
Abstract
Cork oak is a tree species with ecological importance that contributes to economic and social development in the Mediterranean region. Cork oak decline is a major concern for forest sustainability and has negative impacts on cork oak growth and production. This event has been increasingly reported in the last decades and seems to be related with climate changes. Biscogniauxia mediterranea is an endophytic fungus of healthy cork oak trees that turns into a pathogen in trees weaken by environmental stress. Understanding the drivers of B. mediterranea populations diversity and differentiation is expected to allow a better control of cork oak decline and preserve forest sustainability. Endophyte isolates from different cork oak forests were identified as B. mediterranea and their genetic diversity was evaluated using phylogenetic and microsatellite-primed PCR analyses. Genetic diversity and variability of this fungus was correlated with environmental/phytosanitary conditions present in forests/trees from which isolates were collected. High genetic diversity and variability was found in B. mediterranea populations obtained from different forests, suggesting some degree of isolation by distance. Bioclimate was the most significant effect that explained the genetic variability of B. mediterranea, rather than precipitation or temperature intensities alone or disease symptoms. These findings bring new implications for the changing climate to cork oak forests sustainability, cork production and quality.
Similar content being viewed by others

Introduction
Cork oak (Quercus suber L.) is an evergreen oak species with high economic, ecological, and social importance in the Mediterranean region1,2. This species is mainly distributed throughout the Mediterranean Basin, where is well adapted to the climate. Despite that, global climate changes can induce abiotic stress on trees, impacting negatively cork oak growth and productivity3,4. Indeed, cork oak decline has been increasingly reported in the last decades and seems to be linked to climate changes, which can increase susceptibility to pathogen attack and facilitate infection by opportunistic pathogens5,6. Biscogniauxia mediterranea is an opportunistic pathogen with an endophytic lifestyle that has been associated with cork oak decline in the Mediterranean region7,8,9. This fungus causes charcoal disease that leads to an extensive inner bark and xylem necrosis, often associated with a blackish exudation on the outer bark10,11. As the appearance of B. mediterranea-related symptoms are mainly associated with weakened trees (e.g., by drought), the incidence of this disease increased over the last years and was recently reported on young trees12,13.
Molecular markers are widely used to study population genetics of phytopathogenic fungi14. Early studies on B. mediterranea populations used random amplified polymorphic DNA (RAPD), being able to detect a high genetic variability for geographically close populations9,15. These results indicated a high rate of sexual reproduction and heterothallic mating system displayed by this species. More recently, high genetic variability was reported within a single stromata using microsatellite-primed PCR primers16. Also, B. mediterranea populations from different Mediterranean countries displayed high genetic diversity (by using sequence analysis of Internal Transcribed Spacer of ribosomal RNA gene, translation elongation factor 1-α and β-tubulin, as well as microsatellite-primed PCR primers)17, but studied isolates were not phylogenetic associated with host species or age, geographic position, or charcoal canker expression. However, the analysis of Tunisian populations of B. mediterranea by using microsatellite-primed PCR primers suggested a positive association of their morphological variability and ecological factors, such as altitude, rainfall, and temperature18. Altogether, these findings pointed to the high plasticity and adaptability of this species to the environment15,17. In this work, we aimed to understand the correlation of B. mediterranea genetic populations with environmental factors and disease symptoms found on cork oak trees from different bioclimates. For increasing the significance of molecular characterization of B. mediterranea populations we used two different typing analyses, a multilocus (6 loci) and a microsatellite (4 primers) approach. This work will increase the current knowledge about drivers of B. mediterranea genetic variability and contribute for the formulation of appropriate disease management.
Results
The molecular characterization of B. mediterranea endophytic isolates from cork oak trees was performed using a multilocus sequence analysis and microsatellite-primed PCR fingerprinting. From all analyzed loci, partial glutamine synthetase (GS) revealed the highest variability with 19.2% of identical sites (including the outgroup sequences, B. atropunctata, B. nummularia and Xylaria hypoxylon), followed by actin (ACT; 27.6%), translation elongation factor 1-α (TEF; 53.2%), β-tubulin 2 (TUB2; 57.7%), chitin synthase 1 (CHS; 61.4%) and internal transcribed spacer (ITS; 66%). As expected, the nucleotide datasets displayed higher percentage of identical sites without outgroups. The corresponding values ranged from 36.9% (GS) to 94.7% (TUB2). The multilocus alignment was in line with this trend (23.4% of identical sites with outgroups; 66.4% without outgroups).
