
Abstract
Infections with multiple strains of Mycobacterium tuberculosis are now widely recognized as a common occurrence. Identification of patients infected with multiple strains provides both insight into the disease dynamics and the epidemiology of tuberculosis. Analysis of Mycobacterial Interspersed Repetitive Unit-Variable-Number Tandem Repeats (MIRU-VNTR) has been shown to be highly sensitive in detecting multiple M. tuberculosis strains even in sputum. The goal of this study was to identify cases of multiple M. tuberculosis strain infections among patients diagnosed with pulmonary tuberculosis in Southwestern Uganda and assessment of factors associated with multiple strain infections. DNA extracted directly from 78 sputum samples, each from an individual patient, was analyzed using the standard 24 loci MIRU-VNTR typing. Five (6.4%) of the 78 patients were infected with multiple strains of M. tuberculosis with all of them being the newly diagnosed cases while two-thirds of them were co-infected with HIV. Exact regression analysis projected that the natives were more likely to harbor multiple strains (OR; 0.981, 95% CI 0–7.926) as well as those with a high microbial load (OR; 0.390, 95% CI 0–3.8167). Despite these findings being not statistically significant due to the small sample size, this points to a critical component of disease dynamics that has clinical implications and emphasizes a need for a study using a larger cohort. It is also essential to study the potential factors associated with higher risk of exposure to newly diagnosed and HIV positive patients at the community level. In addition, our ability to detect multiple M. tuberculosis strains using the standard 24 loci MIRU-VNTR typing especially with allelic diversity in loci 2059 and 3171, which are excluded from the 15-locus MIRU-VNTR, lead us to recommend the use of this genotyping technique, especially in areas with tuberculosis endemicity similar to this study.
Similar content being viewed by others

Introduction
Tuberculosis (TB), caused by members of the Mycobacterium tuberculosis complex (MTBC), is one of humanity's oldest scourges and one of the leading causes of death from a single infectious agent globally, accounting for about 1.5 million fatalities and 10 million new cases each year1,2,3. These cases are as a result of either a primary infection, an endogenous reactivation of a primary infection or exogenous reinfection with a new strain4. Historically, it was presumed that TB was as a result of a single strain and any recurrence was due to reactivation of the same strain that caused the first episode5. Infection due to multiple strains at a single point in time was hardly considered. However, in the mid-1970s using phage typing, it was demonstrated that different strains of MTBC can infect a patient at the same time6 either as a result of a single transmission involving multiple distinct strains or due to multiple transmission events7. Multiple strain infections can either be due to mixed infections or clonal diversity and drawing clear distinctions between the two in clinical settings is somehow difficult. However, there is a significant difference in how these two mechanisms generate within-host diversity. Clonal diversity involves sporadic polymorphism resulting from sequential adaptive mutations (microevolutions)8,9,10, whereas mixed infection involves a host acquiring an entirely new MTBC genome through successive or concurrent exposure to different strains11,12. Considering that members of MTBC have highly conserved genomes13,14, high quality methods are required to identify small alterations within the infecting mycobacterial population. So far, various Polymerase Chain Reaction (PCR) based approaches have been utilized to demonstrate multiple strains within the same sputum sample10,15 or different sputum samples from the same patient8,16. Mycobacterial Interspersed Repetitive Units-variable Number of Tandem Repeats (MIRU-VNTR) analysis, initially developed in 200117, identifies such changes in the genome by varying the copy of repeats in highly variable regions of the MTBC genome18,19. This method was found to be adequate for large-scale prospective studies due to its short turnaround time but still lacked the discriminatory power required for long-term, population-based studies in order to account for a large number of samples and recent strain evolution. However, in 2006, Supply et al. proposed an expanded set of 24 MIRU loci that has high discriminatory power and thus recommended for phylogenetic studies20. This study aimed at identifying multiple MTBC strain infections among patients with pulmonary tuberculosis (PTB) in a high TB incidence area using MIRU-VNTR analysis and determining factors that could be associated with mixed strains infection in this area. TB incidence in Southwestern Uganda is high at 253 cases per 100 000 people per year21,22. It has been shown that multiple MTB strain infections are more common among people living in high TB burdened areas4,7,11,23 and accurate identification of this condition provides not only insight into the disease trends but also helps in the management and control of TB16,24,25,26,27.
