
Abstract
Colloidal quantum-dots (QDs) are highly attractive materials for various optoelectronic applications owing to their easy maneuverability, high functionality, wide applicability, and low cost of mass-production. QDs usually consist of two components: the inorganic nano-crystalline particle and organic ligands that passivate the surface of the inorganic particle. The organic component is also critical for tuning electronic properties of QDs as well as solubilizing QDs in various solvents. However, despite extensive effort to understand the chemistry of ligands, it has been challenging to develop an efficient and reliable method for identifying and quantifying ligands on the QD surface. Herein, we developed a novel method of analyzing ligands in a mild yet accurate fashion. We found that oxidizing agents, as a heterogeneous catalyst in a different phase from QDs, can efficiently disrupt the interaction between the inorganic particle and organic ligands, and the subsequent simple phase fractionation step can isolate the ligand-containing phase from the oxidizer-containing phase and the insoluble precipitates. Our novel analysis procedure ensures to minimize the exposure of ligand molecules to oxidizing agents as well as to prepare homogeneous samples that can be readily analyzed by diverse analytical techniques, such as nuclear magnetic resonance spectroscopy and gas-chromatography mass-spectrometry.
Similar content being viewed by others
Introduction
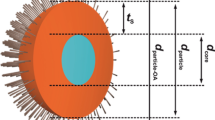
Colloidal quantum-dots (QDs) are nano-sized crystalline semiconductors. By modulating their size, composition, and surface-passivating molecules, various semiconducting features can be imparted to these materials, thus rendering them versatile for many applications such as light-emitting devices, display devices, solar cells, and sensors1,2,3,4,5. QDs are synthesized by modulating the inorganic precursors and the associated organic molecules, reaction temperature, and time; the resulting QD is composed of an inorganic nano-crystalline core surrounded by the organic ligand molecules (Fig. 1)6. The ligands of QDs play important roles of shaping the structure of the core, passivating and protecting the cores after their growth, and providing additional physical and chemical functionalities to QDs7. Many studies have been conducted on developing novel procedures of modulating the ligand composition of QDs to impart or maximize the desired physicochemical properties and functionalities7,8.

Schematic structure of an oleic-acid (OA) capped InP/ZnS QD with an inorganic InP/ZnS crystalline core (collection of spheres colored as designated) and OA molecules as passivating ligands (black sticks).
Ligand optimization is one of the most promising strategies to improve the performance and stability of QDs7,8. However, analytical tools available to detect and characterize various ligand molecules are still limited. The procedures of QD syntheses involve harsh conditions (e.g., high temperature, pressure, and highly reactive species) that often lead to the accumulation of by-products on the surface of QD particles9,10. In addition, although ligand-exchange processes are frequently used to impart novel chemical functionalities to QDs, it is still difficult to examine the final status of ligands due to the lack of appropriate analytical methodologies.
Various techniques and protocols11,12 have been developed for ligand analysis to date; among them, nuclear magnetic resonance (NMR) spectroscopy13,14 and gas-chromatography mass-spectrometry (GC–MS)15 have been notably powerful to elucidate the identity and composition of organic ligands on the QD surface. However, information from either technique is often limited owing to its intrinsic technical hurdles. Signal quality of solution-state NMR spectroscopy depends on isotropic tumbling rates of target molecules. Although ligand molecules are as such small and tumble fast enough to yield sharp signals, the tumbling of ligand molecules bound on QD is much slower, making the corresponding NMR signals broad and unsuitable even for simple 2-dimensional (2D) NMR experiments13. Similarly, because GC–MS detects organic molecules in a gas phase, ligand molecules first need to be easily transformed into the gaseous state for GC–MS analysis to be feasible. QD ligands are, however, often tightly bound to the nanoparticle surface, which prevents them from being gasified. This challenge is sometimes overcome by inducing ligand dissociation through chemical modification (e.g., ligand derivatization) prior to GC analysis; however, this procedure compromises the original chemical integrity of the ligands16.