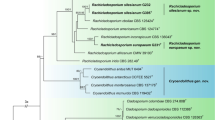
Phylogenetic trees of each single locus were not able to individually resolve B. mediterranea isolates for any factors of interest (forest, bioclimate, disease severity index or disease symptoms; results not shown). The concatenated dataset comprised sequences from 52 B. mediterranea isolates and from the three outgroup species (B. atropunctata, B. nummularia and Xylaria hypoxylon). From a total of 2874 characters, 1775 were constant, 815 parsimony-uninformative and 284 parsimony-informative. Maximum Likelihood (ML) tree produced clades of B. mediterranea isolates with moderate [ML bootstrap (BS) 70–80; BI posterior probability (PP) 0.8–0.9], high (BS 81–95; PP 0.91–0.95) or very high (BS > 95; PP > 0.95) support values19,20, but no evident patterns regarding factors of interest were exposed using this approach (Fig. 1). Isolates obtained from cork oaks growing in different locations and distinct bioclimates did not clustered together; and visible tree disease symptoms did not contribute for the distribution of isolates. For example, Clade A comprised isolates (7; e.g. Bm25, Bm57, Bm37) from different forests and bioclimates (from the most humid to the driest), also presenting isolates obtained from trees with different disease severity levels. The same was observed for Clade C (with 6 isolates; e.g. Bm36, Bm66, Bm08), which even included an isolate (Bm79) obtained from an olive tree. Moreover, some information can be inferred from the tree. Group III (with moderate ML support) displayed a higher number of isolates from trees with mild symptoms (21 out of the 31 found in Group III, contrasting with 7 isolates in the remaining 19). Clade B (in Group I) only included isolates (Bm50, Bm54 and Bm69) from declining trees, while Clade D (Group III) only included isolates (Bm55, Bm11 and Bm03) collected from mild diseased trees, though both clades presented isolates from different forests and bioclimates. Overall, isolates from different locations were distributed along the phylogenetic tree, with few of them being placed together. The same distribution pattern was observed regarding isolates from different bioclimates, even though the sequences in Group VII refer to isolates (Bm49, Bm47, Bm46 and Bm64) obtained from two forests (GV and GR) with the same bioclimate (sub-humid). Also, a clear distribution pattern was not observed concerning the presence of exudates on the B. mediterranea host trees. However, subclade A1 only encompassed isolates (Bm25, Bm23, Bm57 and Bm37) obtained from cork oak trees not producing trunk exudates. The opposite was observed for Clade B (Bm50, Bm54 and Bm69) and Group VI (Bm55, Bm11, Bm03 and Bm15), where all isolates were recovered from trees producing exudates, even if from dissimilar bioclimatic locations (sub-humid and humid). In line with the low resolution of B. mediterranea isolates by any of the studied factors (forest, bioclimate, disease severity index or disease symptoms), the majority of B. mediterranea isolates collected from the same tree were not clustered together. For instance, although pairwise identity within those isolates was high, isolates from a single tree, such as Bm60, Bm61, Bm63 and Bm67 (pairwise identity of 95.4%) and Bm46, Bm56 and Bm58 (99.6%), were found to cluster better with isolates obtained from other forests and/or were placed apart in the phylogenetic tree. Similarly, isolates collected on olive trees (Bm79 and Bm80) were distantly placed from each other, in separate lineages.

Cladogram showing the phylogenetic relationship between the 52 B. mediterranea isolates (50 from cork oaks and 2 from olive trees*) and three outgroup species (B. atropunctata, B. nummularia and Xylaria hypoxylon). This multilocus ML tree was constructed from the concatenated alignment of ITS, TEF, GS, ACT, CHS and TUB2 sequences. Numbers shown at nodes are support branch values for ML bootstraps (only BS values ≥ 70 are shown) and BI posterior probabilities (only PP values ≥ 0.8 are shown), respectively. Disease severity levels are highlighted in green, grey, or red, representing isolates collected from healthy, mild, and declining trees, respectively. Black dots refer to isolates collected from trees showing exudation. Forests are highlighted in different colors, according to their bioclimate, as follow: dark blue for hyper-humid (PG-RC and PG-ER), light blue for humid (AL and LI), green for sub-humid (GV and GR) and dark yellow for semi-arid (HC-MA and HC-CT). Clades (A–F) and some groups of sequences (I–VII) are exposed for clarity.
Microsatellite-primed PCR fingerprinting of 68 B. mediterranea isolates generated different banding patterns. Primer (GTG)5 generated 15 bands (from 0.25 to 1.5 kb), (CAG)5 generated 17 bands (ranging from 0.3 to 2 kb, in which one was monomorphic), (ACAC)5 generated 17 bands (from 0.25 to 1 kb) and M13 generated 26 bands (from 0.15 to 1.5 kb). The monomorphic band was removed and a binary dataset with the remaining (74) band positions was used for molecular analysis. Using this approach, the genetic diversity of B. mediterranea varied within different factors of interest/populations (Table 1). Considering total populations, the number of alleles (Na) varied from 1.40 to 1.97 and the number of effective alleles (Ne) from 1.45 to 1.57, which were found considering ‘forest’ and ‘exudates’ factors, respectively. These results were corroborated by the genetic diversity found within populations (Hs; Table 2). ‘Exudates’ factor (total population) also displayed the highest Shannon’s information index (I = 0.50), Nei’s gene diversity (h = 0.33) and percentage of polymorphic loci (PPL = 98.7), while ‘forest’ factor shown the lowest genetic diversity (I = 0.38; h = 0.26; PPL = 67.9). In addition to ‘exudates’, other disease-related factors also revealed high genetic diversity levels for total populations, in particular when considering ‘cankers’ and ‘disease severity levels’ factors (Tables 1 and 2). The genetic diversity of B. mediterranea also differed among populations when considering individually each environmental/disease-related factor (Table 1). Considering the ‘bioclimate’ factor, isolates obtained from sub-humid forests revealed the highest genetic diversity and those from hyper-humid the lowest. While Grândola (GR, a sub-humid forest) was the forest with the highest B. mediterranea genetic diversity, PG-ER (hyper-humid) and AL (humid) were the forests with the lowest diversity. Isolates collected from healthy trees (low disease severity level) revealed the lowest genetic diversity and declining trees were associated with more diverse B. mediterranea isolates. This result is in line with the highest genetic diversity found among B. mediterranea isolates collected from trees producing trunk exudates. However, the opposite was found for trees with trunk cankers, in which higher genetic diversity was associated with isolates from trees without this symptom. Regarding defoliation, the isolates obtained from trees with very accentuated damage were also less genetically diverse, while those from trees with moderate and light damages presented higher genetic diversity.