Results
Prevalence of multiple strains infection
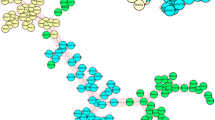
MIRU-VNTR typing was performed on 78 sputum samples, each from an individual PTB patient. Majority of these samples (91%;71/78) were from newly diagnosed cases while 9% (7/78) were relapse patients. Ten (12.8%) patients were from refugees residing in the resettlement camps, 6 (7.7%) were from patients in prison. According to the HIV status records, 39.7%, 24.4%, and 35.9% were HIV positive, negative or unknown respectively. Five of the 78 patients (6.4%; 95% CI 0.864–0.976) were found harbor more than one strain of M. tuberculosis with all cases of patients infected with multiple strains being the newly diagnosed patients (p, 0.468) whereas three of them were HIV positive (see Tables 1 and 2). Two of the five samples showed double alleles at three loci (patient # 202 and # 10,546), while two other samples had three alleles in 1 locus and two alleles at another locus (patient # 228 and # 2264) with allelic diversity being noted in loci 424, 1644, 2059, 3171, 3192 and 3690 (see Table 2). Based on the unweighted pair group method with arithmetic mean (UPGMA) analysis of the standard 24 loci MIRU-VNTR, a total of 12 different strains with 58 unique patterns were identified; Uganda I, Uganda II, EAI, LAM, Haarlem, Cameroon, Ghana, URAL, TUR, S, Bovis and Caprae. Six (7.7%) samples did not match any strain in the database hence regarded as unique. Uganda I and Uganda II sub-lineages accounted for 23.1% and 19.2% of the strains respectively whereas Ghana, Tur and S strains were individually identified in 1.3% of the samples. Animal strains; M. bovis and M. caprae were present in 2.6% and 1.3% of the samples (Table 3). MIRU-VNTRplus similarity search indicated that four of the patients with multiple strains had two distinct strains while only one patient (#2264) had three strains. Furthermore, one of the patients (#63) with two distinct strains these strains belonged to two distinct sub-lineages whereas the rest of the patients had strains belonging to the same sub-lineage (Fig. 1). Patient #202 harbored strains that were resistant to isoniazid that had mutations in both katG and inhA regions (Table 2).

UPGMA tree based on the standard 24 loci MIRU-VNTR of MTB recovered from PTB patients in Southwestern, Uganda. Lineage sub-type identified by MIRU-VNTRplus similarity search.
Exact regression analysis of factors associated with multiple strains infection
The conditional maximum likelihood from the bivariable exact regression revealed that none of the patients’ demographic variables such as age, sex was linked to multiple strain infections (Table 4). The model projected that patients who tested with high/ > 10AFB/OIF (OR 0.390, 95% CI 0–3.8167) and medium/1–10 AFB/OIF (OR; 0.300, 95% CI 0–2.9200) MTB loads in their sample were more likely to be diagnosed with multiple strains compared to those who had tested with Very low/1-9AFB/100 OIF when all other factors were held constant. Furthermore, it was also revealed that the natives were more likely to harbor multiple strains (OR; 0.981, 95% CI 0–7.926) as compared to the refugees. However, because the number of refugees and individuals with multiple strain infections is so small, drawing firm inferences from these findings is difficult.
Discussion
Multiple strain infections in TB are now recognized as common occurrences and identifying patients with multiple MTB strains is critical in clinical practice, public health and molecular epidemiology. This is because not only does it provide insight into the disease patterns but also aids in the management and control of TB. This study revealed that one out of every sixteen PTB patients (6.4%) was infected with multiple strains of MTB. This prevalence is almost similar to the 7.1% reported in Kampala, Uganda11 but much lower than the 11% observed in Mubende, Uganda23. The disparities in estimations are probably due to discrepancies in the sensitivities of the genotyping techniques used to differentiate between MTB strains. While there are diverse genotyping approaches employed in the identification of mixed infections, the degree of sensitivity of each method varies. While the Mubende and Kampala investigations used 15 loci MIRU-VNTR typing, this study utilized 24 loci MIRU-VNTR typing with a single target conventional PCR. This method has been demonstrated to be very sensitive and discriminative, rendering it the gold standard in the diagnosis of multiple strain infections29,30. Other significant discrepancies can be a result of the laboratory methods utilized. As with any other genotyping approaches, detection of multiple strains can only be established when there are sufficient DNA copies of that strain in the sample being studied. Many studies, including the Kampala and Mubende studies, use culture to increase the mycobacterial population11,23,31, however, the culture step can drastically change the clonal composition thus influencing the frequency with which multiple strains are detected9,29,32. In our study, we utilized DNA isolated directly from processed sputum samples. Detection of multiple strains directly from sputum samples has successfully been documented29. However, Our findings are also much higher than the 2.8%12 reported in Malawi and 3.2% in Zambia33 but lower than the 9.6%34 and 10%35 reported in Botswana. Differences between the study settings may partly account for this difference whereby for instance the annual risk of TB infection in Malawi is approximately 1%3,36 while in Botswana it is 3%37.
