was also amplified without the blocking primer. PCR cycling conditions consisted of an initial denaturation step (95 °C, 5 min) followed by 22 cycles of denaturation (98 °C, 20 s), blocking primer annealing (53 °C, 15 s), COI-specific primer annealing (47 °C, 15 s) and extension (72 °C, 40 s), followed by a final extension at 72 °C for 5 min.
-
2.
Amplification by primers including tails compatible with sequencing adaptors, using a PCR mixture comprising 5 µl of PCR mastermix, 2.8 µl of ddH2O, 0.6 µM of forward and reverse COI primers flanked by tails complementary to Access Array sequencing adaptors (Fluidigm Corporation, USA) and 1 µl of PCR product from the 1st PCR round. PCR cycling conditions comprised an initial denaturation step (95 °C, 5 min) followed by 15 cycles of denaturation (98 °C, 20 s), primer annealing (50.5 °C, 15 s) and extension (72 °C, 40 s), followed by a final extension at 72 °C for 5 min.
-
3.
PCR-based ligation of sequencing adaptors, using a reaction mixture comprising Access Array sequencing adaptors (4 µl) along with PCR mastermix (10 µl), 4 µl of ddH2O and 2 µl of 25 × diluted PCR product from the 2nd PCR round. PCR cycling consisted of an initial denaturation step (95 °C, 5 min) followed by 16 cycles of denaturation (98 °C, 20 s), primer annealing (55.5 °C, 20 s), and extension (72 °C, 40 s), followed by a final extension at 72 °C for 5 min.
Kappa HIFI HotStart polymerase mastermix (Kapa Biosystems, USA) was used in all PCR reactions. Technical PCR duplicates were prepared for all samples. Products from the 3rd PCR round were quantified by GenoSoft software (VWR International, Belgium) based on band intensities after electrophoresis on 1.5% agarose gel and mixed at equimolar concentration. The final library was purified using Agencourt AmpureXP beads (Beckman Coulter Life Sciences). Products of the desired size were extracted by PipinPrep (Sage Science Inc., USA) and sequenced on Illumina Miseq (v3 kit, 300 bp paired-end reads) at the Central European Institute of Technology (CEITEC, Masaryk University, Brno, Czech Republic).
Bioinformatic analysis of diet profiles
Regions corresponding to gene-specific primers were removed from fastq files using skewer46. Subsequently, the fastq files were quality-filtered (< 2 expected error per read) and denoised using R version 3.4.447, with the dada2 package48 used to define reliable COI amplicon sequence variants (ASVs). Technical duplicates showed significant consistency in Shannon diversities (Pearson correlation: r = 0.982, p < 0.0001) and composition of COI profiles (Procrustean analysis: r = 0.996, p < 0.0001). Consequently, we merged COI profiles for sample duplicates to obtain sample-specific COI profiles. To suppress any effect of PCR and sequencing artefacts, ASVs that were not consistently present in both technical duplicates were eliminated from the dataset e.g.,49. For a limited number of samples, we failed to sequence both duplicates (n = 3). In these cases, we eliminated all ASVs whose presence was not confirmed in samples for which both duplicates were available.
For the purpose of taxonomic classification, 200 top blastn hits for each COI ASV were downloaded from the NCBI nt database and used for the construction of a reference database. Dada2 implementation of RDP classifier50 was subsequently applied for taxonomic assignment of COI ASVs at an 80% posterior confidence threshold. Abundances matrix, representing read counts for individual ASVs in each sample, along with sample metadata, taxonomic annotations and ASVs sequences were merged into a phyloseq database51.
Bioinformatic analysis of faecal microbiota
To assess the effect of diet on faecal microbiota, we used sequencing data previously published in Kreisinger et al.41. The steps for quality filtering, data denoising, and ASV frequency matrix generation were the same as described above. Chimeric ASVs were detected and eliminated using UCHIME52 and the gold.fna reference (available at https://drive5.com/uchime/gold.fa). The taxonomy of non-chimeric ASVs was assigned using the RDP classifier50 and the Silva database (version 138)53 as a reference. We also excluded ASVs corresponding to mitochondria, chloroplasts, or those that were not assigned to any bacterial phylum. Similarly to diet profiling, technical PCR duplicates were made for each faecal microbiota sample. We checked the consistency of their content using Procrustean analysis and eliminated ASVs that were not consistently present in both technical duplicates. Finally, the sequences of the ASVs were aligned using R package DECIPHER54 and a phylogenetic tree was constructed using FastTree255.
Bacterial metagenome functional predictions were conducted using PICRUSt2 pipeline56 using default setup, and predicted metagenomes were categorized into MetaCyc pathways57. Their predicted abundances were used in later statistical analyses.
Statistical analyses
Krona pie-charts58 were used to visualise the taxonomic content of the whole COI dataset. Next, all non-insect ASVs (i.e. not corresponding to putative dietary items) were eliminated. Congruence in Shannon diversities (calculated after the exclusion of non-insect ASVs; hereinafter termed dietary profile) between sample pairs amplified either with or without the blocking primer were assessed as intra-class correlations calculated using the rptR function in the R statistical environment assuming Gaussian error distribution59. We also evaluated congruence in the composition of insect dietary profiles using Procrustean analysis, with Hellinger dissimilarity matrices scaled by Principal Coordinate Analysis (PCoA) used as inputs.