In agreement with phylogenetic tree analysis from multilocus sequence analysis, principal coordinates analysis (PCoA) did not revealed clusters of cork oak isolates, according to the factors of interest (Fig. S1, both axes only capturing around 15% of data variation). Also, isolates from olive tree did not cluster all together and were closer to isolates from healthy and mild diseased trees. However, certain populations revealed a higher differentiation in allele frequencies than others, as evaluated by Gst coefficient (Table 2). There was a higher genetic differentiation among ‘forest’ (Gst = 0.239), followed by ‘defoliation’ (Gst = 0.151), and ‘bioclimate’ (Gst = 0.097) populations. The lowest genetic differentiation was found in ‘cankers’ (Gst = 0.046), ‘disease severity levels’ (Gst = 0.045) and ‘exudates’ (Gst = 0.032) populations. As expected, gene flow (Nm) values were opposite to Gst, being higher among populations with lower genetic differentiation and vice-versa (Table 2). These results agree with the genetic pairwise distances found in such populations (Table S1). Pairwise genetic distances in ‘forest’ achieved higher values (ranged from 0.244 to 0.067), than ‘defoliation’ (0.200 to 0.019), or bioclimate (0.085 to 0.053). Other disease-related variables (‘cankers’, ‘disease severity levels’ and ‘exudates’) never achieved more than 0.048 genetic distances. Among populations, isolates obtained from sub-humid bioclimate were the most genetically close from the ones obtained in the other forests (Table S1). Indeed, GR (a sub-humid forest) displayed the least genetic distance from all other forests. In contrast, isolates from humid bioclimate (in particular from AL forest) displayed the highest genetic distance from all other forests. Interestingly, isolates obtained from very accentuated defoliated trees revealed a high genetic distance from other isolates. AMOVA results revealed that variation within populations was always higher than among populations, which reinforces the high variability of B. mediterranea composition. In any case, variation among populations was higher for ‘forest location’ (10%), followed by ‘bioclimate’ (6%), ‘cankers’ (4%) and ‘exudates’ (3%), all at p < 0.001 (Table 3), suggesting higher genetic differentiation between regions and bioclimates. Accordingly, Mantel test shown significant correlation between B. mediterranea genetic diversity and geographic distance of cork oak forests (R = 0.105, p = 0.001). For further understanding the relative contribution of forest location, environmental, and disease-related factors in driving the genetic diversity of B. mediterranea populations, a redundancy analysis was performed (Fig. 2). The variables that explained the variation of B. mediterranea genetic diversity were ‘forest location’ and ‘bioclimate’ factors, followed by ‘exudates’ and ‘cankers’ (all at p < 0.001), being all the others (temperature, precipitation, disease severity levels, and defoliation) not significant. The combination of all significant variables explained 9.602% (p = 0.001) of B. mediterranea genetic variance. Most of this variation is due to the ‘bioclimate’ and ‘forest location’ factors that together contribute to 8.831% of variation (p = 0.001), in contrast with the shared contribution of ‘exudates’ and ‘cankers’ variables of only 2.722% of variation (p = 0.001). Indeed, when excluding all other variables, ‘exudates’ and ‘cankers’ do not present a significant contribution for B. mediterranea genetic diversity (p > 0.05). The production of exudates showed higher correlation with ‘bioclimate’ (0.188%) than with ‘forest location’ (0.081%). The opposite was found for the presence of cankers, in which no correlation was found with ‘bioclimate’, but a 0.943% correlation was found for ‘forest location’ variable. The index of association for the estimation of linkage disequilibrium revealed that LI and GV forests, as well as forests from the humid, sub-humid and semi-arid bioclimates was significantly different from 0, which indicates linkage disequilibrium (Table S2).

Contribution of the most significant variables for explaining genetic variability in B. mediterranea populations. The unique and shared contribution of variables is detailed. Percentage of variation was calculated by adjusted R2. ***represent p < 0.001 and **p < 0.01 for statistical significance of model.
Discussion
In this study, the genetic diversity of endophytic B. mediterranea isolates was evaluated by multilocus phylogeny and microsatellite fingerprinting (MSP-PCR). The evaluated isolates comprised those obtained from cork oak trees, thriving in different forests, bioclimates, and displaying distinct charcoal disease severity levels and symptoms. This experimental approach is expected to provide new information about which factors/variables contribute the most to this species diversity and variability. Other isolates obtained from olive trees were also analyzed to evaluate host-specificity. As expected, all studied B. mediterranea isolates were phylogenetically apart from other Biscogniauxia species included in this study as outgroups, revealing the genetic divergence of B. mediterranea species. In general, individual (i.e., single-locus) and concatenated (i.e., multi-locus) phylogenetic analyses did not correlate B. mediterranea isolates with any of the studied environmental/disease variables. Also, after determining the microsatellite polymorphic patterns, B. mediterranea isolates did not clustered according to the factors of interest. These results suggest a high genetic diversity and variability of B. mediterranea populations, which is concordant with other reports9,15,17,18, even when isolates come from the same stroma16. Furthermore, the inability to resolve B. mediterranea isolates obtained from cork oak and those obtained from olive tree reinforces current knowledge about the high adaptability of this fungus to different hosts, as suggested by17.
