
Abstract
We aimed to elucidate the differences in genomic methylation patterns between ADLI and non-ADLI patients to identify DNA methylation-based biomarkers. Genome-wide DNA methylation patterns were obtained using Infinium MethylationEPIC (EPIC) BeadChip array to analyze 14 peripheral blood samples (7 ADLI cases, 7 non-ADLI controls). Changes in the mRNA and DNA methylation in the target genes of another 120 peripheral blood samples (60 ADLI cases, 60 non-ADLI controls) were analyzed by real-time polymerase chain reaction and pyrosequencing, respectively. A total of 308 hypermethylated CpG sites and 498 hypomethylated CpG sites were identified. Significantly, hypermethylated CpG sites cg06961147 and cg24666046 in TANC1 associated with ADLI was identified by genome-wide DNA methylation profiling. The mRNA expression of TANC1 was lower in the cases compared to the controls. Pyrosequencing validated these two differentially methylated loci, which was consistent with the results from the EPIC BeadChip array. Receiver operating characteristic analysis indicated that the area under the curve of TANC1 (cg06961147, cg24666046, and their combinations) was 0.812, 0.842, and 0.857, respectively. These results indicate that patients with ADLI have different genomic methylation patterns than patients without ADLI. The hypermethylated differentially methylated site cg06961147 combined with cg24666046 in TANC1 provides evidence for the diagnosis of ADLI.
Similar content being viewed by others
Introduction
Tuberculosis (TB) remains one of the top 10 causes of death worldwide1. Directly observed treatment short-course chemotherapy (DOTS), which emphasizes the use of a combination of isoniazid (H), rifampicin (R), pyrazinamide (Z), and ethambutol (E) for 6 to 8 months, was the most effective strategy2. The combination of these medications was most likely to result in an increase in the incidence of ADLI3. The incidence of ADLI currently ranges from 2 to 28%, depending on the definition of liver toxicity and the population being studied4,5. ADLI has significant side effects, such as interruption of treatment, prolonged disease progression, and development of drug resistance, and may even lead to death6. There are currently no sensitive and specific biomarkers for the diagnosis of ADLI, and the diagnosis of ADLI still depends on serum biochemical tests. This is because the identification novel potential non-invasive biomarker for the early diagnosis of ADLI in China, as well as across the world, remains a challenge.
DNA methylation is one of the most stable epigenetic modifications in mammalian cells. It controls a variety of cellular and developmental processes, including embryonic development, X-inactivation, chromosome stability, and imprinting7. Over the last decade, aberrant DNA methylation has been shown to be a candidate biomarker of cancer and occurs very early in cancer development8. Moreover, it has become clear that DNA methylation is reversible and dynamic as a result of enzymatic DNA de-methylation9; therefore, aberrant DNA methylation modifications have attracted increased interest as potential drug targets10.
Numerous studies have demonstrated that the hypermethylation of CpG islands in the promoter regions of CYP2E1, CYP2D6, and GSTP1 is associated with the occurrence of ADLI11,12. However, these studies have only focused on single or multiple genes, and the research conclusions drawn remain limited. At present, there have been studies using Agilent Human DNA Methylation Microarray 1 × 244 K array for genome-wide DNA methylation. However, the Agilent Human DNA Methylation Microarray 1 × 244 K array focuses only on CGI and differentially methylated regions. It only covers 27,627 human CpG islands and 5081 UMR regions, and cannot reach the resolution of a single base. The EPIC BeadChip not only contains over 850,000 probes and covers the entire CpG islands, promoter, coding region, open chromatin, and enhancer, but also includes CpG sites outside the CpG islands, known differentially methylated region sites, and microRNA promoter regions. Moreover, it has the advantage of single base resolution, which can directly detect the exact site of methylation. It is currently the most suitable DNA methylation research technology for apparent genome-wide association analysis (EWAS) research. At present, there is no comprehensive and systematic genome-wide DNA methylation analysis using EPIC BeadChip array in peripheral blood of ADLI patients As such, in the present study, we chose to use EPIC BeadChip to characterize genome-wide DNA methylation profiles in the peripheral blood of ADLI and non-ADLI patients. The differentially methylated CpG sites (dmCpGs) were identified by differential methylation analysis, and pyrophosphate sequencing was used to verify the selected gene sites. In summary, this study aimed to identify epigenetic changes in peripheral blood samples in order to identify potential biomarkers of ADLI.