Analysis of covariance (ANCOVA) was used to test whether Shannon diversity (square-root-transformed) of dietary profiles was affected by locality, Julian date of sample collection, age class (i.e. adult vs. young) and by two-way interactions between these variables. Julian date was centred60, both in this statistical model and all later analyses. We also checked whether diversity varied with sequencing depth (log-scaled). Significant predictors of dietary diversity were identified via step-wise backward elimination of nonsignificant variables from the initial full model (i.e. containing all the above-mentioned predictors). After visual exploration of divergence in dietary profile composition by PCoA, variation in dietary composition due to the effect of locality, age class and Julian date of sample collection was analysed by distance-based redundancy analysis (db-RDA)61 running on Hellinger and binary Jaccard dissimilarities among samples. We considered linear, quadratic and cubic effect of Julian date to account for potentially non-monotonic abundance changes in dietary items during the breeding season. Hellinger dissimilarities automatically account for the different number of sequences between samples. Jaccard dissimilarities were calculated after rarefaction of the abundance matrix (n = 536 sequences per sample, i.e. the minimum sequencing depth achieved). To demonstrate that rarefaction has a negligible effect on overall beta diversity, we calculated Jaccard dissimilarities for a subset of samples with > 5000 sequences (n = 47), which were rarefied to either 500 or 5000 seqs./sample. The resulting strength of correlation between these two distance matrices was high (Procrustean analysis: r = 0.934, p = 0.0001). The db-RDA model selection strategy was based on the forward step-wise approach implemented in the ordiR2step function (vegan package in R)62. The abundances of dietary taxa that varied due to the effects of predictors suggested by db-RDA were identified using generalised linear models with negative binomial distribution in the DESeq2 package63. False discovery rates (FDR)64 were used for multiple testing corrections.
To examine beta diversity of the faecal microbiota, we used phylogenetically controlled weighted and unweighted UniFrac distances in addition to Jaccard and Hellinger dissimilarities. UniFrac only marginally accounts for variation caused by phylogenetically related ASVs. The unweighted UniFrac, which only accounts for the absence/presence of ASVs, was calculated after the abundance matrix rarefaction (threshold = 2315 sequences per sample). The weighted UniFrac (accounting for variation in the abundance of ASVs) was calculated based on the proportions of ASVs in each sample. To infer variation in predicted metagenome content, only Hellinger dissimilarities in functional profiles were used. Procrustean analysis was applied to test for correlation between interindividual divergence in diet and faecal microbiota or predicted metagenome profiles. Furthermore, we employed db-RDA and variation partitioning analysis (varpart function in the R package vegan) to account for direct and indirect effects of environmental factors, where dissimilarities in faecal microbiota composition (i.e. Hellinger , Jaccard, or UniFrac) or predicted metagenomes (only Hellinger dissimilarities) were considered as a response and divergence in diet composition (i.e. PCoA axis scores for Hellinger or Jaccard divergences in diet profiles) and matrix of other variables (including linear, quadratic and cubic effects of Julian date, locality and age class) were included as explanatory and/or conditional variables. To prevent db-RDA model overfitting, we only considered PCoA axes for diet that exhibited a significant correlation with microbiota composition, selected using a forward selection approach (ordiR2step function from R package vegan). Finally, we applied the joint species distribution model (JSDM) from the boral package65 to estimate pair-wise residual correlations between diet taxa and bacterial ASVs of predicted metagenome pathways after accounting for the effects of explanatory variables. A similar approach was previously applied to search for cross-domain correlations between faecal microbiota and the intestinal helminth community66. To run JSDM, we merged community matrices for insect genera and 16S rRNA ASVs or predicted metagenome profiles and used them as model responses. To account for uneven sequencing depth, we used a model offset equal to the log-transformed number of sequences for a given sample and marker gene (or total predicted abundance of all metagenomic pathways in given sample). Dietary genera and bacterial ASVs detected in < 10 samples were excluded. Similarly, metagenome features with a relative abundance < 0.01% were not considered. Locality, age class and Julian date of sample collection were considered as explanatory variables. We considered JSDM versions, with the effect of Julian date modelled as either, a linear and quadratic term, or a linear, quadratic and cubic term and reported residual correlations that received substantial support based on 95% posterior credible intervals for both these JSDM versions. The models were fitted using default priors (described in boral documentation) and assuming negative binomial distribution of read counts for bacterial and dietary taxa. Diet vs. microbiota taxa correlations were estimated based on a Markov Chain Monte Carlo simulation consisting of 50,000 iterations. The thinning interval was set to 40 iterations, with the first 1000 iterations discharged as burn-in.
Ethics declarations
All field procedures were conducted in accordance with European Union Guidelines for Animal Care and Treatment and approved by the Animal Care and Use Committees at the Czech Academy of Sciences (041/2011) and Charles University in Prague (4789/2008-0).
Results
Effect of blocking primers on COI amplification
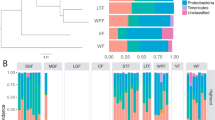
We analysed faecal samples from 140 individuals (47 adults and 93 juveniles). PCR amplification failed in 34 samples; hence, we only sequenced 106 individuals (23 adults and 83 juveniles) for the COI profile. We obtained 2,369,181 high-quality reads that were grouped into 1,591 COI ASVs. Median sequencing depth corresponded to 13 258 sequences per sample (range = 536–60,484). Insects representing putative dietary components formed the dominant fraction of COI profiles (47% of all reads, 961 ASVs). Non-target taxa were represented by avian ASVs (18% of reads), plants (10% of reads), fungi (namely Oomycetes, 3% of reads) and putative symbiotic Arachnida (Trombidiformes and Dermanyssidae, 6% reads; Supporting information Fig. A1). The relative abundance of avian ASVs was significantly higher when blocking primer was not included in the first PCR reaction (62% of reads per sample vs. 1.3% of reads; Wilcoxon rank sum test: W = 11, p < 0.0001).