In order to overcome this challenge, we devised a novel protocol of using oxidizing agents to detach organic molecules from the inorganic core or shell surface. Although several procedures in which ligands on QD are detached for their analysis were reported17,18,19, their applications were often limited to certain types of nanoparticles or ligand molecules. Based on previous studies that have demonstrated that oxidation disrupts the interaction between the ligands and nanocrystal20, we developed a protocol of oxidizing the nanocrystals and subsequently separating the ligand-containing fraction. Among various oxidizing agents, we found that hydrogen peroxide (H2O2) is an efficient heterogeneous catalyst to detach most ligands from inorganic parts, while being mild enough to minimize the disruption of their chemical integrity. Owing to phase difference, H2O2 could be easily removed from the ligand-containing organic phase by a simple fractionation step, further ensuring minimal exposure of ligands to oxidation. Previous studies have shown that H2O2 is capable of etching nanoparticles to enhance their functionalities without compromise in performance21,22. We applied our method to analyze the ligands of InP/ZnS QD samples. Despite their overall inferior performance and stability, InP-based QDs have attracted significant interest due to their suitability for environment-friendly application23,24.
Results and discussion
The ligand-analysis procedure (Fig. 2) was initiated with the addition of H2O2 to an oleic-acid capped InP/ZnS QD (InP/ZnS-OA) sample dispersed in organic solvents, such as CDCl3. Upon oxidation, discoloration of the sample and subsequent precipitation of an insoluble material were observed (Fig. 2c). After bleaching, D2O was added to the sample, and the sample was centrifuged to obtain three different fractions: a CDCl3 fraction, a D2O fraction, and an insoluble layer (Fig. 2d and Fig. S1). By X-ray photoelectron spectroscopy (XPS), we confirmed that the insoluble layer mostly contained oxidized inorganic material from the core and shell (such as ZnSO4 and InPO4; Fig. S2). We also found that the D2O addition and fraction separation steps are critical for high-resolution ligand analysis. D2O plays an important role to retrieve excess H2O2 from the CDCl3 fraction and contain it to the D2O fraction in a diluted state, thus minimizing the exposure of ligands to H2O2 as organic molecules were partitioned into the CDCl3 fraction. Moreover, D2O addition increased the effective volume of H2O2-containing fraction, making its removal easy with a simple pipetting action. Fraction separation procedure is important to remove most H2O2 and H2O molecules from the CDCl3 fraction, which prevents signal overlapping and broadening for high-resolution 1D/2D NMR analyses. It is also advantageous that water-soluble materials, if any, were separated from CDCl3-soluble ligands, simplifying the subsequent ligand characterization (Fig. 3).

The ligand analysis procedure involving the use of H2O2 as a heterogeneous oxidizing agent. First, the QDs dispersed in CDCl3 (a) are treated with H2O2 (b) and vortexed vigorously. Green color originating from the QDs is retained until step (b). Upon reaction under ambient condition, the sample loses its green color, leading to an insoluble layer (precipitate). In order to separate the fraction containing organic ligands from that containing H2O2 and H2O, D2O is added to the sample, and the sample is again vortexed vigorously (c). Subsequently, the sample is separated into three fractions via centrifugation: the D2O fraction, insoluble fraction, and CDCl3 fraction (d). The final soluble fractions are transparent and colorless.

1H NMR spectra of InP/ZnS-OA: (a) as-prepared sample in CDCl3, (b) after treatment with 50 μL of H2O2, (c) CDCl3 fraction, and (d) D2O fraction. The CDCl3 and D2O fractions were obtained after the centrifugation of H2O2-treated samples. The 1H signals at 5.35 ppm (vinyl protons of OA) for the as-prepared sample in CDCl3 (e) and for the CDCl3 fraction sample separated after H2O2 treatment (f) are shown to stress that H2O2 treatment and phase fractionation effectively reduced the signal width of ligand molecules. Asterisks (*) indicate the signals of residual ethanol from purification procedure of QD.
