
Abstract
Breast cancer metastasis accounts for most of the deaths from breast cancer. Identification of germline variants associated with survival in aggressive types of breast cancer may inform understanding of breast cancer progression and assist treatment. In this analysis, we studied the associations between germline variants and breast cancer survival for patients with distant metastases at primary breast cancer diagnosis. We used data from the Breast Cancer Association Consortium (BCAC) including 1062 women of European ancestry with metastatic breast cancer, 606 of whom died of breast cancer. We identified two germline variants on chromosome 1, rs138569520 and rs146023652, significantly associated with breast cancer-specific survival (P = 3.19 × 10−8 and 4.42 × 10−8). In silico analysis suggested a potential regulatory effect of the variants on the nearby target genes SDE2 and H3F3A. However, the variants showed no evidence of association in a smaller replication dataset. The validation dataset was obtained from the SNPs to Risk of Metastasis (StoRM) study and included 293 patients with metastatic primary breast cancer at diagnosis. Ultimately, larger replication studies are needed to confirm the identified associations.
Similar content being viewed by others


Introduction
Breast cancer is the most common female cancer in the Western world and one of the most common causes of cancer death in women globally1. Early detection and better treatments have helped to reduce breast cancer mortality in recent decades2. Yet, when breast cancer metastasizes to distant sites, prognosis continues to be poor and for most cases treatment is only palliative3. Metastases in breast cancer can remain undetectable for many years after initial diagnosis, leading to incurable lesions4. Approximately 15% of patients with breast cancer will develop distant metastases within 3 years after diagnosis of the primary tumor5. Therefore, it is important to have the tools able to detect breast cancer metastases at earlier stages, in order to better manage and predict breast cancer progression. Prognostication models could benefit from the inclusion of germline genetic biomarkers that are capable of predicting tumor recurrence, second tumors or prognosis of second tumors. However, so far, it has been difficult to identify individual common germline variants associated with primary breast cancer survival due to the small effect size these variants are likely to have6,7. Likewise, evidence as to whether or not germline variants can increase the probability of metastatic progression is currently limited to a few studies4,8. For example, a candidate gene study identified common single nucleotide polymorphisms (SNPs) located within SIPA1 that were associated with metastasis and poor breast cancer prognosis9. Other studies have identified other metastasis susceptibility genes such as RRP1b10. Germline variants could specifically provide metastatic predisposition by affecting treatment response11 or promoting tumor initiating events and providing new metastatic functions to tumor cells4.
The aim of this study was to identify associations between common germline variants and breast cancer-specific survival in patients with metastasis at primary breast cancer diagnosis. We hypothesized that germline variants might predispose to poorer survival after breast cancer metastasis, and that analyzing a set of patients with similar stage of the disease might help identify variants that do not show evidence of association in larger but more heterogeneous datasets.
Results
We used data from the Breast Cancer Association Consortium (BCAC): the dataset comprised data from 50 studies from which follow-up information for women diagnosed with distant metastases at primary breast cancer diagnosis was available. The results were based on the meta-analysis of two genome-wide SNP arrays (iCOGS12 and OncoArray13 (see “Methods”). We analyzed variants that had a minor allele frequency (MAF) > 0.01 and an imputation quality r2 > 0.7 for at least one of the two arrays. Details about the individual studies, the genotyping array used and number of patients included are given in Supplementary Table 1. We analyzed the genotypes and clinico-pathological data of a total of 1062 breast cancer patients, 606 of whom died of breast cancer within 15 years of follow-up. Of these, 721 of the patients had estrogen receptor (ER)-positive disease (388 deaths) and 227 had ER-negative disease (148 deaths). All patients were women of European descent. The patients were diagnosed from 1979 to 2014 (median: 2004) and aged 26–92 (median: 60) years.
Manhattan plots showing the association between germline variants and breast cancer-specific survival of all, ER-positive and ER-negative metastasized breast cancers are shown in Fig. 1. We identified two genome-wide significant (P < 5 × 10−8) variants (SNPs: rs138569520 and rs146023652) on chromosome 1 associated with breast cancer-specific survival for all metastasized breast cancers (Table 1, Supplementary Table 2). The two variants were part of a set of six highly correlated SNPs (Table 1, r2 > 0.88) based on European subjects in phase 3 of the 1000 Genomes Project14. No variant reached genome-wide significance for ER-positive or for ER-negative breast cancer tumors alone (Supplementary Tables 3 and 4).

Manhattan plots of the meta-analysis of OncoArray and iCOGS datasets for the association of common germline variants and breast cancer-specific survival for patients with metastases at primary breast cancer diagnosis for (A) all breast tumors, (B) ER-positive tumors, and (C) ER-negative tumors. The y axis shows the − log10 P values of each variant analyzed, and the x axis shows their chromosome position. The red horizontal line represents P = 5 × 10−8.