Next, we evaluated the potential effects of using the blocking primer on alpha diversity and composition of dietary profiles (i.e., insect ASVs only). Because the number of insect reads was very low for a subset of the samples amplified without blocking primer, we performed these comparisons for 21 pairs of samples, both of which contained at least 400 insect reads. Sample pairs that were amplified both with and without blocking primers (n = 21 pairs) exhibited high consistency in Shannon diversities (Intra-class correlation = 0.961, 95% bootstrap confidence intervals = 0.927–0.987, permutation-based p = 0.0001) and relative abundance of individual ASVs (Procrustean analysis: r = 0.999, p < 0.0001; Fig. 1). For the purpose of all subsequent analyses, diet profiles for sample duplicates generated with and without blocking primers were merged and insect ASVs were grouped into genus-level bins.

Consistence between diet profiles generated with or without blocking primer, assessed based on (A) Shannon diversity correlations and (B) procrustean superimposition for Hellinger dissimilarities in diet profiles.
Diet profile variation
Because the total number of insect reads was low for a subset of individuals, which could negatively affect the robustness of dietary profiles, we based all analyses on diet variation and faecal microbiota-diet correlations on 82 individuals (17 adults and 68 juveniles; Supporting information Table A1) with > 400 insect reads, while 24 individuals with < 400 insect reads were excluded. Importantly, our later missing data analyses suggest that the excluded samples with small numbers of insect reads did not differ in diet composition.
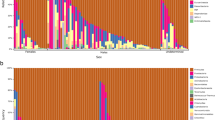
We detected 171 insect genera or higher insect taxa (in the case of insufficient support for genus-level delimitation). In terms of reads counts, the most abundant insect order was Diptera (61% of reads per sample on average, dominated by the genera Chironomus and Nephrotoma). A considerable proportion of the dietary profile comprised Hemiptera (17% of reads, dominated by the genus Lygus), Coleoptera (14% of reads, dominated by the genera Aphodius and Psylliodes) and Hymenoptera (6% of reads, dominated by ants of the genus Lasius; Supporting information Fig. A2). Other taxa were represented by < 1% reads per sample on average. The number of insect genera per sample ranged between one and 18 (median = 4, mean = 5.22). Individual samples were mostly dominated by a single insect genus (Fig. 2). Subsequently, rarefaction analysis for 1–5001 randomly selected reads per sample revealed that sequencing coverage corresponding to ~ 500 sequences per sample was sufficient to capture the majority of genus-level diversity (Supporting information Fig. A3).

Insect genera detected by diet profiling of barn swallow faecal samples. The average proportion of reads is shown. Taxa present at low abundances (< 1% of all reads in the entire dataset) are indicated as "others".
Using ANCOVA, we found that the Shannon diversity of dietary profiles differed between localities (F(1,80) = 5.087, p = 0.0268). However, there was no difference in diversity between adults and juveniles (F(1,79) = 1.831, p = 0.180, mean Shannon diversity [± S.E] = 0.4047 ± 0.087 for adults and 0.663 ± 0.065 for juveniles) and we found no support for any other predictor (i.e. by Julian date of sample collection and by two-way interactions between predictors) of diet diversity (p > 0.05 in all cases).
Explorative insights provided by PCoA for Jaccard and Hellinger dissimilarities suggested an effect of both locality and Julian date on variation in dietary composition (Fig. 3). Specifically, scores for the second PCoA axis separated samples from different localities (Wilcoxon test: W = 459, p < 0.0001 for Jaccard and W = 514, p = 0.005 for Hellinger dissimilarities) and were correlated with Julian date of sample collection (Spearman correlation, rho = 0.248, p = 0.025 for Jaccard and rho = 0.413, p = 0.0001 for Hellinger dissimilarities). Constrained db-RDA models running on Hellinger and Jaccard dissimilarities provided comparable results (Table 1). However, neither PCoA nor db-RDA supported a difference in dietary composition between adults and juveniles. Inclusion of polynomial terms into the final db-RDA models suggested non-monotonic variation in dietary items during the breeding season. Subsequently, DESeq2 analysis aimed at identifying particular insect genera involved in this variation included both the effect of locality and Julian date. In the case of Julian date, we tested for the effect of cubic and quadratic polynomes via likelihood ratio tests. While no insect genera exhibited cubic association with Julian date of sample collection, the abundance of 14 insect genera exhibited quadratic correlation with sampling date (Supporting information Fig. A4 and Table A2). For example, flies from the genera Pollenia and Hybomitra and from the family Tabaninae, as well as crane flies (genus Nephrotoma) and ants (genus Lasius), were most commonly detected in the middle of the breeding season. Conversely, beetles from the genus Aphodius were more common at the beginning of the breeding season, while mosquitoes from the genera Culiseta and Ochlerotatus, as well as Hemiptera from the genus Lygus, were more prevalent late in the breeding season. No insect genus exhibited significant variation between localities after statistical control for within-season variation (i.e. quadratic effect of sample collection date) and multiple testing corrections.

Variation in diet profile composition based on PCoA running on (A) Hellinger and (B) Jaccard dissimilarities. Samples from adult versus young are indicated by different plotting characters. Samples taken during the breeding season are indicated with different shades of grey. Data for different localities are in different facets.
Missing data analyses
As already mentioned, we excluded samples in which PCR failed (n = 34) and in which the number of target insect reads was low (n = 24). Here we investigated the reasons for the relatively high frequency of these failures, as their non-random distribution could potentially influence interpretations of diet variation and diet-microbiota correlation analyses.