The final CDCl3 fraction provided sharp 1H NMR signals (Fig. 3e vs f.) that were further analyzed by multi-dimensional NMR techniques (see below). Notably, we were still able to retrieve active and stable QDs by conducting fraction separation shortly after H2O2 addition, indicating that H2O2 had indeed been removed from the QD-containing CDCl3-fraction; the same H2O2-treated QD sample was found to completely bleach out when incubated without fraction separation. In addition, we observed that the 1H NMR signal originating from H2O was significantly weak for the CDCl3 fraction, while the NMR signal of H2O was clearly observed with the H2O2-treated QD samples not subjected to fraction separation (Fig. 3b vs c). Consistently, only the D2O fraction exhibited the 1H NMR signal for H2O2 (Fig. 3d)25. We also confirmed that the intensities of 1H OA signals from the CDCl3-extracted fraction correlated well with the added amount of QDs (Fig. S3). With the calibration curves of OA (Fig. S3b), it is possible to quantify ligand molecules and determine the surface coverage or ligand composition at a higher accuracy (the procedure for surface coverage analysis and the detailed calculation results on the QD sample are described in supplementary method).
Next, we examined whether the ligand signals of the CDCl3 fraction were indeed from free ligands (not from QD-bound ligands). 2D DOSY is a useful tool to measure the hydrodynamic radius of molecules in the solution and distinguish the molecular weight or size of molecules in different mixtures26,27. By employing this technique, we confirmed that the bound OA ligands (log D, − 9.64) on the QD surface in the as-prepared sample diffuse slower than the free ligands (log D, − 9.05) in the CDCl3 fraction of the H2O2-treated QD sample (Fig. 4). This result indicates that H2O2 treatment liberated the ligands from the high molecular-weight inorganic particle, which is also consistent with significant NMR signal sharpening and discoloration of the H2O2-treated sample. Subsequently, we compared the spectral quality of the series of CDCl3-fraction samples obtained after treatment with different amounts of H2O2. We added 0, 5, 15, 50, and 100 μL of H2O2 to a series of QD samples (same amount), and collected the 1H NMR spectra of their CDCl3-fraction samples (Fig. 5). This spectral comparison revealed that the relative portion of free ligands increased with the amount of H2O2 up to 50 μL H2O2, while there was no significant difference in the spectra with further increase in its quantity (Fig. 5d vs. e), implying minimal artifact from excess treatment of H2O2, if any. In addition, we tested whether the incubation reaction time with H2O2 (i.e., the time interval between H2O2 addition and fraction separation) affects the ligand dissociation and NMR spectral quality. As expected, ligand signals sharpened with the reaction time (Fig. S4), which is again consistent with time-dependent QD oxidation and subsequent ligand dissociation. Notably, we also tested whether addition of D2O may affect H2O2-mediated oxidation of QD (Fig. S5). From this trial, we confirmed that H2O2 cannot efficiently oxidize QD in a D2O-diluted condition, validating that the addition of D2O contributes to regulation of H2O2-mediated oxidation as well as prevention for over-oxidation of ligand molecules.

2D-DOSY NMR spectra of InP/ZnS-OA: (a) before and (b) after treatment with 50 μL of H2O2 and CDCl3 fraction separation. The vertical axis shows the diffusion coefficient (log D). The measured diffusion coefficients for OA and solvent signals are denoted in the figure.

1H NMR spectra of InP/ZnS-OA with adding different amounts of H2O2, (a) no H2O2, (b) 5 μL, (c) 15 μL, (d) 50 μL, and (e) 100 μL, and then extracted into the CDCl3 fraction.