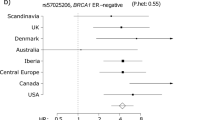
The variant with the strongest association was the SNP rs138569520 (HR = 3.67, 95% CI 1.86–7.23 and P = 3.19 × 10−8). The HR estimates for rs138569520 in the ER-positive (HR = 3.38, 95% CI 1.48–7.70 and P = 4.37 × 10−4) and ER-negative (HR = 2.76, 95% CI 1.16–6.64 and P = 8.70 × 10−3) were similar (P = 0.97 for difference).
Several genes (SDE2, LEFTY2, PYCR2 and H3F3A) were located within 100 kb of the most significant SNP rs138569520. We interrogated functional genomic data including annotations of enhancers, promoters and transcription factor binding sites and found evidence consistent with gene regulation in the regions containing the associated variants (Fig. 2). Hi-C analysis in HMEC cells15 showed that the lead variant rs138569520 is located in a genomic region interacting with the promoter region of H3F3A. SNPs rs146023652 and rs114512448 overlapped with transcription factor (TF) binding sites which might reflect the active transcription of SDE2. ChIP-seq signals from primary breast sub-populations16 also showed potential regulatory regions containing rs114512448. ChIA-PET analysis in MCF-7 cells from ENCODE17, detected an interaction between rs114512448 and the PYCR2 gene. Finally, ChIA-PET also detected an interaction between rs72757046 and SDE2 and H3F3A.

Functional annotation of the six highly correlated SNPs: rs138569520, rs146023652, rs114512448, rs143653255, rs115086585 and rs72757046. TF transcription factor.
Using KMplotter (kmplot.com/analysis)18, we tested the association of the mRNA tumor expression of SDE2 and H3F3A, the genes in closest proximity to rs138569520, with overall survival in grade 3 breast tumors (to select the most aggressive subtype; selection for stage 4 was not available). Low mRNA expression levels of SDE2 gene were significantly associated (P = 0.01) with poorer breast cancer survival (Fig. 3a), while, in contrast, high expression of H3F3A was associated with lower survival (P = 6.7 × 10−5) (Fig. 3b). These associations were not statistically significant, neither for grade 1 or for grade 2 disease (P > 0.21).

Kaplan–Meier overall survival plot for high versus low expression level of the genes (A) SDE2 (n = 204) and (B) H3F3A (n = 503) restricted to patients with a grade 3 tumor and 15 years of follow-up. The differential expression analysis was performed in KMplotter.
Lastly, we aimed to evaluate the significance of the two genome-wide significant SNPs using an independent set of 293 breast cancer patients with metastatic primary breast cancer at diagnosis from the SNPs to Risk of Metastasis (StoRM) study19. All patients were diagnosed in France from March 2012 to May 2014, aged 18 years or older (median: 59 years) and followed up to July 2017. A total of 293 patients were available for the validation study, 239 of whom had events, defined as progression and/or death occurring during follow-up. Both SNPs had good imputation quality (r2 ~ 0.7) and similar MAFs to those in the BCAC dataset (~ 2%). However, neither of the two SNPs replicated in the survival analysis with the StoRM dataset (Table 2): rs138569520 (HR = 1.49, 95% CI 0.60–3.71, P = 0.34) and rs146023652 (HR = 1.25, 95% CI 0.46–3.37, P = 0.66). Although the HR estimates in the StoRM validation dataset were smaller than those from the BCAC analyses (HR = 3.67 and 3.64), the confidence limits overlapped.
Because the BCAC dataset also included prevalent cases (n = 466), we repeated the analysis with incident cases (n = 596) to match the study design in StoRM more closely. The HR estimates were similar to those for the overall analysis (rs138569520: HR = 3.77, 95% CI 1.71–8.30, P = 3.12 × 10−5 and rs146023652: HR = 3.75, 95% CI 1.70–8.29, P = 3.60 × 10−5). Finally, since the maximum follow-up in the StoRM dataset was shorter (5 years, compared with a maximum of 15 years in the BCAC dataset), we repeated the main analysis in BCAC using a follow-up of 5 years (n = 1031, 476 deaths). The associations for the two SNPs were slightly less significant (rs138569520: HR = 3.43, 95% CI 1.74–6.80, P = 1.83 × 10−7 and rs146023652: HR = 3.41, 95% CI 1.72–6.76, P = 2.55 × 10−7) but the HR estimates were similar to those from the main analysis.
Discussion
In this analysis of breast cancer patients with metastatic primary breast cancer at diagnosis, involving 1062 patients with 606 breast cancer-specific deaths, we identified two variants on chromosome 1 (rs138569520 and rs146023652) associated with survival, at genome-wide levels of statistical significance. The most significant association was for the SNP rs138569520 (P = 3.19 × 10−8). The HR estimates were similar in patients with ER-positive and ER-negative disease.