Materials and methods
Study population and ethics
Patients with newly diagnosed TB over 18 years old hospitalized in the Tangshan Fourth Hospital between March 2016 and July 2017 were recruited. TB was diagnosed based on previously described guidelines13. All patients received standardized daily treatment with isoniazid (H), rifampicin (R), pyrazinamide (Z), and ethambutol (E) in the first two months, and H and R on a daily basis in the following four months14. The follow-up period for patients was from the beginning of treatment until 6 months later. During this period, patient compliance, the choice of treatment options, tuberculosis-related symptoms, and adverse drug reactions were strictly monitored by trained staff. Liver enzymes and bilirubin, as biomarkers of liver function, were monitored. To this end, 10 mL Peripheral blood samples were collected every two weeks after starting antituberculosis treatment for the initial 2 months and every 4 weeks for the next 4 months, or any time when symptoms and signs of hepatitis developed during treatment15. We collected peripheral blood samples from patients at 8–9 am and asked patients to fast for 8–12 h before collecting blood samples to ensure the accuracy of the test results. Detailed demographic and clinical characteristics of the patients were obtained from electronic medical records.
We selected 67 patients, who developed ADLI within 2 to 8 weeks after receiving anti-TB treatment as the liver injury group, and then identified 67 patients without ADLI based on similar characteristics of age, sex, and admission time during the same period15. A total of 134 TB patients met the inclusion and exclusion criteria and were included in the study. We first performed EPIC BeadChip array of 7 ADLI patients and 7 age- and sex-matched non-ADLI patients. Subsequently, an independent cohort of 60 patients with ADLI and 60 patients with non-ADLI was used for pyrosequencing. This study was reviewed and approved by the Ethics Committee of North China University of Science and Technology (process no. 14-016). Informed consent was obtained from patients prior to any experimental procedures.
Inclusion/exclusion criteria
The inclusion criteria were as follows: (1) over 18 years old; (2) newly diagnosed with TB; (3) normal liver function before anti-tuberculosis treatment. The exclusion criteria were as follows: (1) patients with acute hepatitis, cirrhosis of the liver, encephalopathy, or cancer; (2) patients taking concomitant hepatotoxic medications; (3) heavy alcohol intake; (4) patients with severe cardiovascular, cerebrovascular, renal, or thyroid disease.
Diagnosis of anti-tuberculosis drug-induced liver injury
Hepatotoxicity due to anti-TB drug treatment was not only based on the liver enzyme results, according to the criteria of the American Thoracic Society (ATS)16, but also took into account the diagnostic criteria developed by the Centre for Drug Re-evaluation (CDR) of the Chinese State Food and Drug Administration, as well as various previously definitions2,17. Specifically, ADLI cases need to meet one of the following criteria: (1) an increase in serum alanine aminotransferase (ALT) or aspartate aminotransferase (AST) that is over threefold2 the upper limit of normal (ULN) in the presence of liver injury symptoms; (2) an increase in total bilirubin (TBIL) that is over twofold the ULN in the presence of liver injury symptoms; 3) a fivefold increase in the ULN of serum ALT, AST, or TBIL with or without liver injury symptoms.
Illumina Infinium MethylationEPIC BeadChip array
Microarray-based DNA methylation profiling was performed using the Illumina Infinium MethylationEPIC (EPIC) BeadChip (Illumina, Inc., San Diego, CA, USA) on 7 paired blood samples. Genomic DNA from peripheral blood samples was extracted using a DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). Bisulfite conversion of isolated genomic DNA (500 μg) was performed using the EZ DNA methylation Gold Kit (Zymo Research, Irvine, USA). Bisulfite-converted DNA was then whole-genome amplified, enzymatically fragmented, and hybridized to the array as per the EPIC BeadChip protocol18. Subsequent scanning of chips was performed using an Illumina HiScan2000. The raw intensity of the data was determined using GenomeStudio methylation module version 1.9.0 (Illumina, Inc.).