The genetic variability of B. mediterranea isolates was mostly explained by ‘forest location’ (6.9%) and ‘bioclimate’ (4.6%). Indeed, when these factors were combined, they explained 8.8% of B. mediterranea genetic variation. Considering ‘forest location’, B. mediterranea populations demonstrated the lowest genetic diversity within population, while also revealing the highest genetic differentiation and lowest gene flow among populations. These results suggest some isolation by distance of B. mediterranea communities, which agrees with the significant correlation (Mantel test) found between the B. mediterranea genetic diversity and forest geographic location of cork oak forests from which they were obtained. This contrasts with previous studies, where B. mediterranea intraspecific polymorphism and genetic diversity were not associated with geographic position of host trees17. However, the contribution of geographic isolation in B. mediterranea genetic differentiation was suggested for cork oak populations in Tunisia18. The high genetic variability of B. mediterranea has been related with the heterothallic mating system of this species15 and sexual reproduction with the production of a high number of variable ascospores21. Our results suggest that global B. mediterranea population displays linkage disequilibrium and a clonal genetic structure. Despite that, populations from the majority of forests (PG-ER, PG-RC, AL, GR, HC-CT and HC-MA) demonstrated to have random mating with frequent sexual reproduction. B. mediterranea ascospores are primarily dispersed by wind after the occurrence of precipitation21,22, although insects could also play a role for their spreading in short- and long- distances, depending on their bioecology21,22. Our results suggest low migration rates between geographically distant B. mediterranea populations, indicating short-distance dispersal. The clonal structure of the populations from the majority of bioclimates and the finding that PG-ER and AL forests presented the lowest genetic diversity among all other sampled forests reinforces this suggestion. In contrast with other sampled forests, both are characterized by a high density of mixed forest trees and low anthropogenic interference, which may restrain spore dispersal to and from distant locations. Indeed, canopy architecture and the use of mixed tree species have been reported as a management strategy to reduce spore dispersal of pathogenic fungi23,24,25. In addition, high genetic differentiation among forest populations can be a result of random events but there is enough gene flow to refute the effects of genetic drift26. While a significant variation was found among forest populations, high variability of B. mediterranea was also described within populations. This result concurs with the high intraspecific genetic variability found within populations described by Henriques et al.27, indicating an adaptation of B. mediterranea to the environment and ensuring species long-term survival.
Besides ‘forest location’ factor, a significant variation on B. mediterranea populations occurred in response to ‘bioclimate’, although precipitation and temperature alone were not significantly correlated with B. mediterranea variability, as revealed by variation partitioning redundancy analysis. In contrast, other studies indicated a positive correlation between B. mediterranea genetic diversity with temperature and rainfall18. The ability of B. mediterranea to develop in a wide range of temperatures27, associated with the significant variability of this fungus in different bioclimates may represent a problem for charcoal cork oak disease management. This will be further challenged by the effect of combined bioclimate and charcoal symptoms (exudates in cork oak trunk) in increasing B. mediterranea variability. Indeed, in Tunisia, the correlation between bioclimate and charcoal disease development has been suggested7. In any case, the variability promoted by disease symptoms (exudates and cankers presence) was better explained when taking into consideration the forest location, suggesting that other factors characteristic of each forest (i.e., silvicultural practices not included in this study) are contributing to B. mediterranea variability. Moreover, B. mediterranea isolates collected from declining trees or trees with charcoal symptoms, like trunk exudates, presented a higher genetic diversity than those collected from healthier trees.
Conclusions
Several reports suggested that Quercus suber populations and their associated microbiota are vulnerable to different bioclimates28,29,30 and will be affected by the predicted climate changes31. Therefore, as cork oak forests currently displaying a moist bioclimate become more arid, they will be increasingly affected by environmental stressors. Taking all together, the results obtained in this work support the previous suggestions that B. mediterranea isolates have facilitated adaptation. Our findings reinforce the previous knowledge of B. mediterranea opportunistic behavior and reveal the importance of bioclimate as a source of B. mediterranea variability, exacerbating the implications of a changing climate on cork oak forests sustainability which will affect cork production and quality.