Two genes, SDE2 and H3F3A, were in closest proximity of rs138569520. Both genes have been previously associated with oncogenic processes relevant for metastatic progression: the SDE2 gene (“silencing defective 2”) is known to be involved in DNA replication, telomere maintenance and cell cycle control20,21. The functional roles of SDE2 have been studied in a proteome dynamics analysis in prostate cancer cells; the results suggested that alterations of the gene might diminish the error-prone DNA repair pathway activation and promote missense mutations22. The gene H3F3A encodes for histone H3.3, and mutations in this protein have been linked to multiple cancer processes23, including breast invasive ductal carcinoma24. Additionally, the differential expression of these two genes was significantly associated with survival in grade 3 tumors based on KMplotter. Previous studies have also linked the expression of these genes to oncogenic processes. For example, downregulation of SDE2 was associated with mutation disease phenotype as well as poorer mortality outcomes22. Likewise, overexpression of H3F3A was associated with lung cancer progression and promotion of lung cancer cell migration by activation of metastasis-related genes25. Unfortunately, in KMplotter it was not possible to specifically select stage 4 tumors, which limits the interpretation of our findings. Future studies are needed in order to corroborate the association of SDE2 and H3F3A expression with survival in this group of patients.
Additionally, there was predicted genomic activity in the locus based on the intersection of multiple genomic regulatory features in breast tissue. Although the SNPs appeared to cluster around SDE2, there was also in-silico evidence for two other potential target genes at this locus (H3F3A and PYCR2). PYCR2 encodes for a mitochondrial protein involved in proline biosynthesis. While little is known about this proline form, studies for the close family member PYCR1 have found that higher levels of mRNA were associated with reduced survival from breast cancer patients26. To support further our hypothesis that the two genome-wide significant SNPs (rs138569520 and rs146023652) were specific for survival in patients with metastatic disease, we confirmed that there were no associations (HR = 1.04, P = 0.58, MAF = 0.02 and HR = 1.03, P = 0.60, MAF = 0.02 respectively) with breast cancer-specific survival in the most recent BCAC dataset for all invasive early (stages I–III) breast cancers (OncoArray and iCOGS, n = 86,627)27.
On the other hand, the two genome-wide significant variants, rs138569520 and rs146023652, were not replicated (P = 0.34 and P = 0.66, respectively) using an independent dataset of patients with metastatic primary breast cancer diagnosis (n = 293). The imputation quality and the minor allele frequency of the SNPs in the replication cohort were comparable to those in the BCAC analyses (MAF = 2% and r2 > 7%), therefore the negative result could not be attributed to those factors. Age of the patients could also not explain the difference since both datasets had comparable median ages at diagnosis, 60 years for BCAC and 59 years for StoRM. On the other hand, it is important to state that there were several factors that varied between the datasets. First, the sample size differed considerably between BCAC (n = 1062) and the StoRM study (n = 293), the latter having a relatively small sample size which limits the power to detect associations. Total follow-up time also varied: for the BCAC dataset, patients were followed for a maximum of 15 years, while for the StoRM study the follow-up ended at 5 years. However, the results from the complementary analysis using the BCAC dataset and 5-year follow-up were comparable to the initial 15 years follow-up results. This finding suggests that the disparity in estimates between the two analyses is not due to shorter follow-up. There were several other differences between the main BCAC dataset and the StoRM cohort used for validation. For example, the BCAC dataset included multiple studies from several countries while the StoRM cohort included solely patients from France. Moreover, StoRM was a recent cohort with the earliest reported diagnosis starting in 2012. On the other hand, in BCAC, the year of patients’ diagnosis ranged between 1979 and 2014 and included prevalent cases. While the analysis in BCAC using exclusively incident cases gave comparable estimates to the main analysis, the difference in the years of diagnosis could be related to differences in treatment strategies that were not considered in the current analysis. The lack of information about detailed treatment is a potential weakness of the current analysis and validation. Treatment strategy, together with characteristics of the tumor, will also influence the final prognosis of metastatic breast cancer28. It is important to note that the associations observed in the BCAC study may be false positives, and that further large replication studies will be required to confirm or refute the associations.
In conclusion, this analysis of BCAC patients with metastatic primary breast cancer at diagnosis from the BCAC dataset identified a new region in chromosome 1 associated with breast cancer-specific survival. The region includes six highly correlated SNPs that are predicted to be in an active region of the genome based on in-silico evidence from breast cancer tissues and that are located in close proximity to genes involved in oncogenic processes. However, we were unable to validate the association using a smaller, independent set of patients. Overall, the role of germline variants in metastasis and progression remains unclear. Further analyses with larger datasets including treatment information and functional analysis are needed to better understand the underlying biological processes and the links between this locus and the nearby genes. Prior validation of the reported associations is needed before these findings can be used in clinical-decision making. Therefore, a next step is to study these SNPs in a, preferably, prospective large series of metastasized breast cancer patients. Ultimately, germline variants could help identifying tailored treatments for patients with metastatic disease or better strategies for risk management stratification of aggressive forms of breast cancer.