Our study also revealed that unlike the relapse patients, who were not infected with multiple strains, all (100%) of the patients in our study with multiple strain infections were newly diagnosed cases. This is consistent with the findings of the Mubende study23, which observed that the majority (87.5%) of patients with multiple strains were newly diagnosed cases. This might reflect a high level of transmission and heterogeneity of strains in this category of patients (Cohen et al., 2012). This hypothesis is supported by the proportion of newly diagnosed cases that exhibited the multiple strain infection phenomena, an attribute that is reported to indicate high transmission rates38,39,40. Furthermore, a third (9.7%) of the patients with multiple strain infections were also HIV-infected. This finding is consistent with other studies, in which nearly all multiple strain TB infected people were HIV positive11,23. This appears to support the notion of the link between multiple strain TB infection and HIV/TB co-infection16,23,34,41,42. Given the high prevalence of HIV and HIV/TB co-infection in this region43, it is plausible to suggest that HIV-induced immune deficiency exposes patients to the risk of concurrent infections. HIV removes the security of being reinfected as one battles an ongoing infection thereby creating a scenario where one can be infected even before they clear an ongoing infection (Elizabeth Glaser Foundation, 2015).
Study limitations
A notable limitation of this study is the small sample size of patients with multiple strains which may have limited the precision with which we could estimate the relationship between the feature of interest and the likelihood of outcome/exposure However, exact logistic regression was selected to make such estimates since this type of analysis provides the highest chance of an event occurring within the sub-population formed by the various factors included in the model. Another limitation of this study is that since genotyping was done on genomic DNA extracted directly from MTB positive sputum samples, low levels of DNA could have been obtained especially from samples with very low/trace Mtb load thus insufficient DNA to detect multiple strains. However, doing PCR on isolates is associated with a drastic change of clonal composition9,29,32.
Conclusion
The findings of this study reveal that the rate of multiple strains infection in SWU is at 6.4% with all the patients being diagnosed with TB for their first time having this condition and two-third of them being HIV positive. Despite these findings being not statistically significant due to the small sample size, this points to a critical component of disease dynamics that has clinical implications thus emphasizing a need for a study using a larger cohort. It is also essential to establish the potential factors associated with the high risk of exposure to newly diagnosed patients at the community level. Our ability to detect multiple M. tuberculosis strains using the standard 24 loci MIRU-VNTR typing with allelic diversity in loci such as 2059 and 3171, which are excluded from the 15-locus MIRU-VNTR, lead us to recommend the use of this genotyping technique, especially in areas with M. tuberculosis endemicity similar to that observed in this study.
Materials and methods
Ethical consideration
This study was approved by the Institutional Review Board of Mbarara University of Science and Technology (MUST-IRB), the Uganda National Council for Science and Technology (with UNCST reference number HS 2379). The health facility administrators and the prime minister granted permission to access their facilities and refugee camps, respectively. Written informed consent was obtained from each patient who participated in the study.
Patients, sample collection and processing
All methods of this study were carried out in accordance with the approved guidelines. Sputum samples evaluated in this study were from individuals involved in an ongoing epidemiological study in Southwestern, Uganda, from which some papers have been published44,45. Sputum samples were collected between May 2018 and April 2019 from patients seeking health care services at either Nakivale HC111, Kabale Regional Referral Hospital and Mbarara Regional Referral Hospital who consented to the study after filling out an informed consent form. The patients were over the age of 18 years who were diagnosed with PTB using either smear microscopy or Cepheid GeneXpert and reported to have not undergone TB treatment in the preceding month.