Methylation EPIC BeadChip data pre-processing
The raw intensity data (IDAT) were imported into R version 3.4.2 and processed using the R/Bioconductor package minfi (version 1.22.1)19. Low-quality data (probes with detection P-value > 0.05), probes from the X and Y chromosome, and probes overlapped with single-nucleotide polymorphisms were removed20. The background data were normalized using the Noob method21 to generate methylation beta (β) values, which were used for subsequent analysis.
Differential DNA methylation analyses
The β-values were used as an indicator of the methylation of each locus in each sample. Delta beta (Δβ) is defined as the difference in the β values between the two groups, in which the absolute value is directly proportional to the degree of difference. We calculated the mean detection P-value to check the overall data quality. In the present study, dmCpGs between the groups were identified with P < 0.05 and |Δβ|> 0.10. Subsequently, dmCpGs located in the promoter regions (promoters were defined as regions located between 1500 bp upstream of TSS and 500 bp downstream of transcriptional start sites (TSS)) and genes containing multiple differentially methylated probes were selected as our candidate CpG sites.
Real-time quantitative polymerase chain reaction
RNA was extracted using TRIzol reagent (Invitrogen, Grand Island, NY, USA). cDNA was generated using the PrimerScript RT Kit with the gDNA Eraser (DRR047A, Takara, Dalian, China). Subsequently, the cDNA was amplified using SYBR Primix EX Taq II (RR820A; Takara, Dalian, China). The data were normalized to the reference gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The primers are shown in Supporting Information Table S2. The 2−∆∆Ct22 method was used to calculate the relative level of target gene expression.
DNA pyrosequencing
DNA samples were bisulfite converted using the Zymo EZ DNA Methylation Kit (Zymo Research) as mentioned previously. PCR reaction (25 μL) was performed using TaKaRa EpiTaq HS (for bisulfite-treated DNA) (R110A; Takara, Dalian, China), according to the manufacturer’s instructions. The primer sequences and reaction conditions for all pyrosequencing assays are listed in Supporting Information Table S3. Biotin-labeled PCR products were captured using Streptavidin Sepharose beads (GE Healthcare Life Sciences, Pittsburgh, PA, USA), and single-strand DNA (ssDNA) was made with a Pyrosequencing Vacuum Prep Tool (Qiagen, Hilden, Germany). Sequencing primers were subsequently annealed to the ssDNA template and pyrosequenced using the PyroMark Q96 system (Qiagen, Hilden, Germany). All pyrosequencing data were analyzed using CpG run-analysis on PyroMark Q96 2.5.8 software.
Ingenuity pathway analysis
Ingenuity pathway analysis (IPA) software (QIAGEN Inc. software version 65367011, https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis) was used to analyze significant differentially methylated genes to elucidate the canonical pathways, upstream regulators, disease and biological functions, and networks. The algorithms developed for use in IPA have been described by Kramer et al.23.
Statistical analysis15
SPSS software version 22.0 (IBM Corp, Armonk, NY, USA) was used for data analysis. Continuous variables were described as the mean ± standard deviation (SD) or as the median with interquartile range (IQR). Differences between groups were analyzed using the paired-samples t-test or Wilcoxon signed-rank test. Categorized variables are expressed as numbers and percentages and were analyzed using the chi-squared (χ2) test or Fisher’s exact test. Receiver operating characteristic (ROC) curve analysis was used to determine the AUC, sensitivity, and specificity of peripheral blood DNA methylation. Logistic regression with forward stepwise analysis was used to further establish the diagnostic panel. A two-tailed P-value < 0.05 was considered statistically significant.
Ethics approval and consent to participate
Approval was obtained from the ethics committee of the North China University of Science and Technology. The procedures used in this study adhere to the tenets of the Declaration of Helsinki. Informed consent was obtained from all individual participants included in the study.