Methods
Breast cancer samples and genotype data: Breast Cancer Association Consortium (BCAC)
We used genotype and clinico-pathological data (database version 12) data from the Breast Cancer Association Consortium (BCAC). The dataset included 1062 breast cancer patients with metastatic primary breast cancer at diagnosis that were genotyped using one of the two different genotyping platforms: iCOGS12 and OncoArray13, providing genome-wide coverage of common variants. The main analyses were based on imputed variants using the Haplotype Reference Consortium29 as reference panel. All patients were women of European ancestry, aged 26–92 years (median: 60) years with metastasized breast cancer at diagnosis. Women were diagnosed between 1979 and 2014, with a median follow-up was three and a half years. Additional details about the genotype data and sample quality control have been described previously7,27,30. We only analyzed variants that had a minor allele frequency (MAF) > 0.01 and an imputation quality r2 > 0.7 for at least one of the two genotyping platforms (iCOGS or OncoArray). Details about the individual studies included in the analyses, including the array used, associated country and number of patients with metastatic primary breast cancer at diagnosis are given in Supplementary Table 1. The secondary use of data for the study was approved by the Data Access Committee of the BCAC, under the legal provisions of the Memorandum of Understanding and Data Transfer Agreements of Cambridge University which all the contributing institutions, which includes that all contributing institutions provided the data with the appropriate approval of their institutional review boards and informed consent of the participants of the individual studies.
Statistical and bioinformatic methods
We estimated the association of the germline variants with breast-cancer specific survival using Cox proportional hazards regression. We analyzed separately the OncoArray and iCOGS datasets and combined the estimates using fixed-effect meta-analyses. Follow-up was right censored on the date of death, last date known alive if death did not occur, or at 15 years after diagnosis, whichever came first27. Time at risk was calculated from the date of diagnosis with left truncation for prevalent cases. The models were stratified by country and included the first two ancestry informative principal components12. We performed the analysis for all breast cancers and for ER-positive and ER-negative tumors separately. To identify evidence of potential cis-regulatory activity, we intersected germline variants with numerous sources of genomic annotation information from primary breast cells (e.g., chromosome conformation, enhancer–promoter correlations, transcription factor and histone modification ChIP-seq). To assess the effect of gene expression on survival we used the Kaplan–Meier plotter on breast tissue data, grade 3 tumors and 15 years of follow-up (180 months)18.
Validation dataset: SNPs to risk of metastasis (StoRM)
To attempt to validate our results we used data from the SNPs to Risk of Metastasis (StoRM) study. StoRM is a multicentric, prospective, cohort study of metastatic breast cancer patients in France that was originally designed to identify genetic and other factors associated with metastatic relapse and survival19. Patients aged 18 years or older, with a histologically proven breast cancer that was metastatic for less than 1 year were included. All patients that had another coexisting cancer or another cancer diagnosed within the last 5 years, were excluded from the study. Patients were followed from March 2012 to July 2017. Time to progression on the first metastatic treatment was recorded and patients were followed until death, every 6 months for 3 years, and then annually until July 2017. A total of 293 patients were available for the validation. The median follow-up was of 3.2 years. Because of the short total follow-up time (5 years) and the advanced disease stage of the patients in the cohort, both a recorded progression and/or death were considered as an event in the survival analyses. Of the whole set of 293 patients, 239 had a progression and/or died during the follow-up period.
Ethical approval
The study was performed in accordance with the Declaration of Helsinki. All individual studies, from which data was used, were approved by the appropriate medical ethical committees and/or institutional review boards. All study participants provided informed consent.
References
Torre, L. A., Islami, F., Siegel, R. L., Ward, E. M. & Jemal, A. Global cancer in women: Burden and trends. Cancer Epidemiol. Biomark. Prev. 26, 444–457 (2017).
Narod, S. A., Iqbal, J. & Miller, A. B. Why have breast cancer mortality rates declined?. J. Cancer Policy 5, 8–17 (2015).
Sledge, G. W. Curing metastatic breast cancer. J. Oncol. Pract. 12, 6–10 (2016).
Nguyen, D. X. & Massagué, J. Genetic determinants of cancer metastasis. Nat. Rev. Genet. 8, 341–352 (2007).
Weigelt, B., Peterse, J. L. & van’t Veer, L. J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 5, 591–602 (2005).
Pharoah, P. D. P. et al. Polygenic susceptibility to breast cancer and implications for prevention. Nat. Genet. 31, 33–36 (2002).
Escala-Garcia, M. et al. Genome-wide association study of germline variants and breast cancer-specific mortality. Br. J. Cancer 120, 647–657 (2019).
Priestley, P. et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216 (2019).
Crawford, N. P. S. et al. Germline polymorphisms in SIPA1 are associated with metastasis and other indicators of poor prognosis in breast cancer. Breast Cancer Res. 8, R16 (2006).
Crawford, N. P. S. et al. Rrp1b, a new candidate susceptibility gene for breast cancer progression and metastasis. PLoS Genet. 3, e214 (2007).
O’Donnell, P. H. & Ratain, M. J. Germline pharmacogenomics in oncology: Decoding the patient for targeting therapy. Mol. Oncol. 6, 251–259 (2012).
Michailidou, K. et al. Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94 (2017).
Amos, C. I. et al. The OncoArray Consortium: A network for understanding the genetic architecture of common cancers. Cancer Epidemiol. Biomark. Prev. 26, 126–135 (2017).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Rao, S. S. P. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Pellacani, D. et al. Analysis of normal human mammary epigenomes reveals cell-specific active enhancer states and associated transcription factor networks. Cell Rep. 17, 2060–2074 (2016).
Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Györffy, B. et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 123, 725–731 (2010).
Delrieu, L. et al. Analysis of the StoRM cohort reveals physical activity to be associated with survival in metastatic breast cancer. Sci. Rep. 10, 10757 (2020).
Jo, U. et al. PCNA-dependent cleavage and degradation of SDE2 regulates response to replication stress. PLOS Genet. 12, e1006465 (2016).
Rageul, J. et al. SDE2 integrates into the TIMELESS-TIPIN complex to protect stalled replication forks. Nat. Commun. 11, 5495 (2020).
Luo, A., Gong, Y., Kim, H. & Chen, Y. Proteome dynamics analysis identifies functional roles of SDE2 and hypoxia in DNA damage response in prostate cancer cells. NAR Cancer 2, zcaa010 (2020).
Yuen, B. T. K. & Knoepfler, P. S. Histone H3.3 mutations: A variant path to cancer. Cancer Cell 24, 567–574 (2013).
Sweeney, S. M. et al. AACR project genie: Powering precision medicine through an international consortium. Cancer Discov. 7, 818–831 (2017).
Park, S.-M. et al. Histone variant H3F3A promotes lung cancer cell migration through intronic regulation. Nat. Commun. 7, 12914 (2016).
Ding, J. et al. Human mitochondrial pyrroline-5-carboxylate reductase 1 promotes invasiveness and impacts survival in breast cancers. Carcinogenesis 38, 519–531 (2017).
Escala-Garcia, M. et al. Breast cancer risk factors and their effects on survival: A Mendelian randomisation study. BMC Med. 18, 327 (2020).
Deluche, E. et al. Contemporary outcomes of metastatic breast cancer among 22,000 women from the multicentre ESME cohort 2008–2016. Eur. J. Cancer 129, 60–70 (2020).
Frye, F. L. & Cucuel, J. P. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Escala-Garcia, M. et al. A network analysis to identify mediators of germline-driven differences in breast cancer prognosis. Nat. Commun. 11, 312 (2020).
Acknowledgements
BCAC: We thank all the individuals who took part in these studies and all the researchers, clinicians, technicians and administrative staff who have enabled this work to be carried out. ABCFS: Maggie Angelakos, Judi Maskiell, Gillian Dite. ABCS: Frans Hogervorst, Sten Cornelissen and Annegien Broeks. BBCS: Eileen Williams, Elaine Ryder-Mills, Kara Sargus. BCINIS: Dr. K. Landsman, Dr. N. Gronich, Dr. A. Flugelman, Dr. W. Saliba, Dr. E. Liani, Dr. I. Cohen, Dr. S. Kalet, Dr. V. Friedman, Dr. O. Barnet. BIGGS: Niall McInerney, Gabrielle Colleran, Andrew Rowan, Angela Jones. BREOGAN: Manuela Gago-Dominguez, Jose Esteban Castelao, Angel Carracedo, Victor Muñoz Garzón, Alejandro Novo Domínguez, Maria Elena Martinez, Sara Miranda Ponte, Carmen Redondo Marey, Maite Peña Fernández, Manuel Enguix Castelo, Maria Torres, Manuel Calaza, José Antúnez, Máximo Fraga; Joaquín González-Carreró and the Department of Pathology and Biobank of University Hospital Complex of Vigo, Instituto de Investigacion Biomedica Galicia Sur, SERGAS. BSUCH: Peter Bugert, Medical Faculty Mannheim. CCGP: Styliani Apostolaki, Anna Margiolaki, Georgios Nintos, Maria Perraki, Georgia Saloustrou, Georgia Sevastaki, Konstantinos Pompodakis. CGPS: Dorthe Uldall Andersen, Maria Birna Arnadottir, Anne Bank, Dorthe Kjeldgård Hansen and the Danish Cancer Biobank. CPS-II: Centers for Disease Control and Prevention National Program of Cancer Registries. The National Cancer Institute Surveillance Epidemiology and End Results program. Participants and the investigators of EPIC (European Prospective Investigation into Cancer and Nutrition). ESTHER: Hartwig Ziegler, Sonja Wolf, Volker Hermann, Christa Stegmaier, Katja Butterbach. GC-HBOC: Stefanie Engert, Heide Hellebrand, Sandra Kröber and LIFE. Markus Loeffler, Joachim Thiery, Matthias Nüchter, Ronny Baber. GENICA: Christian Baisch, Hiltrud Brauch, Thomas Brüning, Hans-Peter Fischer, UH, Volker Harth, RH, Yon-Dschun Ko, Wing-Yee Lo, Anne Lotz, Beate Pesch, Sylvia Rabstein. HABCS: Michael Bremer, Natalia Bogdanova, Peter Schürmann, Johann H. Karstens, Peter Hillemanns. HEBCS: Carl Blomqvist, Kristiina Aittomäki, Rainer Fagerholm, Kirsimari Aaltonen, Karl von Smitten, Irja Erkkilä. HUBCS: Darya Prokofyeva, Shamil Gantsev. KARMA and SASBAC: Swedish Medical Research Counsel. KBCP: Eija Myöhänen, Helena Kemiläinen. LMBC: Gilian Peuteman, Thomas Van Brussel, EvyVanderheyden and Kathleen Corthouts. MARIE: Petra Seibold, Nadia Obi, Sabine Behrens, Ursula Eilber and Muhabbet Celik. MBCSG: Siranoush Manoukian, Bernard Peissel, Jacopo Azzollini, Erica Rosina, Daniela Zaffaroni, Bernardo Bonanni, Irene Feroce, Mariarosaria Calvello, Aliana Guerrieri Gonzaga, Monica Marabelli, Davide Bondavalli and the personnel of the Cogentech Cancer Genetic Test Laboratory. The following are NBCS Collaborators: Kristine K. Sahlberg (PhD), Anne-Lise Børresen-Dale (Prof. Em.), Lars Ottestad (MD), Rolf Kåresen (Prof. Em.), Dr. Ellen Schlichting (MD), Marit Muri Holmen (MD), Toril Sauer (MD), Vilde Haakensen (MD), Olav Engebråten (MD), Bjørn Naume (MD), Alexander Fosså (MD), Cecile E. Kiserud (MD), Kristin V. Reinertsen (MD), Åslaug Helland (MD), Margit Riis (MD), OSBREAC and Grethe I. Grenaker Alnæs (MSc). NHS/NHS2: the following state cancer registries: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. OBCS: Arja Jukkola-Vuorinen, Mervi Grip, Saila Kauppila, Meeri Otsukka, Leena Keskitalo and Kari Mononen. ORIGO: E. Krol-Warmerdam, and J. Blom. PBCS: Louise Brinton, Mark Sherman, Neonila Szeszenia-Dabrowska, Beata Peplonska, Witold Zatonski, Pei Chao, Michael Stagner. The ethical approval for the PREFACE: Sonja Oeser and Silke Landrith. RBCS: Petra Bos, Jannet Blom, Ellen Crepin, Elisabeth Huijskens, Anja Kromwijk-Nieuwlaat, Annette Heemskerk, the Erasmus MC Family Cancer Clinic. SBCS: Sue Higham, Helen Cramp, Dan Connley, Ian Brock, Sabapathy Balasubramanian and Malcolm W.R. Reed. We thank the SEARCH and EPIC teams. SKKDKFZS: Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, Germany [UH, MM], Staedtisches Klinikum Karlsruhe, Institute of Pathology [TR]. SZBCS: Ewa Putresza. UCIBCS: Irene Masunaka. UKBGS: Breast Cancer Now and the Institute of Cancer Research and NHS funding to the Royal Marsden/ICR NIHR Biomedical Research Centre.
Funding
BCAC is funded the European Union's Horizon 2020 Research and Innovation Programme (Grant numbers 634935 and 633784 for BRIDGES and B-CAST respectively), and PERSPECTIVE I&I, funded by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, the Ministère de l’Économie et de l'Innovation du Québec through Genome Québec, the Quebec Breast Cancer Foundation. The EU Horizon 2020 Research and Innovation Programme funding source had no role in study design, data collection, data analysis, data interpretation or writing of the report. Additional funding for BCAC is provided via the Confluence project which is funded with intramural funds from the National Cancer Institute Intramural Research Program, National Institutes of Health. Genotyping of the OncoArray was funded by the NIH Grant U19 CA148065, and Cancer UK Grant C1287/A16563 and the PERSPECTIVE project supported by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research (Grant GPH-129344) and, the Ministère de l’Économie, Science et Innovation du Québec through Genome Québec and the PSRSIIRI-701 grant, and the Quebec Breast Cancer Foundation. Funding for iCOGS came from: the European Community's Seventh Framework Programme under Grant agreement n° 223175 (HEALTH-F2-2009-223175) (COGS), Cancer Research UK (C1287/A10118, C1287/A10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007, C5047/A10692, C8197/A16565), the National Institutes of Health (CA128978) and Post-Cancer GWAS initiative (1U19 CA148537, 1U19 CA148065 and 1U19 CA148112—the GAME-ON initiative), the Department of Defence (W81XWH-10-1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, and Komen Foundation for the Cure, the Breast Cancer Research Foundation, and the Ovarian Cancer Research Fund. ABCFS was supported by Grant UM1 CA164920 from the National Cancer Institute (USA). The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centres in the in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the USA Government or the BCFR. The ABCFS was also supported by the National Health and Medical Research Council of Australia, the New South Wales Cancer Council, the Victorian Health Promotion Foundation (Australia) and the Victorian Breast Cancer Research Consortium. J.L.H. is a National Health and Medical Research Council (NHMRC) Senior Principal Research Fellow. M.C.S. is a NHMRC Senior Research Fellow. The ABCS study was supported by the Dutch Cancer Society [Grants NKI 2007-3839; 2009-4363; 2015-7632]. The work of the BBCC was partly funded by ELAN-Fond of the University Hospital of Erlangen. The BBCS is funded by Cancer Research UK and Breast Cancer Now and acknowledges NHS funding to the NIHR Biomedical Research Centre, and the National Cancer Research Network (NCRN). For the BCFR-PA this work was supported by Grant UM1 CA164920 from the National Cancer Institute. For BIGGS, ES is supported by NIHR Comprehensive Biomedical Research Centre, Guy's & St. Thomas' NHS Foundation Trust in partnership with King's College London, United Kingdom. IT is supported by the Oxford Biomedical Research Centre. The BREOGAN is funded by Acción Estratégica de Salud del Instituto de Salud Carlos III FIS PI12/02125/Cofinanciado FEDER; Acción Estratégica de Salud del Instituto de Salud Carlos III FIS PI17/00918/Cofinanciado FEDER; Acción Estratégica de Salud del Instituto de Salud Carlos III FIS Intrasalud (PI13/01136); Programa Grupos Emergentes, Cancer Genetics