DNA extraction
The DNA was extracted directly from clinical sputum samples using the cetyltrimethylammonium bromide (CTAB)-based method described in CLSI46 with minor modifications since these were sputum already processed for GeneXpert analysis. The, ZN microscopy-diagnosed samples underwent similar processing as the cepheid Gene Xpert-diagnosed samples by adding the Gene Xpert MTB/RIF sample reagent (Cepheid, Sunnyvale, CA, USA) to the sputum at a 2:1 ratio. The mixture was then manually agitated twice during a 15-min room temperature incubation period. Briefly, sputum samples (containing the GeneXpert processing fluid) were centrifuged for 15 min at 14,000 rpm and the supernatant discarded. The pellet was resuspended in 400μL of TE buffer before being heat-killed at 95 °C for 30 min. Subsequently, 50μL of lysozyme (10 mg/mL in Tris–EDTA [TE] buffer) was added to the sample which was then incubated overnight at 37 °C. After incubation, 70% of sodium dodecyl sulfate and 5μL of proteinase K (20 mg / mL) were added, and the sample incubated at 65 °C for 15 min. One hundred microliters of the pre-heated CTAB / NaCl solution mixture (prepared by mixing 10% CTAB with 0.7 M NaCl and heated at 65 °C for 30 min) was added to the sample in a tube together with 100μL of 5 M NaCl and gently mixed by pipetting. The mixture was then incubated for 10 min at 65 °C, after which 750μL of chloroform: isoamyl alcohol (24:1) was added and gently swirled. The mixture was then centrifuged for 5 min at 14,000 rpm after which the aqueous layer was carefully transferred to a new microcentrifuge tube containing 450μL ice-cold isopropanol and incubated for 30 min at − 20 °C. The mixture was then centrifuged for 15 min at 14,000 rpm and the supernatant discarded. The sample was then rinsed with 70% ice-cold ethanol and centrifuged for 5 min at 14,000 rpm following which ethanol was removed and the sample allowed to air dry for approximately 15 min. The pellet was then resuspended in 50μL TE buffer and incubated at 65 °C to allow resuspension after which the sample was then ready for downstream use.
The quantity of the extracted DNA was obtained using NanoDrop 3300 Fluorospectometer (ThermoFisher Scientific) after which all the samples were confirmed as MTB by PCR-detection of a 123 bp fragment of the IS6110, which is common in the members of the MTB complex. Drug susceptibility testing to screen for rifampicin and isoniazid was carried out using high resolution melting curve analysis as described in Micheni et al.45.
Single nucleotide polymorphic (SNP) typing
SNP typing was then performed on these samples using lineage 3 and 4 markers as earlier described in Micheni et al.44 to screen for the commonly occurring MTBC lineages in this region while SNP typing was performed as previously described44 using lineage 3 and 4 specific primers (Rv004C for MTB L4-U, Rv2962C for MTB L4-NU and Rv0129C for MTB L3) and their accompanying hybridization probes. Briefly, the assays were performed in 20 µl reaction mixture containing 3.75 μl of PCR water, 1.25 μl (0.5 μM final concentration) of each primer, 0.625 μl (0.25 μM final concentration) of each probe, 9.5 μl of 2X Lunar® Universal genotyping master mix, and 3 μl (5–50 ng) of extracted genomic DNA. RT-PCR was carried out in a Bio-Rad CFX96 Touch™ that was programmed for PCR amplification and a melting curve stage. For each of the three uniplex assays, the amplification stage consisted of a pre-PCR stage performed at 95 °C for 10 min, an amplification stage with denaturation at 95 °C for 30 s, primer annealing (50 °C for Rv004C or 52 °C for Rv0129C or 51 °C for Rv2962C) for 30 s and extension at 60 °C for 30 s for 45 cycles. The melting curve analysis consisted of denaturation of the amplicons at 95 °C for 10 s to produce single-stranded DNA, probe annealing temperature at 65 °C for 05 s with a continuous acquisition mode to allow capture the fluorescence and probe melting temperature ranging from 40–80 °C. The MTBC lineages were identified based on differences in melting temperature (Tm). H37Rv (L4-NU), kc32969 (L4-U) and delicus (L3) genomic DNA were used as positive control while non-template mix as a negative control.