This novel protocol of ligand sample preparation is also good for GC–MS analysis. The routine GC–MS ligand analysis procedure often involves the compromise of the chemical integrity of ligand molecules. In contrast, the sample prepared with the present protocol only contains free organic ligands with minimal modification, if any, enabling straightforward and reliable GC–MS analysis (Fig. S6). Notably, GC–MS could be used to detect palmitic acid (PA), stearic acid (SA), and trioctylamine (TOA) in addition to OA. Due to the chemical similarity between these molecules, 1D 1H NMR could not be used to resolve PA, SA, or TOA signals from the OA signals. By employing this GC–MS-based approach, we found that two distinctive compositions of the ligands (particularly, PA and OA) in two different batches (QD 1 and QD 2) of InP/ZnS-OA could be identified (Fig. S6). In addition, we were able to determine relative response factors28 for OA, PA, and TOA (Fig. S7), which could be subsequently applied to analyze the GC–MS chromatograms to have accurate quantification analysis of QD ligands (Fig. S6e).
Finally, we decided to evaluate whether the new approach can be applied to characterize ligands de novo. Due to the lack of an appropriate analytical methodology, identification and characterization of ligands without prior information (e.g., precursors used for QD synthesis) has been challenging. As we were able to consistently observe a set of distinctive 1H signals near the signals of OA with our QD samples (e.g., the overlapped signal on the shoulder of the OA signal at ~ 5.3 ppm; Fig. S8), we decided to identify the ligand molecule presenting these signals. While the signals from ligands, in their intact QD-bound state, are broad and not amenable for further analysis, the novel protocol enabled us to obtain sharper and higher-resolution signals from the CDCl3 fraction of the H2O2-treated QD samples (Fig. 6). We collected a series of 2D NMR spectra, such as 1H–1H TOCSY and 1H–13C HSQC (Fig. S9), with the CDCl3 fraction, and found that this spectral feature arises from the trans-isomer of OA, i.e., elaidic acid (EA; Fig. 6).

1H NMR spectra of the CDCl3 fractions separated from the H2O2-treated samples of (a) elaidic acid (EA), (b) oleic acid (OA), (c) CdS-OA QD, (d) InP/ZnS-OA QD batch #1, and (e) InP/ZnS-OA QD batch #2. The extra peaks (marked as ‘EA’) near the signals of OA correspond to the trans-isomer of OA, i.e., EA [refer Fig. S8 for 1H NMR spectra of the as-prepared (without H2O2 treatment) samples].
We confirmed the presence of EA by comparing the data with the NMR data of a sample containing only EA (Fig. 6a) and by obtaining the ion chromatograms and mass spectra of extracted EA and OA using a highly polar column (Fig. S10)29. In order to determine how EA was formed, we tested whether high temperature, as in the QD synthesis procedures, facilitated OA isomerization. Indeed, we observed that the nucleation/growth of QDs at 120 °C induced time-dependent formation of EA, suggesting that EA on QDs might have been originated from OA (data not shown)30.
Conclusion
We developed a novel methodology of H2O2-induced oxidation by which we induced the efficient liberation of ligands, which upon subsequent phase fractionation becomes a sample suitable for various analytical techniques. We confirmed that H2O2 as a heterogeneous catalyst did not compromise the chemical integrity of ligand molecules, thus demonstrating the accuracy and versatile applicability of the present method. We also found that the D2O addition and fraction separation step was effective to remove H2O2 from the ligand-containing apolar fraction, thus preventing any possible deformation of organic ligands, as well as to fractionate polar and apolar ligand molecules into separate fractions, which is advantageous to reduce the complexity in the ligand mixtures. Overall, our ligand sample preparation procedure effectively enhances the overall resolution and sensitivity at the subsequent ligand analysis. We would like to stress that this strategy is wildly compatible with various ligand molecules having different chemical properties. In our follow-up ligand analysis studies that will be reported elsewhere, we also found that amine and sulfhydryl ligands can be analyzed by the present method; these ligands were detected as a form of amide and disulfide, sulfoxide, or sulfonate, respectively.