Results
Characteristics of the study population
The characteristics of the two study populations used in EPIC BeadChip for discovery, pyrosequencing, and validation are presented in Table 1. All participants were native Han Chinese. In the discovery and validation groups, there were no significant differences between the cases and controls in terms of smoking status, drinking status, ALP levels, TBIL levels, albumin (ALB) levels, or TP levels. However, statistically significant differences in the levels of ALT and AST were found when the cases were compared to controls.
Differential DNA methylation patterns between ADLI cases and controls
To clarify the difference in the levels of methylation between the ADLI patients and the control group, we analyzed the methylation status of 866,091 CpG sites in 7 paired blood samples from 14 subjects using the EPIC BeadChip array. The high quality of the samples is reflected in the pattern and highly comparable distribution shown by the density plot of the β-value of the probe (Fig. 1a). Principal component analysis (PCA) of the full methylomes clearly differentiated between ADLI patients and the controls, as shown in Fig. 1b. Subsequent to data pre-processing and quality filtering, a final data matrix comprised β-values (methylation levels) across 841,456 loci in 14 blood samples was created for further statistical analyses.

Statistical methylation patterns of cases and controls. (a) Density plot of β-values for all probes in the EPIC arrays for all 14 individuals. (b) PCA of whole DNA methylomes allows for the differentiation between ADLI cases (N = 7, red shapes) and matched controls (N = 7, green shapes).
Next, a pooled t-test was used to identify the differentially methylated CpG loci between the ADLI patients and the matched control blood samples, resulting in the identification of 806 significantly differentially methylated CpG loci (P < 0.05 and |Δβ|> 0.10) (Fig. 2a). Among the 806 dmCpGs, 308 were hypermethylated and 498 were hypomethylated (Fig. 2a). Furthermore, unsupervised hierarchical clustering of the 806 dmCpGs showed a clear segregation between ADLI and without ADLI (Fig. 2b), confirming that the DNA methylation patterns of leukocytes in ADLI patients differ from those of non-ADLI patients.

Visualization of differentially methylated probes. (a) Volcano plot showing differentially methylated CpG sites. The x-axis represents the magnitude of the difference in signal intensity between the groups for each probe in the microarray, expressed as: ∆β = β (cases) − β (controls). The y-axis represents the − log10 (P‑value), with a P‑value of 0.05. Significant difference sites (P < 0.05 and |Δβ|> 0.10) are highlighted in red and blue. Sites highlighted in red and blue are those that were hypermethylated and hypomethylated compared to the controls, respectively. (b) Unsupervised hierarchical clustering of the 806 most variable CpG sites derived from samples distinguishes ADLI (N = 7) cases and matched controls (N = 7). Two different groups are represented: group 1 (X1546, X299, X306, X680, X383, X612, and X503), i.e. the case group (columns marked in red), and group 2 (X1245, X416, X1139, X670, X667, X896, and X1073), i.e. the control group (columns marked in blue). Each column represents one sample (sample names below), and each horizontal line represents the methylation levels of a given CpG locus across samples. Methylation levels are expressed as β-values from 0 to 1 (blue and red, un-methylated, and completely methylated, respectively).
Figure 3a depicts the genomic distribution of dmCpGs and distinguishes between CpG island-related regions and gene-related regions. Both hypomethylated and hypermethylated dmCpGs were found to be enriched in open sea regions rather than the CpG islands (Fig. 3a). In terms of gene-related locations, hypermethylated and hypomethylated CpGs were preferentially situated in gene body regions and intergenic regions, respectively, and both were impoverished at the 3’UTR regions (Fig. 3b).

Stacked bar plots depicting the percentage of differentially methylated CpG sites according to their CpG island-related or gene location-related status. (a) Percentage of dmCpGs according to their location in relation to CpG islands. (b) Percentage of dmCpGs according to genic location.