Methods
Biscogniauxia mediterranea isolates
Biscogniauxia mediterranea isolates were obtained from cork oak twigs as endophytes32. Twigs were sampled, during 2017, from eight Portuguese cork oak forests with different locations and bioclimate classifications (Fig. S2; Table S3). Bioclimate for each forest was determined using Emberger index and Emberger climatogram33,34 and ranged from hyper-humid to semi-arid bioclimates [hyper-humid (PG-RC and PG-ER), humid (AL and LI), sub-humid (GV and GR) and semi-arid (HC-MA and HC-CT)]. If existent, trees with different disease severity levels were collected from each location. Evaluated disease symptoms included defoliation (5 levels: 0–10%—no damage; 11–25%—light damage; 26–50%—moderate damage; 51–90%—severe damage; > 90%—extreme damage), as well as canopy and trunk damages (3 levels: 0—no damage; 1—moderate damage; 2—severe damage) (Table S4). Disease damages included dried, wilting and decolorated leaves, presence of cankers, decolorated trunk and presence of exudates (Fig. 3). Disease severity levels were grouped into three categories (healthy, mild, and declining), considering the combination of different symptoms and corresponding levels, as described elsewhere32. Endophytic isolates were obtained by sterilizing the surface of cork oak twigs, plating twigs onto Potato Dextrose Agar (PDA) medium and obtaining pure cultures through re-plating outgrowing fungi into fresh PDA medium30. DNA of pure cultures was extracted using Quick-DNA Fungal/Bacterial Miniprep Kit (Zymo Research, Irvine, CA, USA) and B. mediterranea isolates were identified using universal primer pairs ITS1-F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′)35. A total of 74 B. mediterranea isolates obtained from cork oak were distinguished and identified by sequencing (Table 4). Three other isolates were obtained from Olea europaea twigs (Bm7836) and olives (Bm79 and Bm8037), collected from cvs. Cobrançosa (Bm78 and Bm80) and Madural (Bm79). These O. europaea-derived isolates were included in this study to evaluate host-specificity in B. mediterranea. All methods complied with relevant institutional, national, and international guidelines and legislation.

Charcoal disease severity levels (A–C) and disease symptoms (D,E), displayed by sampled cork oak trees. (A) Healthy tree; (B) Tree with mild symptoms; (C) declining tree; (D) presence of canker and exudates; (E) presence of exudates on uncorked tree; (F) presence of decolorated and dried leaves.
Multilocus sequence analysis
Multilocus phylogenetic analysis was performed by targeting several loci: internal transcribed spacer (ITS), translation elongation factor 1-α (TEF), partial glutamine synthetase (GS), actin (ACT), chitin synthase 1 (CHS) and β-tubulin 2 (TUB2). PCR amplifications were performed according to each pair of primers and target region (Table 5). PCR reactions were prepared for all loci in 10 µl volume using 0.2 U/µl of NZYTaq II 2× Green Master Mix (NZYTech, Portugal), 0.5 µM of each primer and 1 µl of DNA. PCR products were run on a 1% (w/v) agarose gel, stained with Green Safe Premium (NZYTech, Portugal). Isopropanol 75% (v/v) was used to purify PCR products and sequencing was performed by Macrogen, Inc services (Madrid, Spain).
DNA sequences were processed using AB1 trace files in Geneious version 2010.4.8.5 (Biomatters, New Zealand), unless stated otherwise. For each isolate and molecular marker, forward and reverse sequences were trimmed (0.05 error probability limit), assembled and consensus sequences were created. Consensus sequences obtained in this study were deposited in GenBank (for accession numbers, see Table 4). Multiple sequence alignments of each region were performed by using MUSCLE version 3.5 algorithm with a maximum of 10 iterations. Distance measure used for 1st iteration was kmer4_6 and subsequent iterations were run with pctid_kimura. All iterations were performed using UPGMB clustering method and CLUSTALW sequence weighting scheme. If needed, alignments were manually edited and Gblocks (web-based, version 0.91b, January 2002, http://molevol.cmima.csic.es/castresana/Gblocks_server.html, last accessed date: 08/02/2021) was used to eliminate poorly aligned positions and divergent regions, allowing smaller final blocks38. Geneious version 2010.4.8.5 was then used to concatenate alignments. Some isolates were not sequenced with enough quality in some targeted loci and were excluded from individual alignments before concatenation. DNA sequences from closely related taxa—Biscogniauxia atropunctata, B. nummularia and Xylaria hypoxylon39,40—were used as outgroups and were retrieved from GenBank (Table 4).
Phylogenetic trees for each individual locus were generated using sequences from 74 B. mediterranea isolates for ITS, 70 for TEF, 68 for GS, 66 for ACT, 69 for CHS and 71 for TUB2 (Table 4). The final concatenated alignment used to build the multilocus phylogenetic tree included sequences from 52 B. mediterranea isolates and from the three outgroup species (isolates marked with * in Table 4). PartitionFinder2 version 2.1.1 was run on CIPRES Science Gateway (web-based, version 3.3, https://www.phylo.org/portal2/, last accessed date: 17/02/2021)41 to find best-fit partition schemes of each loci42. Bayesian inference (BI) trees were computed using Markov chain Monte Carlo (MCMC) algorithm in MrBayes version 3.2.743. Models (lset) and prior probability distributions (prset) were set according to PartitionFinder2 results. Two independent runs were performed with one million generations and four chains in each run. The two runs were converged with a burnin of 25% and tree with posterior probabilities was generated. Maximum Likelihood (ML) trees were generated using W-IQ-TREE (http://iqtree.cibiv.univie.ac.at/, last accessed date: 17/02/2021)44, a web server for IQ-TREE45. Best-fit model was computed using ModelFinder version 1.4.246, with an edge-linked partition model47. Branch support analysis was performed using 1000 ultrafast bootstrap replicates48 and minimum correlation coefficient of 0.99. Phylogenetics trees were visualized in FigTree version 1.4.449. ML bootstrap (BS) and BI posterior probability (PP) values and topologies obtained by both phylogenetic inference methods were compared using TreeGraph 2 version 2.0.4050. This allowed the distinction of clades with reliable support value19,20 or groups of sequences showing limited support. Additional data from environmental/disease factors of interest were added using the vector image editor Inkscape version 0.9251.