In this work, we particularly focused on applying NMR spectroscopy and GC–MS for sample analysis to prove that, upon being combined with the novel sample preparation procedures, these two techniques are capable of exerting their full analytical strengths for the identification and characterization of diverse organic molecules. Notably, we were able to determine the structural isomerization of OA molecules possibly during the QD synthesis, exemplifying the good sensitivity and resolvability of the developed methodology for organic ligands. Notably, the samples prepared with the presented procedure can be analyzed not only with NMR and GC–MS but also with other analytical techniques such as LC–MS, ICP, and IR. Compared to previous attempts to analyze ligands in the QD-bound state, our new analytical procedure may greatly advance our understanding of the ligand composition of QD samples, ligand degradation during the QD synthesis and subsequent manipulation, and the correlation between the nature of ligands and the QD performance.
Methods
Synthesis of InP/ZnS nanocrystals. Indium acetate and oleic acid were dissolved in 1-octadecene in a 200 mL reaction flask under vacuum at 120 °C for 1 h. The molar ratio of indium to oleic acid (OA) was set to 1:3. Then, N2 gas was flown into the flask and the reaction mixture was heated to 280 °C. Subsequently, a mixed solution of tris(trimethylsilyl) phosphine (TMS3P) and trioctylphosphine was injected quickly, and the reaction was allowed to proceed for 20 min. The reaction mixture was then cooled rapidly to room temperature and acetone was added to precipitate the nanocrystals, which were then separated by centrifugation and dispersed in toluene to obtain a suspension of the InP core nanocrystals. The amount of TMS3P used was ~ 0.75 mol per mole of indium. For coating a ZnS shell, in a 200 mL reaction flask, zinc acetate and OA were dissolved in trioctylamine and the solution was heated under vacuum at 120 °C for 10 min. Then, N2 was flown into the reaction flask. While the resulting solution was heated to 280 °C, a toluene dispersion of the InP cores was injected into it followed by the injection of 1-dodecanethiol. The reaction was carried out for 60 min. The total amount of S used per mole of indium was set to 6 mol. An excess amount of ethanol was added to the final reaction mixture containing the resulting InP/ZnS nanocrystals, which was then centrifuged, the supernatant was discarded, and the precipitate was dried to obtain the QD sample in a powder form. The UV–Vis absorption and photoluminescence spectra and the transmission electron microscopy image of the as-prepared InP/ZnS-OA QD samples are shown in Fig. S11.
Oxidative treatment of QDs for ligand analysis. Ligand analysis procedure was initiated with the dispersion of 10 mg of an oleic-acid capped InP/ZnS QD (InP/ZnS-OA) sample in 500 μL of CDCl3. This sample was treated with a desired amount of H2O2, vigorously vortexed, and incubated at the ambient condition. During reaction with H2O2 incubation, the sample was repeatedly vortexed in order to mix the reaction mixture thoroughly. Once the QD sample was incubated for a desired period, 500 μL of D2O was added to the sample. The sample was again vortexed vigorously and subsequently centrifuged at 13,000 rpm for 10 min on a table-top centrifuge. After centrifugation, the contents of the tube separated into three fractions: an upper D2O layer, a middle insoluble precipitate, and a lower CDCl3 layer. Each fraction was separated with a micropipette, and used for subsequent analyses.
XPS analysis. The XPS analysis was carried out using Quantum 2000 Scanning ESCA microprobe (manufactured by ULVAC-PHI) with monochromatic Al Kα source (hυ, 1486.60 eV).
GC–MS analysis. GC–MS was performed on Thermo Scientific™ ISQ LT GC–MS and Agilent 5977A series MSD. Two columns were used to analyze the ligands: DB5-MSUI (30 m × 250 μm × 0.25 μm) was employed to determine the composition of the ligand mixture, while a highly polar column such as Select FAME (50 m × 250 μm) was employed to separated cis and trans isomers29. Quantitative analyses were carried out by determining the relative response factors of ligand molecules and applying the internal standard method28.