MIRU-VNTR typing
The MIRU-VNTR PCRs were performed on genomic DNA extracted from the sputum samples using primers specific for sequences flanking the MIRU units (see Table 5). The PCR was designed to amplify a standard set of 24 MIRU-VNTR loci from genomic DNA retrieved from each sample. Each MIRU locus was amplified individually using a reaction mix and amplification profile described by Supply28 with slight modifications. Briefly, assays for the various simplex reactions were prepared according to Supply (2005). Two microliters (5–50 ng) of extracted genomic DNA was added to the PCR pre-mix and amplified in the MultiGene™ OptiMax Thermal Cycler (Massachusetts, USA) which was programmed for PCR amplification with a pre-PCR stage performed at 95 °C for 15 min, an amplification stage with denaturation at 94 °C for 60 s, primer annealing at 59 °C for 60 s, extension at 72 °C for 90 s for 40cycles and a final extension at 72 °C for 10 min. In all the assays, M. tuberculosis H37Rv was used as positive controls and sterile water as the negative control. Ten microliters of each PCR product were separated electrophoretically on 2% agarose gels for 3 h, with a 100-bp DNA ladder (Solis Biodyne™, Estonia) serving as size markers. The corresponding MIRU-VNTR bands in the gel images were reported as Roman numerals representing the number of repeats per loci as described in the protocol reference table by Supply (2005)28. For any sample that revealed multiple bands at any of the MIRU loci, the PCR was repeated to confirm the results. Multiple strains were concluded as being present if a sample had double alleles at more than one locus while those samples that had varying copy numbers at a single locus were considered as single strain evolution rather than multiple strains.
Statistical analysis
Patients’ biodata and the presence or absence of multiple strain infection results were entered and validated in Microsoft Excel® 2013. The data was then exported to Stata (Stata/SE 14.2 for windows, Stata Corp, College Station, TX) for statistical analysis. Chi-square test was used to compute proportions and determine the relationship between independent factors and dependent variables (presence multiple strain infection) with statistical significance considered at a 95% level of confidence. Since the feature of interest (multiple strain infections) was found in a small number of patients, an exact bivariate logistic regression analysis was performed to obtain odds ratios for factors that could be associated with the occurrence of multiple strains of M. tuberculosis among PTB patients in our setting. Exact logistic regression was selected because it calculates the conditional maximum chance of an event occurring within the sample population described by the model's varying factors. We did not specify a statistical significance threshold as per the recent statistical guidelines47,48.
References
WHO. Global Tuberculosis Report 2016 (World Health Organization, 2016). https://doi.org/10.16309/j.cnki.issn.1007-1776.2003.03.004.
WHO. Global Tuberculosis Report 2018 (World Health Organization, 2018). https://doi.org/10.1177/2165079915607875.
WHO. Global Tuberculosis Report 2019 (World Health Organization, 2019).
McIvor, A., Koornhof, H. & Kana, B. D. Relapse, re-infection and mixed infections in tuberculosis disease. Pathog. Dis. 75, 1–16 (2017).
Stead, W. W. Pathogenesis of a first episode of chronic pulmonary tuberculosis in man: recrudescence of residuals of the primary infection or exogenous reinfection?. Am. Rev. Respir. Dis. 95, 729–745 (1967).
Bates, J. H., Stead, W. W. & Rado, T. A. Phage type of tubercle bacilli isolated from patients with two or more sites of organ involvement. Am. Rev. Respir. Dis. 114, 353–358 (1976).
Tarashi, S., Fateh, A., Mirsaeidi, M., Siadat, S. D. & Vaziri, F. Mixed infections in tuberculosis: The missing part in a puzzle. Tuberculosis 107, 168–174 (2017).
Cohen, T., Wilson, D., Wallengren, K., Samuel, E. Y. & Murray, M. Mixed-strain Mycobacterium tuberculosis infections among patients dying in a Hospital in KwaZulu-Natal, South Africa. J. Clin. Microbiol. 49, 385–388 (2011).
Martin, A. et al. The clonal composition of Mycobacterium tuberculosis in clinical specimens could be modified by culture. Tuberculosis 90, 201–207 (2010).
Shamputa, I. C. et al. Mixed infection and clonal representativeness of a single sputum sample in tuberculosis patients from a penitentiary hospital in Georgia. Respir. Res. 7, 1–10 (2006).