Microsatellite-primed PCR fingerprinting
The molecular diversity of 68 B. mediterranea isolates was also evaluated by using microsatellite-primed PCR (MSP-PCR) with four sets of primers, (GTG)5, (CAG)5, (ACAC)5 and M13 (phage M13 core sequence; 5′-GAGGGTGGNGGNTCT)17,18. PCR reactions were prepared in 10 µl volume using 0.2 U/µl of NZYTaq II 2× Green Master Mix (NZYTech, Portugal), 1 µM of each primer and 1 µl of DNA. The amplifications with (GTG)5 were performed using the following PCR program: initial denaturation at 94 °C, for 4 min; 35 cycles of 45 s at 94 °C, 45 s at 56 °C and 30 s at 72 °C; and final elongation at 72 °C for 10 min. Amplifications with (CAG)5, (ACAC)5 and M13 primers comprised the following PCR program: initial denaturation at 94 °C, for 2 min; 40 cycles of 30 s at 93 °C, 60 s at 53 °C and 30 s at 72 °C; and final elongation at 72 °C for 10 min. Amplifications with each primer were performed in duplicate for reproducibility. PCR products were run on a 1.5% (w/v) agarose gel, stained with Green Safe Premium (NZYTech, Portugal). The visualization and image acquisition were performed using an UV transilluminator (VWR Genosmart, United Kingdom). DNA fingerprinting gel images were analyzed using GelAnalyzer version 19.1 (http://www.gelanalyzer.com) and each band was scored as 0 (absence) or 1 (presence).
Molecular diversity of B. mediterranea isolates was evaluated by grouping samples into populations, based on their host origin, such as climate parameters (including bioclimate), forest provenience, or disease-related parameters (including severity level, defoliation, and presence of exudates). Frequency- and distance-based genetic diversity of each population was evaluated using GenAlEX version 6.51b252. Frequency-based genetic diversity was evaluated considering the number of different alleles (Na), number of effective alleles (Ne), Shannon’s Information index (I), diversity [h = 1 − (p2 + q2)] and percentage of polymorphic loci (PPL). Distance-based genetic diversity was accessed using different analyses: Principal Coordinates Analysis (PCoA) to find patterns within dataset; Nei’s pairwise genetic distance to calculate genetic distance between populations; Analysis of Molecular Variance (AMOVA) to calculate hierarchical partitioning of genetic variation within and among populations (999 permutations); and Mantel test to calculate statistical correlation between genetic diversity and geographic distance (999 permutations). F-statistics to analyze diversity and genetic differentiation among populations were calculated in POPGENE version 1.3253. Given the reduced sample size, B. mediterranea isolates collected from olive tree (Bm78, Bm79 and Bm80) were included only in phylogenetic and PCoA analyses.
Redundancy analysis (RDA) was used to explore responses of B. mediterranea composition to environmental (bioclimate, mean maximum and minimum temperatures and mean total precipitation for the 10 years previous to sampling collection) and disease variables (disease severity level, exudates, cankers, and defoliation), by making use of the R version 4.0.254. Analyses were performed using the package vegan version 2.5-755, except when stated otherwise. Spatial trend was included in RDA using a trend surface analysis. Latitude–longitude data was transformed into flat Cartesian coordinates using geoXY() of SoDA package (version 1.0-6.1)56. To compute polynomials of degree 3, poly() of STATS package (version 4.0.2) was used. Spatial and environmental variables were analyzed separately on a first approach. RDA was performed using rda(), while anova.cca() was used to perform Monte Carlo permutation test (1000 permutations) and test significance of global model. Forward selection of explanatory variables was performed using ordistep() with 999 permutations, using spatial and environmental variables to find most parsimonious RDA. Then, another RDA was performed, as described before, for selected variables. The contribution to variation of those variables was performed using RsquareAdj() and Monte Carlo permutation test (1000 permutations) to determine significance. For explaining the variation on B. mediterranea composition, variation partitioning was performed for the best model, using the most explanatory variables (combination of spatial and environmental variables), and making use of varpart(). Partial-RDA was performed to evaluate the influence of conditional variables obtained from variation partition. Statistical significance was performed as described for RDA. Spatial variable was referred as ‘forest location’ variable in sections related with this analysis. Linkage disequilibrium of B. mediterranea populations was evaluated by the index of association (IA) and the unbiased index of association (\(\overline{r}\)d), in which clonal populations have results significantly different from 0 and sexual populations do not have statistical significant IA and \(\overline{r}\)d61. Analysis was performed using popsub() to specify population and ia() with 999 permutations for calculation using poppr package (version 2.9.0)62.
References
FAO and Plan Bleu. State of Mediterranean Forests 2018 (Food and Agriculture Organization of the United Nations, 2018).
Gauquelin, T. et al. Mediterranean forests, land use and climate change: A social-ecological perspective. Reg. Environ. Change. 18, 623–636 (2018).
Oliveira, V., Lauw, A. & Pereira, H. Sensitivity of cork growth to drought events: Insights from a 24-year chronology. Clim. Change 137, 261–274 (2016).
Acácio, V., Dias, F. S., Catry, F. X., Rocha, M. & Moreira, F. Landscape dynamics in Mediterranean oak forests under global change: Understanding the role of anthropogenic and environmental drivers across forest types. Glob. Change Biol. 23, 1199–1217 (2017).