We found that PCR failure was more common in adults (51.1%) than in juveniles (10.8% chi-squared test: d.f. = 1, χ2 = 27.591, p < 0.0001). Based on electrophoresis gel band intensity, we observed lower PCR outputs for bacterial 16S rRNA amplicons in adults than in juveniles (Welsh t-test: d.f. = 124.44, t = − 3.458, p = 0.001; amplicons prepared in parallel for the same samples). Hence, we suggest that the lower PCR success in adults was caused by an overall lower quantity and quality of DNA template, probably due to lower amount of faecal material (Schmiedova et al. unpublished). On the other hand, we did not detect any effect of sample location (chi-squared test: d.f. = 1, χ2 = 0.037, p = 0.848) or Julian date (Welsh t-test: d.f. = 45.882, t = 0.315, p = 0.755) on the probability of PCR failure. In addition, we found no difference in microbiota composition between samples that failed vs. those that passed the PCR step with diet primers (PERMANOVA: pseudo-F(1,137) = 0.9156, R2 = 0.006, p = 0.459 for weighted UniFrac and pseudo-F(1,137) = 1.1648, R2 = 0.008, p = 0.157 for Hellinger dissimilarities) after statistical control for the effect of age and locality (i.e. predictors that impacted microbiota composition in our population)41.
Frequency of samples with a low vs. sufficient number of insect reads did not vary between adults and juveniles (chi-squared test: d.f. = 1, χ2 = 0.199, p = 0.656) or Julian date (Welsh t test: d.f. = 24.088, t = − 0.550, p = 0.588). However, there was a higher percentage of samples with a low number of insect reads at Saloun farm (40%) than Hamr farm (4%; chi-squared test: d.f. = 1, χ2 = 19.666, p < 0.0001), which was paralleled by a higher fraction of no-target reads at Saloun farm (58.8%) than at Hamr farm (31.3%) across all sequenced samples (Welsh t test: d.f. = 103.75, t = 3.904, p = 0.0002). This difference was mainly associated with increased percentage of symbiotic Arachnida (36.9% vs. 4.3% of reads) at the former location, suggesting that higher abundance of these nontarget taxa could compromise efficient amplification of insect DNA. Alternatively, it could also be that insect taxa that were poorly captured by our wet-lab protocol were more abundant at Saloun Farm. It is likely that this difference would affect resulting PCR yields. Contrary to this prediction, the concentration of PCR products, as determined by the gel band intensities of the gel bands for diet amplicons, was the same at both sites (Welsh t test: d.f. = 103.57, t = − 0.446, p = 0.656). Importantly, we also found no difference in microbiota composition (PERMANOVA : pseudo-F(1,103) = 0.969, R2 = 0.010, p = 0.284 for weighted UniFrac and pseudo-F(1,103) = 0.904, R2 = 0.009, p = 0.498 Hellinger distances) or diet composition (PERMANOVA for Hellinger distances: pseudo-F(1,101) = 1.052, R2 = 0,01, p = 0.376, only samples with at least one insect read) between samples with low versus sufficient number of insect reads while we accounted for locality-specific variation in microbiota or diet content.
Association between diet variation and faecal microbiota composition
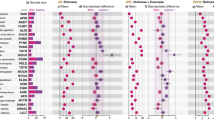
Bivariate Procrustean analysis revealed a significant congruence between dietary profiles and faecal microbiota or metagenome content composition with Procrustean correlation coefficients between 0.51 and 0.80 (Supporting information Fig. A5 and Table A3). Furthermore, db-RDA and subsequent variation partition analyses indicated that diet had a low, though significant, effect on faecal microbiota, independent of other covariates (R2adjusted ranging between 0.033 and 0.149, Supporting information Table A4). Db-RDA models also revealed considerable effect of diet on predicted metagenome functions (Supporting information Table A4). Moreover, the most important principal components of dietary variation typically correlated with microbiota composition and its predicted functions (Supporting information Table A5). Faecal microbiota was also significantly affected by an independent effect of environmental covariates (R2adjusted ranging between 0.022 and 0.048, Supporting information Table A4). Finally, variation partitioning revealed a fraction of faecal microbiota variation explained by both diet and environment (R2adjusted ranging between 0.005 and 0.027, Supporting information Table A4). JSDM indicated 23 highly supported (posterior confidence > 0.95 for both fitted JSDM models) residual correlations between bacterial ASVs and insect genera present in the dietary profile (Fig. 4) and 14 highly supported residual correlations between insect genera and predicted metagenome pathways (Supporting information Fig. A6).

Residual correlations between bacterial ASVs and insect genera detected in faecal samples based on JSDM. Shown are correlations with posterior support > 0.95.
Discussion
Variation in barn swallow diet
Diptera represented the largest fraction of reads in our study, followed by Coleoptera, Hemiptera, and Hymenoptera. Interestingly, dietary profiles were mostly dominated by a single insect genus. We suspect this is due to the short retention and digestion time typical for birds67,68. The order-level taxonomic composition of diet in our population was roughly comparable to the dietary content reported in previous studies based on macroscopic examination of undigested food remains or faecal samples28,69,70,71,72. However, the dominance of individual insect groups tends to differ between previously published studies, presumably because of dietary variation in space and throughout the breeding season28,69,71. We explicitly addressed this possibility by field sampling within two months of the breeding season at two ca. 4.5 km distant breeding colonies, and were able to show that both spatial variation and collection date affected consumed diet. This illustrates the high spatio-temporal variability of dietary items consumed and shows that the dietary patterns observed during short sampling periods or at single locality cannot be easily generalised, even in aerial insectivores. The temporal variation in consumed diet can be related to the fact that the abundance peaks varied among consumed insects. Similarly, differences in diet composition between the two breeding colonies may be explained by differences in breeding livestock or other environmental factors. Unfortunately, we cannot directly support these explanations because prey availability was not quantified. Obtaining such data can be problematic with regards to aerial foragers, particularly as their hunting strategy, including intensity of hunting, height of hunting trips and their distance from the nest site, may vary dramatically with actual environmental conditions72,73,74.