We found a total of 53 genes, including multiple differentially methylated CpGs. (see Supporting Information Table S1). These genes were located in different gene-related regions. We focused on the analysis of dmCpGs in the promoter since many studies have suggested that promoter methylation significantly affects the levels of gene expression. A total of 10 genes contained two or more dmCpGs in the promoter regions, including 5 hypomethylated genes (C22orf39, HCG27, PKD1L2, BIRC7, and LOC100507140) and 5 hypermethylated genes (C1orf141, CD177, FMOD, LOC102723376, and TANC1) (Table 2).
Expression levels of candidate genes
To determine whether the methylation of a candidate gene affected the gene expression levels, the mRNA expression of these target genes was analyzed using RT-PCR. For one of the hypomethylated differentially methylated genes, LOC100507140, it expression was higher in the ADLI patients compared to the controls (Fig. 4a), while the expression of C22orf39, HCG27, PKD1L2, and BIRC7 showed no significant difference (Fig. 4b–e). Among the hypermethylated differentially methylated genes (DMGs), only TANC1 expression was low in the ADLI patients compared to controls (Fig. 4 f), while the expression levels of the other four genes were not significantly different (Fig. 4g–j). As such, we selected LOC100507140 (2 sites) and TANC1 (2 sites) for the subsequent pyrosequencing experiments.

Expression of selected genes in the blood samples of cases and controls. Relative mRNA quantification of 10 candidate genes using RT-qPCR in cases (N = 60) compared to controls (N = 60) blood samples. Data are represented as mean ± SD. *P < 0.001.
Pyrosequencing validation of differentially methylated ADLI-associated sites
The pyrosequencing results of two CpG sites (cg18472223 and cg20517941) within the promoter region of LOC100507140 and two CpG sites (cg18472223 and cg20517941) within the promoter region of TANC1 displayed lower and higher methylation levels in ADLI samples, respectively, consistent with the EPIC BeadChip array (Fig. 5). Representative images of pyrosequencing for four differentially methylated loci in one patient are shown in Fig. 6.

DNA methylation values of four CpGs sites measured by the EPIC BeadChip (top) and by bisulfite pyrosequencing (bottom). DNA methylation values of cg06961147 (a), cg24666046 (b), cg18472223 (c), and cg20517941 (d) were measured using EPIC BeadChip. DNA methylation values at each CpG site by percentage were measured by pyrosequencing of the ADLI patients (red) and controls (blue) in cg06961147 (e), cg24666046) (f), cg18472223 (g), and cg20517941 (h).

Representative images of pyrosequencing for four differentially methylated loci in one patient. Images of pyrosequencing were generated for cg18472223 and cg20517941 (a), cg06961147 (b), and cg24666046 (c).
Correlation analysis results of various indicators
The mRNA expression levels of TANC1 gene was negatively correlated with ALT and AST (ρ = − 0.818 and − 0.800, respectively; all P < 0.001), while the mRNA expression levels of LOC100507140 was positively correlated with ALT and AST (ρ = − 0.893 and − 0.824, respectively; all P < 0.001). The mRNA expression levels of TANC1 gene was negatively correlated with the methylation levels of the two CpG sites (cg06961147 and cg24666046), and the correlation coefficient ρ was − 0.500 and − 0.515, respectively (all P < 0.001). Similarly, the mRNA expression levels of LOC100507140 gene was negatively correlated with the methylation levels of the two CpG sites (cg18472223 and cg24666046), and the correlation coefficient ρ was − 0.383 and − 0.368, respectively (all P < 0.001).
Diagnostic value of ADLI-associated differentially methylated sites
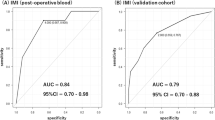
The diagnostic values of TANC1 (cg06961147, cg24666046, and in combination) and LOC100507140 (cg18472223, cg20517941, and in combination) were identified using ROC curves. As shown in Table 3 and Fig. 7, the area under the curve (AUC) of TANC1 (cg06961147, cg24666046, and in combination) were 0.812 (95% CI 0.737–0.887), 0.842 (95% CI 0.774–0.911), and 0.857 (95% CI 0.792–0.923), respectively. The AUCs of LOC100507140 (cg18472223, cg20517941, and in combination) were 0.765 (95% CI 0.680–0.851), 0.754 (95% CI 0.667–0.842), and 0.818 (95% CI 0.739–0.896), respectively. These results indicated that the combination of the two loci of TANC1 genes, cg06961147 and cg24666046, could be used to better distinguish between the ADLI and non-ADLI groups.