Moricca, S. et al. Endemic and emerging pathogens threatening cork oak trees: Management options for conserving a unique forest ecosystem. Plant Dis. 100, 2184–2193 (2016).
Touhami, I. et al. Decline and dieback of cork oak (Quercus suber L.) forests in the Mediterranean basin: A case study of Kroumirie, Northwest Tunisia. J. For. Res. 31, 1461–1477 (2020).
Yangui, I. et al. Occurrence of Biscogniauxia mediterranea in cork oak stands in Tunisia. Phytoparasitica 49, 131–141 (2021).
Linaldeddu, B. T., Sirca, C., Spano, D. & Franceschini, A. Variation of endophytic cork oak-associated fungal communities in relation to plant health and water stress. For. Pathol. 41, 193–201 (2011).
Schiaffino, A., Franceschini, A., Maddau, L. & Serra, S. Molecular characterisation of Biscogniauxia mediterranea (De Not.) O. Kuntze strains isolated from declining trees of Quercus suber L. in Sardinia. In Protection intégrée des forêts de chênes, Vol 27, 26–29 (2001).
Linaldeddu, B. T., Franceschini, A. & Pulina, M. A. Epidemiological aspects of Biscogniauxia mediterranea in declining cork oak forest in Sardinia (Italy). IOBC/WPRS Bull. 28, 75–81 (2005).
Linaldeddu, B. T., Sirca, C., Spano, D. & Franceschini, A. Physiological responses of cork oak and holm oak to infection by fungal pathogens involved in oak decline. For. Pathol. 39, 232–238 (2009).
Henriques, J., Inácio, M. L., Lima, A. & Sousa, E. New outbreaks of charcoal canker on young cork oak trees in Portugal. IOBC/wprs Bull. 76, 85–88 (2012).
Nugent, L. K., Sihanonth, P., Thienhirun, S. & Whalley, A. J. S. Biscogniauxia: A genus of latent invaders. Mycologist 19, 40–43 (2005).
Giraud, T., Enjalbert, J., Fournier, E., Delmotte, F. & Dutech, C. Population genetics of fungal diseases of plants. Parasite 15, 449–454 (2008).
Vannini, A., Mazzaglia, A. & Anselmi, N. Use of random amplified polymorphic DNA (RAPD) for detection of genetic variation and proof of the heterothallic mating system in Hypoxylon mediterraneum. Eur. J. For. Pathol. 29, 209–218 (1999).
Henriques, J., Nóbrega, F., Sousa, E. & Lima, A. Diversity of Biscogniauxia mediterranea within single stromata on cork oak. J. Mycol. 2014, 1–5 (2014).
Henriques, J., Nóbrega, F., Sousa, E. & Lima, A. Analysis of the genetic diversity and phylogenetic relationships of Biscogniauxia mediterranea isolates associated with cork oak. Phytoparasitica 44, 19–34 (2016).
Yangui, I. et al. Biscogniauxia mediterranea associated with cork oak (Quercus suber) in Tunisia: Relationships between phenotypic variation, genetic diversity and ecological factors. Fungal Ecol. 41, 224–233 (2019).
Erixon, P., Svennblad, B., Britton, T. & Oxelman, B. Reliability of bayesian posterior probabilities and bootstrap frequencies in phylogenetics. Syst. Biol. 52, 665–673 (2003).
Hillis, D. M. & Bull, J. J. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst. Biol. 42, 182–192 (1993).
Jiménez, J. & Trapero Casas, A. E. Chancro carbonoso de ‘Quercus’ III: dispersión de ascosporas del agente casual. Boletín Sanid. Veg. Plagas 31, 577–585 (2005).
Henriques, J. et al. Factors affecting the dispersion of Biscogniauxia mediterranea in Portuguese cork oak stands. Silva Lusit. 22, 83–97 (2014).
Field, E. et al. Associational resistance to both insect and pathogen damage in mixed forests is modulated by tree neighbour identity and drought. J. Ecol. 108, 1511–1522 (2020).
Roberts, M. et al. The effect of forest management options on forest resilience to pathogens. Front. For. Glob. Chang. 3, 7 (2020).
Vidal, T. et al. Reduction of fungal disease spread in cultivar mixtures: Impact of canopy architecture on rain-splash dispersal and on crop microclimate. Agric. For. Meteorol. 246, 154–161 (2017).
Wright, S. Evolution in Mendelian populations. Genetics 16, 97 (1931).
Henriques, J. et al. Morphological and genetic diversity of Biscogniauxia mediterranea associated to Quercus suber in the Mediterranean basin. Rev. Ciências Agrárias 38, 166–175 (2015).
Reis, F. et al. Ectomycorrhizal fungal diversity and community structure associated with cork oak in different landscapes. Mycorrhiza 28, 357–368 (2018).
Reis, F. et al. Climatic impacts on the bacterial community profiles of cork oak soils. Appl. Soil Ecol. 143, 89–97 (2019).
Costa, D., Tavares, R., Baptista, P. & Lino-Neto, T. Diversity of fungal endophytic community in Quercus suber L. under different climate scenarios. Rev. Ciências Agrárias 41, 41–50 (2018).
Vanhove, M. et al. Using gradient forest to predict climate response and adaptation in cork oak. J. Evol. Biol. 34, 910–923 (2021).
Costa, D., Tavares, R. M., Baptista, P. & Lino-Neto, T. Cork oak endophytic fungi as potential biocontrol agents against Biscogniauxia mediterranea and Diplodia corticola. J. Fungi 6, 1–21 (2020).