Offspring can be very demanding with regards to the quality and quantity of nutrients required during the early post-hatching phases of development; hence, parents of many animal species supplement the offspring’s diet with specific dietary items75,76 or select breeding microhabitats that satisfy their dietary requirements77,78. Furthermore, adults may switch their typical foraging preferences during the breeding season in order to provide their progeny with a high-quality diet. Previous studies on the barn swallow suggest that parents feed themselves with smaller dietary items than those they provide to nestlings72,79. In the present study, however, both dietary composition and diet alpha diversity failed to provide support for the idea that food composition differs between adults and juveniles.
Correlation between diet and faecal microbiota
While knowledge of gut microbiota in free-living vertebrates is gradually increasing, the extent to which their gut microbiota is affected by variation in diet is still not sufficiently understood. Most studies on wild vertebrate species have applied a comparative approach aimed at detection of microbiota variation between animal species13,20,24,80 or populations18,81. However, to our knowledge, there have been just a few studies attempting to directly integrate metabarcoding data on dietary composition and microbiota profiles on an individual basis19,82,83,84,85.
In comparison with more widely studied mammals, passerine birds have a clearly distinct composition of host-associated microbial communities11,23,80. The microbiota in such communities is characterised by rapid temporal changes at the intra-individual level, with just a few bacteria exhibiting some level of stability over time41. Based on current knowledge, interspecific differences in passerine gut microbiota composition appear to be rather low23,80. Furthermore, it has been shown that passerine gut microbiota structure can be affected by social contacts, age, sex, host immunity or blood concentrations of steroid hormones43,86,87,88. All the above-mentioned factors, however, usually explain just a limited fraction of total gastrointestinal microbiota variation. As such, we hypothesised whether the unexplained variation in gut microbiota may be related to actual diet composition. While bivariate Procrustean analysis indicated a significant correlation between gut microbiota and diet profile composition, use of this approach is problematic as bivariate approaches fail to distinguish direct links from indirect effects mediated by shared correlation of taxa abundance with environmental variables. To address this, we applied db-RDA modelling followed by variance partitioning, which indicated a significant fraction of gut microbiota variation explained by variation in diet, independent of the effect of environmental covariates modulating gut microbiota and/or diet consumed. Interestingly, the db-RDA models showed the strongest association between diet and phylogenetically controlled weighted UniFrac dissimilarities in microbiota composition and predicted metagenome functions. This suggests that individual insect taxa impose similar effects on related bacteria that likely have similar metabolic functions. JSDM identified 23 correlations between prey genera and bacterial ASVs. Most of these correlations were associated with abundance changes in Helina and Sarcophaga flies. In some cases, bacterial ASVs involved in these interactions corresponded to putative insect symbionts (e.g. a positive link between Sarcophaga and Rickettsia ASV_2152), suggesting that gut microbiota can be at least partly affected by bacteria present in the diet. At the same time, however, variation in dietary items was also associated with abundance changes in several bacteria that are widespread residents of vertebrate gut or reproductive tracts (e.g. Candidatus Arthromitus, Enteroccocus, Ureaplasma), suggesting that diet can also modulate proliferation of bacteria already residing in the barn swallow host. According to JSDMs, most changes in metagenome pathways were associated with abundance changes of Sarcophaga in diet. In particular, we observed positive correlations between Sarcophaga and several pathways involved in metabolism of nucleic acids and simple sugars. Despite being significant, the overall effect of diet on the gut microbiota of barn swallows, and in birds in general, appears to be of lower importance than in mammals20, providing further evidence for clear differences in host-microbiota interactions in these two vertebrate clades. Deducing mechanisms behind these differences is rather challenging, given the current state of knowledge. Nevertheless, we speculate that the explanation involves differences in digestion physiology between the two groups. In particular, diet passage through the gut is much faster in passerines than in mammals and, therefore, does not depend largely on bacterial fermentation67,68. Consequently, there would be a limited opportunity for bacterial populations within the gut to be affected by the diet consumed.
Methodical considerations
In our study, we used recently designed universal COI primers that are comparable with existing primers for ribosomal genes in terms of their capability to target a wide range of arthropod taxa45. The broad taxonomic coverage achieved by our protocol was also evident based on our sequencing data, where several plant and fungal taxa were effectively amplified alongside barn swallow COI, resulting in a large proportion of non-target sequences in our dataset. Consequently, researchers intending to adopt these primers should account for this and adjust target sequencing depth accordingly. Further, to uncover potential biases in biological interpretations, researchers should also consider an in-depth missing values analysis, to identify sources of commonly occurring PCR failures and low numbers of target sequences.
To partly overcome the problem with non-target reads, primers blocking passerine COI amplification were added to the PCR reaction. Though this procedure is commonly used in metabarcoding-based diet analyses, it has been noted that blocking primers may systematically bias abundances of taxa in resulting profiles89,90. However, our data were unlikely to be affected as there was a high consistency in diversity and insect COI profile composition for sample duplicates that were prepared with and without blocking primers.