The receiver operating characteristic (ROC) curve for the discrimination of ADLI and non-ADLI. ROC curves were generated for cg06961147 (a), cg24666046 (b), and in combination (c) of the TANC1 gene. ROC curves of the cg18472223 (d), cg20517941 (e), and combination (f) of the LOC100507140 gene were also generated.
Ingenuity pathway analysis of the TANC1 gene
A total of 77 upstream and downstream genes related to the TANC1 gene were enriched in IPA (see Supporting Information Fig. S1). These enriched genes were selected for the analysis of canonical pathways using IPA. The IPA analysis results in the identification of five significant canonical pathways that were significantly enriched with these genes (Fig. 8). The pathways included liver hyperplasia/hyperproliferation, hepatocellular carcinoma, liver inflammation/hepatitis, kidney failure, and heart failure. The results showed that these genes were mainly enriched significantly in several pathways related to liver metabolism, suggesting that TANC1 is related to liver disease.

Significant canonical pathways identified by Ingenuity Pathway Analysis (IPA). Genes related to TANC1 gene (Supplementary Fig. S1) were significantly enriched in the above 5 pathways. The image was generated through the use of IPA (QIAGEN Inc. software version 65367011, https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis)23.
Discussion
In this study, the genome-wide patterns of DNA methylation in the blood of ADLI patients and non-ADLI controls were analyzed. Our results suggest that DNA methylation differs significantly between these two groups. Furthermore, using pyrosequencing in a larger sample size, we demonstrated that the expression and methylation of LOC100507140 and TANC1 could be used to distinguish between ADLI patients and the controls. These findings indicate that changes in DNA methylation levels may be related to alterations in the expression of certain genes during ADLI occurrence. These findings also suggest that the abnormal expression of DNA methylation could be used as an indicator of ADLI, similar to the predictive effect of abnormal DNA methylation in other diseases24,25,26.
To our knowledge, this study is the first to identify distinct differential DNA methylation patterns in blood samples from patients with ADLI and non-liver injury using the EPIC BeadChip array platform. The EPIC BeadChip array has the advantage of covering the whole genome, including the gene promoter region, the gene coding region, CpG islands, and the enhancer regions found in the ENCODE27 and FANTOM528 project. In our results, fundamental differences in DNA methylation patterns between the ADLI and non-ADLI patients were elucidated by PCA (Fig. 1b). Further analysis complemented these results by identifying site-specific CpG percentage (%) methylation changes that were responsible for pattern differentiation between the two groups (Fig. 2). The volcano plot in Fig. 2a shows the full distribution of the observed differential sites. The statistically significant CpG sites (N = 806) in the methylation heatmap (Fig. 2b) show a distinct pattern between the groups. The combined results provide a more comprehensive display of the genome-wide methylation of ADLI. Our understanding these epigenetic changes will enable the use of epigenetic biomarkers for diagnosis of disease in early stages. In general, our results display the genome-wide methylation of ADLI more comprehensively.
The genomic distribution of the dmCpGs suggests that the hypermethylation of dmCpGs may vary depending on the location: based on dmCpGs in CpG island-related or gene location-related status, dmCpGs were mainly observed in the open sea of the CpG-poor regions and the gene bodies, respectively. This phenomenon underscores the value of screening technologies that accurately examine CpG-sparse regions. In addition, the fact that the majority of methylation changes were identified in gene bodies emphasized that we cannot focus solely on methylation sites located in CpG-denser regions, such as CpG islands and gene promoters. Evidence is increasingly suggesting that DNA methylation in gene bodies is able to promote oncogene expression29,30, such that gene body methylation may serve as a therapeutic target for the treatment of cancer31,32. Our results suggest that the DNA methylation of gene bodies will be an important topic for future studies in the field of cancer research.