Emberger, L. Sur une formule climatique et ses applications en botanique. La Meteorol. 92, 423–432 (1932).
Emberger, L. Une classification biogéographique des climats. Recueil des Travaux des Laboratoires de Botanique Vol 7, (1955).
White, T. J., Bruns, T., Lee, S. J. W. T. & Taylor, J. L. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. Guid. Methods Appl. 18, 315–322 (1990).
Gomes, T., Pereira, J. A., Benhadi, J., Lino-Neto, T. & Baptista, P. Endophytic and epiphytic phyllosphere fungal communities are shaped by different environmental factors in a Mediterranean ecosystem. Microb. Ecol. 76, 668–679 (2018).
Martins, F., Mina, D., Pereira, J. A. & Baptista, P. Endophytic fungal community structure in olive orchards with high and low incidence of olive anthracnose. Sci. Rep. 11, 1–11 (2021).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Peláez, F., González, V., Platas, G., Sánchez-Ballesteros, J. & Rubio, V. Molecular phylogenetic studies within the Xylariaceae based on ribosomal DNA sequences. Fungal Divers. 31, 111–134 (2008).
Mazzaglia, A., Anselmi, N., Vicario, S. & Vannini, A. Sequence analysis of the 5.8S rDNA and ITS regions in evaluating genetic relationships among some species of Hypoxylon and related genera. Mycol. Res. 105, 670–675 (2001).
Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Gateway Computing Environments Workshop, GCE 1–8 (2010) https://doi.org/10.1109/GCE.2010.5676129.
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T. & Calcott, B. Partitionfinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773 (2017).
Ronquist, F. et al. Mrbayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235 (2016).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A. & Jermiin, L. S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Chernomor, O., Von Haeseler, A. & Minh, B. Q. Terrace aware data structure for phylogenomic inference from supermatrices. Syst. Biol. 65, 997–1008 (2016).
Hoang, D. T., Chernomor, O., Von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Rambaut, A., Suchard, M., Nenarokov, S. & Klötzl, F. FigTree. (2018).
Stöver, B. C. & Müller, K. F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 11, 1–9 (2010).
Project, I. Inkscape. (2020).
Peakall, R. & Smouse, P. E. GenALEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28, 2537–2539 (2012).
Yeh, F. & Boyle, T. Population genetic analysis of co-dominant and dominant markers and quantitative traits. Belgian J. Bot. 129, 157 (1997).
Team, R. C. R: A language and environment for statistical computing (2019).
Oksanen, J. et al. vegan: community ecology package (R package version 2.5-4) (2019).
Chambers, J. M. SoDA: Functions and Examples for ‘Software for Data Analysis’. (2020).
Carbone, I. & Kohn, L. M. A method for designing primer sets for speciation studies in filamentous ascomycetes. Mycologia 91, 553–556 (1999).
Stephenson, S. A., Green, J. R., Manners, J. M. & Maclean, D. J. Cloning and characterisation of glutamine synthetase from Colletotrichum gloeosporioides and demonstration of elevated expression during pathogenesis on Stylosanthes guianensis. Curr. Genet. 31, 447–454 (1997).
Hoffman, M. T. & Arnold, A. E. Geographic locality and host identity shape fungal endophyte communities in cupressaceous trees. Mycol. Res. 112, 331–344 (2008).
Glass, N. L. & Donaldson, G. C. Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl. Environ. Microbiol. 61, 1323–1330 (1995).
Brown, A. H. D., Feldman, M. W. & Nevo, E. Multilocus structure of natural populations of Hordeum spontaneum. Genetics 96, 523–536 (1980).
Kamvar, Z. N., Tabima, J. F. & Grünwald, N. J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2, e281 (2014).
Acknowledgements
The authors thank Maria Carolina Varela for the help in identification of disease severity levels of trees and Pinto Moreira (HC forests), Eng. Carlos Barroso Dias (PNPG), Eng. Luís Dias (Herdade das Barradas da Serra—GR) and José Lino Neto (GV forest) for providing access to cork oak forests. The authors also thank Ivo Colmonero-Costeira for the help and discussions about population genetics.
Funding
This work was supported by FEDER funds through COMPETE (Programa Operacional Factores de Competitividade) and by national funds by FCT (Fundação para a Ciência e a Tecnologia) in the framework of the project POCI-01-0145-FEDER-028635, as well as for financial support to BioISI (UIDB/04046/2020), CBMA (UIDB/04050/2020), and CIMO (UID/AGR/00690/2020) by national funds by FCT/MCTES/PIDDAC. D. Costa thanks FCT for PhD grant SFRH/BD/120516/2016.
Author information
Authors and Affiliations
Contributions
D.C. contributed to study design, performed the experiments, analyzed data, and contributed to manuscript writing. V.R. contributed to study design, analyzed data, and contributed to manuscript writing. R.M.T. contributed to study design and manuscript editing. P.B. contributed to study design and manuscript editing. T.L.N. contributed to study design and manuscript writing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Costa, D., Ramos, V., Tavares, R.M. et al. Phylogenetic analysis and genetic diversity of the xylariaceous ascomycete Biscogniauxia mediterranea from cork oak forests in different bioclimates. Sci Rep 12, 2646 (2022). https://doi.org/10.1038/s41598-022-06303-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-06303-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