Unit, Instituto de Investigacion Biomedica Galicia Sur. Xerencia de Xestion Integrada de Vigo-SERGAS, Instituto de Salud Carlos III, Spain; Grant 10CSA012E, Consellería de Industria Programa Sectorial de Investigación Aplicada, PEME I + D e I + D Suma del Plan Gallego de Investigación, Desarrollo e Innovación Tecnológica de la Consellería de Industria de la Xunta de Galicia, Spain; Grant EC11-192. Fomento de la Investigación Clínica Independiente, Ministerio de Sanidad, Servicios Sociales e Igualdad, Spain; and Grant FEDER-Innterconecta. Ministerio de Economia y Competitividad, Xunta de Galicia, Spain. The BSUCH study was supported by the Dietmar-Hopp Foundation, the Helmholtz Society and the German Cancer Research Center (DKFZ). CCGP is supported by funding from the University of Crete. The CGPS was supported by the Chief Physician Johan Boserup and Lise Boserup Fund, the Danish Medical Research Council, and Herlev and Gentofte Hospital. The American Cancer Society funds the creation, maintenance, and updating of the CPS-II cohort. The coordination of EPIC is financially supported by the European Commission (DG-SANCO) and the International Agency for Research on Cancer. The national cohorts are supported by: Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale, Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid, German Cancer Research Center (DKFZ), Federal Ministry of Education and Research (BMBF) (Germany); the Hellenic Health Foundation, the Stavros Niarchos Foundation (Greece); Associazione Italiana per la Ricerca sul Cancro-AIRC-Italy and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (The Netherlands); Health Research Fund (FIS), PI13/00061 to Granada, PI13/01162 to EPIC-Murcia, Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, ISCIII RETIC (RD06/0020) (Spain); Cancer Research UK (14136 to EPIC-Norfolk; C570/A16491 and C8221/A19170 to EPIC-Oxford), Medical Research Council (1000143 to EPIC-Norfolk, MR/M012190/1 to EPIC-Oxford) (United Kingdom). The ESTHER study was supported by a grant from the Baden Württemberg Ministry of Science, Research and Arts. Additional cases were recruited in the context of the VERDI study, which was supported by a grant from the German Cancer Aid (Deutsche Krebshilfe). The GC-HBOC is supported by the German Cancer Aid (Grant no 110837, coordinator: Rita K. Schmutzler, Cologne). This work was also funded by the European Regional Development Fund and Free State of Saxony, Germany (LIFE—Leipzig Research Centre for Civilization Diseases, project numbers 713-241202, 14505/2470, 14575/2470). The GENICA was funded by the Federal Ministry of Education and Research (BMBF) Germany Grants 01KW9975/5, 01KW9976/8, 01KW9977/0 and 01KW0114, the Robert Bosch Foundation, Stuttgart, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, the Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, as well as the Department of Internal Medicine, Evangelische Kliniken Bonn gGmbH, Johanniter Krankenhaus, Bonn, Germany. The GESBC was supported by the Deutsche Krebshilfe e. V. [70492] and the German Cancer Research Center (DKFZ). The HABCS study was supported by the Claudia von Schilling Foundation for Breast Cancer Research. The HEBCS was financially supported by the Helsinki University Hospital Research Fund, the Finnish Cancer Society, and the Sigrid Juselius Foundation. The HUBCS was supported by the program for supporting the bioresource collections №007-030164/2, and the study was performed as part of the assignment of the Ministry of Science and Higher Education of Russian Federation (№2020-220-08-2197). Financial support for KARBAC was provided through the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and Karolinska Institutet, the Swedish Cancer Society, The Gustav V Jubilee foundation and Bert von Kantzows foundation. The KARMA study was supported by Märit and Hans Rausings Initiative Against Breast Cancer. The KBCP was financially supported by the special Government Funding (EVO) of Kuopio University Hospital Grants, Cancer Fund of North Savo, the Finnish Cancer Organizations, and by the strategic funding of the University of Eastern Finland. LMBC is supported by the 'Stichting tegen Kanker'. The MARIE study was supported by the Deutsche Krebshilfe e.V. [70-2892-BR I, 106332, 108253, 108419, 110826, 110828], the Hamburg Cancer Society, the German Cancer Research Center (DKFZ) and the Federal Ministry of Education and Research (BMBF) Germany [01KH0402]. MBCSG is supported by grants from the Italian Association for Cancer Research (AIRC). The MCBCS was supported by the NIH Grants R36CA253187, CA192393, CA116167, CA176785 an NIH Specialized Program of Research Excellence (SPORE) in Breast Cancer [CA116201], and the Breast Cancer Research Foundation. MCCS cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further augmented by Australian National Health and Medical Research Council Grants 209057, 396414 and 1074383 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry and the Australian Institute of Health and Welfare, including the National Death Index and the Australian Cancer Database. The MEC was supported by NIH Grants CA63464, CA54281, CA098758, CA132839 and CA164973. The MISS study is supported by funding from ERC-2011-294576 Advanced grant, Swedish Cancer Society, Swedish Research Council, Local hospital funds, Berta Kamprad Foundation, Gunnar Nilsson. The MMHS study was supported by NIH Grants CA97396, CA128931, CA116201, CA140286 and CA177150. The NBCS has received funding from the K.G. Jebsen Centre for Breast Cancer Research; the Research Council of Norway Grant 193387/V50 (to A-L Børresen-Dale and V.N. Kristensen) and Grant 193387/H10 (to A-L Børresen-Dale and V.N. Kristensen), South Eastern Norway Health Authority (Grant 39346 to A-L Børresen-Dale) and the Norwegian Cancer Society (to A-L Børresen-Dale and V.N. Kristensen). The NC-BCFR was supported by Grant UM1 CA164920 from the National Cancer Institute (USA). The NCBCS was funded by Komen Foundation, the National Cancer Institute (P50 CA058223, U54 CA156733, U01 CA179715), and the North Carolina University Cancer Research Fund. The NHS was supported by NIH Grants P01 CA87969, UM1 CA186107, and U19 CA148065. The NHS2 was supported by NIH Grants UM1 CA176726 and U19 CA148065. The OBCS was supported by research grants from the Finnish Cancer Foundation, the Academy of Finland (Grant number 250083, 122715 and Center of Excellence Grant number 251314), the Finnish Cancer Foundation, the Sigrid Juselius Foundation, the University of Oulu, the University of Oulu Support Foundation and the special Governmental EVO funds for Oulu University Hospital-based research activities. The ORIGO study was supported by the Dutch Cancer Society (RUL 1997-1505) and the Biobanking and Biomolecular Resources Research Infrastructure (BBMRI-NL CP16). The PBCS was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. Genotyping for PLCO was supported by the Intramural Research Program of the National Institutes of Health, NCI, Division of Cancer Epidemiology and Genetics. The PLCO is supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics and supported by contracts from the Division of Cancer Prevention, National Cancer Institute, National Institutes of Health. The RBCS was funded by the Dutch Cancer Society (DDHK 2004-3124, DDHK 2009-4318). The SASBAC study was supported by funding from the Agency for Science, Technology and Research of Singapore (A*STAR), the US National Institute of Health (NIH) and the Susan G. Komen Breast Cancer Foundation. SEARCH is funded by Cancer Research UK [C490/A10124, C490/A16561] and supported by the UK National Institute for Health Research Biomedical Research Centre at the University of Cambridge. The University of Cambridge has received salary support for PDPP from the NHS in the East of England through the Clinical Academic Reserve. SKKDKFZS is supported by the DKFZ. The SMC is funded by the Swedish Cancer Foundation and the Swedish Research Council (SIMPLER, VR 2017-00644). STORM was supported by the French Ministry of Health (PHRC K 2011). The SZBCS was supported by Grant PBZ_KBN_122/P05/2004 and the program of the Minister of Science and Higher Education under the name “Regional Initiative of Excellence” in 2019–2022 project number 002/RID/2018/19 amount of financing 12,000,000 PLN. The UCIBCS component of this research was supported by the NIH [CA58860, CA92044] and the Lon V Smith Foundation [LVS39420]. The UKBGS is funded by Breast Cancer Now and the Institute of Cancer Research (ICR), London. ICR acknowledges NHS funding to the NIHR Biomedical Research Centre.
Author information
Authors and Affiliations
Consortia
Contributions
M.K.S. and M.E.G. conceived the study. M.E.G. performed the main data analyses and drafted the initial manuscript. M.K.S., S.C. and M.E.G. were involved in the interpretation of the data. R.K., Q.W., J.D. and M.K.B provided database support. J.B. performed the functional analysis. M.K.S. and S.C. worked on revisions of the manuscript. All authors contributed data from their own studies, helped revise the manuscript, and approved the final version. All authors consented to this publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Escala-Garcia, M., Canisius, S., Keeman, R. et al. Germline variants and breast cancer survival in patients with distant metastases at primary breast cancer diagnosis. Sci Rep 11, 19787 (2021). https://doi.org/10.1038/s41598-021-99409-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-99409-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