Dickman, K. R. et al. Detection of multiple strains of Mycobacterium tuberculosis using MIRU-VNTR in patients with pulmonary tuberculosis in Kampala. Uganda. BMC Infect. Dis. 10, 2 (2010).
Mallard, K. et al. Molecular detection of mixed infections of Mycobacterium tuberculosis strains in sputum samples from patients in Karonga District, Malawi. J. Clin. Microbiol. 48, 4512–4518 (2010).
Manson, A. L. et al. Genomic analysis of globally diverse Mycobacterium tuberculosis strains provides insights into the emergence and spread of multidrug resistance. Nat. Genet. 49, 395–402 (2017).
Gagneux, S. Genetic diversity in mycobacterium tuberculosis. Curr. Top. Microbiol. Immunol. 374, 1–25 (2013).
Warren, R. M. et al. Patients with active tuberculosis often have different strains in the same sputum specimen. Am. J. Respir. Crit. Care Med. 169, 610–614 (2004).
Cohen, T. et al. Mixed-strain Mycobacterium tuberculosis infections and the implications for tuberculosis treatment and control. Clin. Microbiol. Rev. 25, 708–719 (2012).
Supply, P. et al. Automated high-throughput genotyping for study of global epidemiology of Mycobacterium tuberculosis based on mycobacterial interspersed repetitive units. J. Clin. Microbiol. 39, 3563–3571 (2001).
Allix-Béguec, C., Harmsen, D., Weniger, T., Supply, P. & Niemann, S. Evaluation and strategy for use of MIRU-VNTRplus, a multifunctional database for online analysis of genotyping data and phylogenetic identification of Mycobacterium tuberculosis complex isolates. J. Clin. Microbiol. 46, 2692–2699 (2008).
Kremer, K. et al. Discriminatory power and reproducibility of novel DNA typing methods for Mycobacterium tuberculosis complex strains. J. Clin. Microbiol. 43, 5628–5638 (2005).
Supply, P. et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J. Clin. Microbiol. 44, 4498–4510 (2006).
Ministry of Health Uganda. Annual Health Sector performance report 2015/16. Udhs https://doi.org/10.1016/s0033-3506(17)80015-9 (2016).
Ministry of Health Uganda. National Tuberculosis and Leprosy Division July 2017 – June 2018 Report. vol. 48 (2018).
Muwonge, A. et al. Molecular investigation of multiple strain infections in patients with tuberculosis in Mubende district, Uganda. Infect. Genet. Evol. 17, 16–22 (2013).
Zetola, N. M. et al. Clinical outcomes among persons with pulmonary tuberculosis caused by Mycobacterium tuberculosis isolates with phenotypic heterogeneity in results of drug-susceptibility tests. J. Infect. Dis. 209, 1754–1763 (2014).
Sivanand, T. Treatment correlates of successful outcomes in pulmonary multidrug-resistant tuberculosis: an individual patient data meta- analysis. Lancet 392, 821–834 (2018).
Van Rie, A. et al. Reinfection and mixed infection cause changing Mycobacterium tuberculosis drug-resistance patterns. Am. J. Respir. Crit. Care Med. 172, 636–642 (2005).
Kempker, R. R. et al. Acquired drug resistance in mycobacterium tuberculosis and poor outcomes among patients with multidrug-resistant tuberculosis. Emerg. Infect. Dis. 21, 992–1001 (2015).
Supply, P. Multilocus variable number tandem repeat genotyping of mycobacterium tuberculosis. Inst. Pasteur Lille 73, 2 (2005).
Farmanfarmaei, G. et al. Bias in detection of Mycobacterium tuberculosis polyclonal infection: Use clinical samples or cultures?. Mol. Cell. Probes 33, 1–3 (2017).
Bouklata, N. et al. Molecular typing of mycobacterium tuberculosis complex by 24-locus based MIRU-VNTR typing in conjunction with spoligotyping to assess genetic diversity of strains circulating in Morocco. PLoS ONE 10, 1–16 (2015).
Stavrum, R. et al. High diversity of Mycobacterium tuberculosis genotypes in South Africa and preponderance of mixed infections among ST53 isolates. J. Clin. Microbiol. 47, 1848–1856 (2009).