Conclusions
Using COI profiling of faecal samples, we described diet variation in a breeding barn swallow population and demonstrated that diet metabarcoding is a promising non-invasive alternative to traditional diet analysis approaches in insectivorous birds. We also showed that use of blocking primers does not bias the content of diet profiles, probably due to phylogenetic disparity between passerines and their insect prey. The diet of barn swallows showed high interindividual variation, which was partly explained by variation among colonies and systematic changes during the breeding season. Finally, our data provides correlative support for the effect of diet consumed on faecal microbiota composition, independent of environmental factors affecting both diet and faecal microbiota.
Data availability
Sequencing data are available at the European Nucleotide Archive under project accession number PRJEB14586 for the 16S bacterial profile and PRJEB46476 for the COI profile. Accession numbers for each sample are provided in supporting information Table A1. Scripts associated with data analyses are archived in Github repository (https://github.com/jakubkreisinger/Swallow_diet).
References
Büyükdeveci, M. E., Balcázar, J. L., Demirkale, İ & Dikel, S. Effects of garlic-supplemented diet on growth performance and intestinal microbiota of rainbow trout (Oncorhynchus mykiss). Aquaculture 486, 170–174 (2018).
Maklakov, A. A. et al. Sex-specific fitness effects of nutrient intake on reproduction and lifespan. Curr. Biol. 18, 1062–1066 (2008).
Totsch, S. K. et al. Effects of a Standard American Diet and an anti-inflammatory diet in male and female mice. Eur. J. Pain 22, 1203–1213 (2018).
Green, D. A. & Millar, J. S. Changes in gut dimensions and capacity of Peromyscus maniculatus relative to diet quality and energy needs. Can. J. Zool. 65, 2159–2162 (1987).
Jones, V. A. et al. Crohn’s disease: Maintenance of remission by diet. Lancet 2, 177–180 (1985).
Hirai, T. Ontogenetic change in the diet of the pond frog, Rana nigromaculata. Ecol. Res. 17, 639–644 (2002).
Qin, J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010).
Sender, R., Fuchs, S. & Milo, R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell 164, 337–340 (2016).
Reikvam, D. H. et al. Depletion of murine intestinal microbiota: effects on gut mucosa and epithelial gene expression. PLoS ONE 6, e17996 (2011).
Sommer, F. & Bäckhed, F. The gut microbiota-masters of host development and physiology. Nat. Rev. Microbiol. 11, 227–238 (2013).
Ley, R. E. et al. Evolution of mammals and their gut microbes. Science 320, 1647–1651 (2008).
Muegge, B. D. et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332, 970–974 (2011).
Youngblut, N. D. et al. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 10, 2200 (2019).
Ley, R. E., Turnbaugh, P. J., Klein, S. & Gordon, J. I. Microbial ecology: Human gut microbes associated with obesity. Nature 444, 1022–1023 (2006).
Zhu, Y. et al. Beef, chicken, and soy proteins in diets induce different gut microbiota and metabolites in rats. Front. Microbiol. 8, 1395 (2017).
Zimmer, J. et al. A vegan or vegetarian diet substantially alters the human colonic faecal microbiota. Eur. J. Clin. Nutr. 66, 53–60 (2012).
McKenney, E. A., Rodrigo, A. & Yoder, A. D. Patterns of gut bacterial colonization in three primate species. PLoS ONE 10, e0124618 (2015).
Bergmann, G. T. Microbial community composition along the digestive tract in forage- and grain-fed bison. BMC Vet. Res. 13, 253 (2017).
Phillips, C. D. et al. Microbiome structural and functional interactions across host dietary niche space. Integr. Comp. Biol. 57, 743–755 (2017).
Song, S. J. et al. Comparative analyses of vertebrate gut microbiomes reveal convergence between birds and bats. mBio 11, e02901–19 (2020).
Bodawatta, K. H., Sam, K., Jønsson, K. A. & Poulsen, M. Comparative analyses of the digestive tract microbiota of New Guinean passerine birds. Front. Microbiol. 9, 1830 (2018).
Capunitan, D. C., Johnson, O., Terrill, R. S. & Hird, S. M. Evolutionary signal in the gut microbiomes of 74 bird species from Equatorial Guinea. Mol. Ecol. 29, 829–847 (2020).
Hird, S. M., Sánchez, C., Carstens, B. C. & Brumfield, R. T. Comparative gut microbiota of 59 neotropical bird species. Front. Microbiol. 6, 1403 (2015).
Waite, D. W. & Taylor, M. W. Characterizing the avian gut microbiota: membership, driving influences, and potential function. Front. Microbiol 5, 223 (2014).
Loo, W. T., Dudaniec, R. Y., Kleindorfer, S. & Cavanaugh, C. M. An inter-island comparison of Darwin’s finches reveals the impact of habitat, host phylogeny, and island on the gut microbiome. PLoS ONE 14, e0226432 (2019).
Loo, W. T., García-Loor, J., Dudaniec, R. Y., Kleindorfer, S. & Cavanaugh, C. M. Host phylogeny, diet, and habitat differentiate the gut microbiomes of Darwin’s finches on Santa Cruz Island. Sci. Rep. 9, 1–12 (2019).
Murray, M. H. et al. Gut microbiome shifts with urbanization and potentially facilitates a zoonotic pathogen in a wading bird. PLoS ONE 15, e0220926 (2020).
Orłowski, G. & Karg, J. Diet of nestling Barn Swallows Hirundo rustica in rural areas of Poland. Cent. Eur. J. Biol. 6, 1023–1035 (2011).