NMR spectroscopy. The NMR experiments were performed on Bruker NMR spectrometers at 11.7 T and 14.1 T B0 field strengths corresponding to 1H Larmor frequencies of 500.13 MHz (AVANCE HD III) and 600.1 MHz (AVANCE III), respectively. Bruker 5 mm double resonance broadband probes were used for all the experiments. NMR measurements were performed with 500 μL samples in top-sealed tubes (Nihon Seimitsu Kagaku Co. Ltd) so to minimize the effects of sample evaporation and air-exposure. 2D diffusion-ordered spectroscopy (DOSY) experiments were performed using a stimulated-echo sequence incorporating bipolar gradient pulses and a longitudinal eddy current delay (BPP-LED)26,27. The self-diffusion coefficient for residual HDO in D2O was found to be 1.9 × 10–9 m2 s−1 at 298 K (log D = − 8.6), which is consistent with the reported data for the reference27.
References
Talapin, D. V., Lee, J. S., Kovalenko, M. V. & Shevchenko, E. V. Prospects of colloidal nanocrystals for electronic and optoelectronic applications. Chem. Rev. 110, 389–458 (2010).
Kim, J. Y., Voznyy, O., Zhitomirsky, D. & Sargent, E. H. 25th anniversary article: Colloidal quantum dot materials and devices: A quarter-century of advances. Adv. Mater. 25, 4986–5010 (2013).
Shirasaki, Y., Supran, G. J., Bawendi, M. G. & Bulović, V. Emergence of colloidal quantum-dot light-emitting technologies. Nat. Photon. 7, 13–23 (2013).
Nozik, A. J. et al. Semiconductor quantum dots and quantum dot arrays and applications of multiple exciton generation to third-generation photovoltaic solar cells. Chem. Rev. 110, 6873–6890 (2010).
Michalet, X. et al. Quantum dots for live cells, in vivo imaging, and diagnostics. Science 307, 538–544 (2005).
Yin, Y. & Alivisatos, A. P. Colloidal nanocrystal synthesis and the organic-inorganic interface. Nature 437, 664–670 (2005).
Boles, M. A., Ling, D., Hyeon, T. & Talapin, D. V. The surface science of nanocrystals. Nat. Mater. 15, 141–153 (2016).
Heuer-Jungemann, A. et al. The role of ligands in the chemical synthesis and applications of inorganic nanoparticles. Chem. Rev. 119, 4819–4880 (2019).
Kopping, J. T. & Patten, T. E. Identification of acidic phosphorus-containing ligands involved in the surface chemistry of CdSe nanoparticles prepared in tri-N-octylphosphine oxide solvents. J. Am. Chem. Soc. 130, 5689–5698 (2008).
Morris-Cohen, A. J., Donakowski, M. D., Knowles, K. E. & Weiss, E. A. The effect of a common purification procedure on the chemical composition of the surfaces of cdse quantum dots synthesized with trioctylphosphine oxide. J. Phys. Chem. C 114, 897–906 (2010).
Morris-Cohen, A. J., Malicki, M., Peterson, M. D., Slavin, J. W. J. & Weiss, E. A. Chemical, structural, and quantitative analysis of the ligand shells of colloidal quantum dots. Chem. Mater. 25, 1155–1165 (2013).
Colangelo, E. et al. Characterizing self-assembled monolayers on gold nanoparticles. Bioconjugate Chem. 28, 11–22 (2017).
Hens, Z. & Martins, J. C. A solution NMR toolbox for characterizing the surface chemistry of colloidal nanocrystals. Chem. Mater. 25, 1211–1221 (2013).
Marbella, L. E. & Millstone, J. E. NMR techniques for noble metal nanoparticles. Chem. Mater. 27, 2721–2739 (2015).
Fisher, E. A., Duffy, S. J. & Meli, M. V. The determination of ligand shell composition of bifunctional alkanethiol-capped gold nanoparticles using GC/MS/MS. RSC Adv. 5, 33289–33293 (2015).