Hanekom, M. et al. Population structure of mixed mycobacterium tuberculosis infection is strain genotype and culture medium dependent. PLoS ONE 8, 5–10 (2013).
Mulenga, C. et al. Diversity of Mycobacterium tuberculosis genotypes circulating in Ndola. Zambia. BMC Infect. Dis. 10, 2 (2010).
Shin, S. S. et al. Mixed mycobacterium tuberculosis-strain infections are associated with poor treatment outcomes among patients with newly diagnosed tuberculosis, independent of pretreatment heteroresistance. J. Infect. Dis. 218, 1974–1982 (2018).
Zetola, N. M. et al. Mixed Mycobacterium tuberculosis complex infections and false-negative results for rifampin resistance by genexpert MTB/RIF are associated with poor clinical outcomes. J. Clin. Microbiol. 52, 2422–2429 (2014).
Vorkas, C. et al. Tuberculosis drug resistance and outcomes among tuberculosis inpatients in Lilongwe, Malawi. Malawi Med. J. 24, 21–24 (2012).
Tuelo, M. et al. Genetic diversity of Mycobacterium tuberculosis strains circulating in Botswana. PLoS ONE 4, 1–14 (2019).
Perez-Lago,. Co-infection with drug-susceptible and reactivated latent MDR Mtbletter. Emerg. Infect. Dis. 21, 20–22 (2015).
Zamani, S. et al. Determination of circulating Mycobacterium tuberculosis strains and transmission patterns among TB patients in Iran, using 15 loci MIRU-VNTR. Int. J. Mycobacteriol. 4, 119 (2015).
Tessema, B. et al. Molecular epidemiology and transmission dynamics of Mycobacterium tuberculosis in Northwest Ethiopia: New phylogenetic lineages found in Northwest Ethiopia. BMC Infect. Dis. 13, 2 (2013).
Cohen, T. et al. Within-host heterogeneity of mycobacterium tuberculosis infection is associated with poor early treatment response: A prospective cohort study. J. Infect. Dis. 213, 1796–1799 (2016).
Shin, S. S. et al. Advanced immune suppression is associated with increased prevalence of mixed-strain mycobacterium tuberculosis infections among persons at high risk for drug-resistant tuberculosis in Botswana. J. Infect. Dis. 211, 347–351 (2015).
Elizabeth Glaser Foundation. Strengthening the Tuberculosis and HIV / AIDS Response in the Southwest Region of Uganda ( STAR-SW ) Project. (2015).
Micheni, L. N., Kassaza, K., Kinyi, H., Ntulume, I. & Bazira, J. Diversity of mycobacterium tuberculosis complex lineages associated with pulmonary tuberculosis in Southwestern, Uganda. Tuberc. Res. Treat. 2021, 1–6 (2021).
Nkatha, L. et al. Rifampicin and isoniazid drug resistance among patients diagnosed with pulmonary tuberculosis in southwestern Uganda. PLoS ONE 16, 1–9 (2021).
CLSI. Laboratory Detection and Identification of Mycobacteria; Approved Standard (Clinical and Laboratory Standards Institute, 2008).
Greenland, S. et al. Statistical tests, P values, confidence intervals, and power: a guide to misinterpretations. Eur. J. Epidemiol. 31, 337–350 (2016).
Wasserstein, R. L. & Lazar, N. A. The ASA’s statement on p-values: Context, process, and purpose. Am. Stat. 70, 129–133 (2016).
Acknowledgements
We gratefully acknowledge the technical help provided by the TB laboratory staff of Kabale and Mbarara regional referral hospitals, Nakivale health centre, and the Genomics and Translational laboratory of Mbarara University of Science and Technology.
Author information
Authors and Affiliations
Contributions
L.N.M. and J.B. designed and funded the study; L.N.M. and K.K. collected the data; L.N.M., K.K. and I.N. conducted data analysis and interpretation. L.N.M., K.K., I.N., H.K., J.B. interpreted results, wrote, revised the initial and final manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Micheni, L.N., Kassaza, K., Kinyi, H. et al. Detection of Mycobacterium tuberculosis multiple strains in sputum samples from patients with pulmonary tuberculosis in south western Uganda using MIRU-VNTR. Sci Rep 12, 1656 (2022). https://doi.org/10.1038/s41598-022-05591-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-05591-3
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