Wiesenborn, W. D. & Heydon, S. L. Diets of breeding southwestern willow flycatchers in different habitats. Wilson J. Ornithol. 119, 547–557 (2007).
Moreby, S. J. An aid to the identification of arthropod fragments in the faeces of gamebird chicks (Galliformes). Ibis 130, 519–526 (1988).
Zeale, M. R. K., Butlin, R. K., Barker, G. L. A., Lees, D. C. & Jones, G. Taxon-specific PCR for DNA barcoding arthropod prey in bat faeces. Mol. Ecol. Resour. 11, 236–244 (2011).
Bolnick, D. I. et al. Individuals’ diet diversity influences gut microbial diversity in two freshwater fish (threespine stickleback and Eurasian perch). Ecol. Lett. 17, 979–987 (2014).
Bolnick, D. I. et al. Individual diet has sex-dependent effects on vertebrate gut microbiota. Nat. Commun. 5, 4500 (2014).
Clarke, L. J., Soubrier, J., Weyrich, L. S. & Cooper, A. Environmental metabarcodes for insects: In silico PCR reveals potential for taxonomic bias. Mol. Ecol. Resour. 14, 1160–1170 (2014).
Deagle, B. E., Jarman, S. N., Coissac, E., Pompanon, F. & Taberlet, P. DNA metabarcoding and the cytochrome c oxidase subunit I marker: Not a perfect match. Biol. Lett. 10, 20140562 (2014).
Elbrecht, V. et al. Testing the potential of a ribosomal 16S marker for DNA metabarcoding of insects. PeerJ 4, e1966 (2016).
Elbrecht, V. & Leese, F. Can DNA-based ecosystem assessments quantify species abundance? Testing primer bias and biomass—Sequence relationships with an innovative metabarcoding protocol. PLoS ONE 10, e0130324 (2015).
Piñol, J., San Andrés, V., Clare, E. L., Mir, G. & Symondson, W. O. C. A pragmatic approach to the analysis of diets of generalist predators: The use of next-generation sequencing with no blocking probes. Mol. Ecol. Resour. 14, 18–26 (2014).
Góngora, E., Elliott, K. H. & Whyte, L. Gut microbiome is affected by inter-sexual and inter-seasonal variation in diet for thick-billed murres (Uria lomvia). Sci. Rep. 11, 1200 (2021).
Teyssier, A. et al. Diet contributes to urban-induced alterations in gut microbiota: Experimental evidence from a wild passerine. Proc. R. Soc. B 287, 20192182 (2020).
Kreisinger, J. et al. Temporal stability and the effect of transgenerational transfer on fecal microbiota structure in a long distance migratory bird. Front. Microbiol. 8, 50 (2017).
Petrželková, A. et al. Brood parasitism and quasi-parasitism in the European barn swallow (Hirundo rustica rustica). Behav. Ecol. Sociobiol. 69, 1405–1414 (2015).
Kreisinger, J. et al. Fecal microbiota associated with phytohaemagglutinin-induced immune response in nestlings of a passerine bird. Ecol. Evol. 8, 9793–9802 (2018).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1 (2013).
Elbrecht, V. & Leese, F. Validation and development of COI metabarcoding primers for freshwater macroinvertebrate bioassessment. Front. Environ. Sci. 5, 11 (2017).
Jiang, H., Lei, R., Ding, S.-W. & Zhu, S. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 15, 182 (2014).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2018).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Meth 13, 581–583 (2016).
Pafčo, B. et al. Metabarcoding analysis of strongylid nematode diversity in two sympatric primate species. Sci. Rep. 8, 5933 (2018).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 (2013).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Wright, E. S. RNAconTest: Comparing tools for noncoding RNA multiple sequence alignment based on structural consistency. RNA 26, 531–540 (2020).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650 (2009).
Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688 (2020).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes—A 2019 update. Nucleic Acids Res. 48, D445–D453 (2020).
Ondov, B. D., Bergman, N. H. & Phillippy, A. M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 12, 385 (2011).
Stoffel, M. A., Nakagawa, S. & Schielzeth, H. rptR: Repeatability estimation and variance decomposition by generalized linear mixed-effects models. Methods Ecol. Evol. 8, 1639–1644 (2017).
Schielzeth, H. Simple means to improve the interpretability of regression coefficients. Methods Ecol. Evol. 1, 103–113 (2010).
Legendre, P. & Anderson, M. J. Distance-based redundancy analysis: Testing multispecies responses in multifactorial ecological experiments. Ecol. Monogr. 69, 1–24 (1999).
Oksanen, J. et al. vegan: Community Ecology Package. R package version 2.5-2. 2018. (2018).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 57, 289–300 (1995).
Hui, F. K. C. boral–Bayesian ordination and regression analysis of multivariate abundance data in R. Methods Ecol. Evol. 7, 744–750 (2016).
Aivelo, T. & Norberg, A. Parasite-microbiota interactions potentially affect intestinal communities in wild mammals. J. Anim. Ecol. 87, 438–447 (2018).
Caviedes-Vidal, E. et al. The digestive adaptation of flying vertebrates: High intestinal paracellular absorption compensates for smaller guts. Proc. Natl. Acad. Sci. U.S.A. 104, 19132–19137 (2007).
McWhorter, T. J., Caviedes-Vidal, E. & Karasov, W. H. The integration of digestion and osmoregulation in the avian gut. Biol. Rev. Camb. Philos. Soc. 84, 533–565 (2009).