Zaikin, V. G. & Halket, J. M. Derivatization in mass spectrometry—8. Soft ionization mass spectrometry of small molecules. Eur. J. Mass Spectrom. 12, 79–115 (2006).
Anderson, N. C., Hendricks, M. P., Choi, J. J. & Owen, J. S. Ligand exchange and the stoichiometry of metal chalcogenide nanocrystals: Spectroscopic observation of facile metal-carboxylate displacement and binding. J. Am. Chem. Soc. 135, 18536–18548 (2013).
Smith, A. M. et al. Quantitative analysis of thiolated ligand exchange on gold nanoparticles monitored by 1H NMR spectroscopy. Anal. Chem. 87, 2771–2778 (2015).
Johnston, K. A., Smith, A. M., Marbella, L. E. & Millstone, J. E. Impact of as-synthesized ligands and low-oxygen conditions on silver nanoparticle surface functionalization. Langmuir 32, 3820–3826 (2016).
Moreels, I., Fritzinger, B., Martins, J. C. & Hens, Z. Surface chemistry of colloidal PbSe nanocrystals. J. Am. Chem. Soc. 130, 15081–15086 (2008).
Liu, L., Peng, Q. & Li, Y. An effective oxidation route to blue emission CdSe quantum dots. Inorg. Chem. 47, 3182–3187 (2008).
Lee, W. et al. An effective oxidation approach for luminescence enhancement in CdS quantum dots by H2O2. Nanoscale Res. Lett. 7 (2012).
Zhang, H. et al. High-efficiency green InP quantum dot-based electroluminescent device comprising thick-shell quantum dots. Adv. Opt. Mater. 7, 1801602 (2019).
Tarantini, A. et al. Physicochemical alterations and toxicity of InP alloyed quantum dots aged in environmental conditions: A safer by design evaluation. NanoImpact 14, 100168 (2019).
Stephenson, N. A. & Bell, A. T. Quantitative analysis of hydrogen peroxide by 1H NMR spectroscopy. Anal. Bioanal. Chem. 381, 1289–1293 (2005).
Johnson, C. S. Diffusion ordered nuclear magnetic resonance spectroscopy: Principles and applications. Prog. Nucl. Magn. Reson. Spectrosc. 34, 203–256 (1999).
Antalek, B. Using pulsed gradient spin echo NMR for chemical mixture analysis: How to obtain optimum results. Concepts Magn. Reson. Part A Bridg. Educ. Res. 14, 225–258 (2002).
Rome, K. & Mcintyre, A. Intelligent use of relative response factors in gas chromatography-flame ionisation detection. Chromatogr. Today 52–56 (2012).
Ledoux, M., Laloux, L. & Wolff, R. L. Analytical methods for determination of trans -C18 fatty acid isomers in milk fat. A review. Analusis 28, 402–412 (2000).
Grisorio, R. et al. The dynamic surface chemistry of colloidal metal chalcogenide quantum dots. Nanoscale Adv. 1, 3639–3646 (2019).
Acknowledgements
The authors thank Hyeokjoo Gwon, Youngsik Shin, Dongmin Kim, and Jaeduck Jang (Samsung Advanced Institute of Technology) for their generous help and insightful discussion. This work was supported by the DGIST Start-up Fund Program of the Ministry of Science and ICT (Grant Number 2020010061).
Author information
Authors and Affiliations
Contributions
J.H.K. and I.-S.J. conceived and conducted majority of NMR experiments. H.P. conducted all GC–MS experiments. T.-G.K. and S.J. prepared quantum dot samples and characterized their basic properties. H.L. assisted sample preparations and NMR experiments. E.L. and W.S.J. assisted data analysis and interpretation. J.C. conducted XPS analysis. The manuscript was written by contributions of all the authors listed. The final version of the manuscript was approved by all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, J.H., Park, H., Kim, TG. et al. Facile and versatile ligand analysis method of colloidal quantum dot. Sci Rep 11, 19889 (2021). https://doi.org/10.1038/s41598-021-99358-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-99358-x
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