Grigolo, C. P. et al. Diet heterogeneity and antioxidant defence in Barn Swallow Hirundo rustica nestlings. Avocetta 43, 1 (2019).
Law, A. A. et al. Diet and prey selection of barn swallows (Hirundo rustica) at Vancouver International Airport. Canadian Field-Naturalist 131, 26 (2017).
McClenaghan, B., Nol, E. & Kerr, K. C. R. DNA metabarcoding reveals the broad and flexible diet of a declining aerial insectivore. Auk 136, uky003 (2019).
Turner, A. K. The use of time and energy by aerial feeding birds (University of Stirling, 1981).
Bryant, D. M. & Turner, A. K. Central place foraging by swallows (Hirundinidae): The question of load size. Anim. Behav. 30, 845–856 (1982).
Møller, A. P. Advantages and disadvantages of coloniality in the swallow, Hirundo rustica. Anim. Behav. 35, 819–832 (1987).
Brodmann, P. A. & Reyer, H.-U. Nestling provisioning in water pipits (Anthus spinoletta): Do parents go for specific nutrients or profitable prey?. Oecologia 120, 506–514 (1999).
Herlugson, C. J. Food of adult and nestling Western and Mountain bluebirds. Murrelet 63, 59–65 (1982).
Batt, B. D. J., Anderson, M. G. & Afton, A. D. Ecology and management of breeding waterfowl (Univ of Minnesota Press, 1992).
Douglas, D. J. T., Evans, D. M. & Redpath, S. M. Selection of foraging habitat and nestling diet by Meadow Pipits Anthus pratensis breeding on intensively grazed moorland. Bird Study 55, 290–296 (2008).
Waugh, D. R. Predation strategies in aerial feeding birds (University of Stirling, 1978).
Kropáčková, L. et al. Co-diversification of gastrointestinal microbiota and phylogeny in passerines is not explained by ecological divergence. Mol. Ecol. 26, 5292–5304 (2017).
Kohl, K. D. et al. Physiological and microbial adjustments to diet quality permit facultative herbivory in an omnivorous lizard. J. Exp. Biol. 219, 1903–1912 (2016).
Baxter, N. T. et al. Intra- and interindividual variations mask interspecies variation in the microbiota of sympatric Peromyscus populations. Appl. Environ. Microbiol. 81, 396–404 (2015).
Holmes, I. A., Monagan, I. V. Jr., Rabosky, D. L. & Davis Rabosky, A. R. Metabolically similar cohorts of bacteria exhibit strong cooccurrence patterns with diet items and eukaryotic microbes in lizard guts. Ecol. Evol. 9, 12471–12481 (2019).
Li, H. et al. Diet diversity is associated with beta but not alpha diversity of pika gut microbiota. Front. Microbiol. 7, 1169 (2016).
Li, H. et al. Diet simplification selects for high gut microbial diversity and strong fermenting ability in high-altitude pikas. Appl. Microbiol. Biotechnol. 102, 6739–6751 (2018).
Ambrosini, R. et al. Cloacal microbiomes and ecology of individual barn swallows. FEMS Microbiol. Ecol. 95, fiz061 (2019).
Kreisinger, J., Čížková, D., Kropáčková, L. & Albrecht, T. Cloacal microbiome structure in a long-distance migratory bird assessed using deep 16sRNA pyrosequencing. PLoS ONE 10, e0137401 (2015).
Noguera, J. C., Aira, M., Pérez-Losada, M., Domínguez, J. & Velando, A. Glucocorticoids modulate gastrointestinal microbiome in a wild bird. R. Soc. Open Sci. 5, 171743 (2018).
Shehzad, W. et al. Carnivore diet analysis based on next-generation sequencing: Application to the leopard cat (Prionailurus bengalensis) in Pakistan. Mol. Ecol. 21, 1951–1965 (2012).
Vestheim, H. & Jarman, S. N. Blocking primers to enhance PCR amplification of rare sequences in mixed samples—A case study on prey DNA in Antarctic krill stomachs. Front. Zool. 5, 12 (2008).
Acknowledgements
We are grateful to Kevin Roche for helpful comments on an earlier version of the manuscript. We thank all those who collaborated with fieldwork, specifically Adéla Petrželková, Marie Kotasová Adámková and Romana Michálková, along with the owners of the Hamr and Šaloun farms for providing access to the breeding swallow populations. Computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA LM2018140), provided within the program ‘Projects of Large Research, Development and Innovation Infrastructures’. We also acknowledge the Core Facility Genomics of the Central European Institute of Technology, Masaryk University, supported by the National Centre for Medical Genomics research infrastructure (LM2015091, funded by the Ministry for Schools, Youth and Sports, Czech Republic), especially Filip Pardy and Boris Tichý for their support with obtaining the scientific data presented in this paper. We are most grateful for the financial and logistic support provided by Czech Science Foundation Projects 15-11782S, 19-19307S and 21-22160S, and Project 281315 of the Grant Agency of Charles University. In addition, LS was supported by SVV Project No. 260571/2021.
Author information
Authors and Affiliations
Contributions
J.K., T.A. study design; T.A., J.K., O.T., L.S. field sampling; L.S., H.P. laboratory analysis; L.S., J.K. data analysis; L.S., T.A., J.K. funding; L.S., J.K. manuscript drafting; all authors provided helpful comments and recommendations and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schmiedová, L., Tomášek, O., Pinkasová, H. et al. Variation in diet composition and its relation to gut microbiota in a passerine bird. Sci Rep 12, 3787 (2022). https://doi.org/10.1038/s41598-022-07672-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-07672-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