Notably, we found that the combination of two CpG sites (cg06961147 and cg24666046) of the TANC1 gene could act as potential biomarkers for the diagnosis of ADLI cases and non-ADLI controls. This is a novel discovery. Although there are currently very few reports on TANC1 research, many studies have reported that TANC1 is an important synaptic scaffold protein that plays a critical role in regulating the density of synaptic spines and excitatory synapse strength33. Studies have also indicated that TANC1 is a candidate gene for neurodevelopmental disorders (NDD)34,35. Furthermore, the TANC1 locus can influence the development of late radiation-induced damage36. In the present study, the TANC1 gene was analyzed with IPA to identify any associated significant pathways, upstream regulators, diseases, and functions. A total of 77 upstream and downstream genes related to the TANC1 gene were found to be enriched in IPA (Fig. 8a). Our results indicated that these genes were significantly enriched in several pathways related to liver metabolism, including liver hyperplasia/hyperproliferation, hepatocellular carcinoma, and liver inflammation/hepatitis (Fig. 8b), suggesting that the TANC1 gene is related to liver disease. To date, the biochemical properties of TANC1 proteins remain largely unknown, and there is currently no research on the relationship between TANC1 methylation and diseases. Since methylated DNA is very stable37 and can be detected in clinical blood samples38,39, it is a promising target for use in disease diagnostics. Taken together, our results indicate that DNA methylation is a promising diagnostic target for ADLI.
Our study contains a number of limitations. First, our sample size was relatively small, with limited power; hence, future studies should investigate whether the CpG sites identified in this study can be replicated in an independent population. In addition, given the large number of dmCpGs found and distributed at different locations in the present study, we were unable to determine all of the dmCpGs. Therefore, we only performed mRNA and pyrosequencing verification of dmCpGs in the TSS1500 region. The dmCpGs located in other regulatory regions will need to be validated in future studies. Finally, although we identified two differentially methylated genes, LOC100507140 and TANC1, we did not study their possible mechanisms of action further. Despite these limitations, this study is the first to use the EPIC BeadChip array platform identify the DNA methylation signatures associated with ADLI.
In summary, the distinctive differences in DNA methylation patterns between ADLI and non-ADLI patients were the main finding of our study. The expression of LOC100507140 and TANC1, as the differentially methylated genes, was found to vary significantly during the occurrence of ADLI. More importantly, we found that the combination of the hypermethylated differentially methylated site cg06961147 and cg24666046 in TANC1 provides a potential target for the diagnosis of ADLI.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ADLI:
-
Anti-tuberculosis drug-induced liver injury
- TB:
-
Tuberculosis
- DOTS:
-
Directly observed treatment short-course chemotherapy
- H:
-
Isoniazid
- R:
-
Rifampicin
- Z:
-
Pyrazinamide
- E:
-
Ethambutol
- EPIC:
-
MethylationEPIC BeadChip
- dmCpGs:
-
Differentially methylated CpG sites
- ATS:
-
American thoracic society
- ULN:
-
Upper limit of normal
- BMI:
-
Body mass index
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- ALP:
-
Aspartate aminotransferase
- TBIL:
-
Total bilirubin
- ALB:
-
Albumin
- TP:
-
Total protein
- SD:
-
Standard deviation
- IQR:
-
Median with interquartile range
- qRT-PCR:
-
Quantitative reverse transcriptase polymerase chain reaction
- PCA:
-
Principal component analysis
- TSS:
-
Transcriptional start site
- UTR:
-
Untranslated region
- CpG:
-
Cytosine-guanine dinucleotide
- IPA:
-
Ingenuity Pathway Analysis
References
Xu, K. et al. Nanomaterials in the prevention, diagnosis, and treatment of mycobacterium tuberculosis infections. Adv. Healthc. Mater. 7(1), 1700509. https://doi.org/10.1002/adhm.201700509 (2018).
Zhang, S. et al. Preventive use of a hepatoprotectant against anti-tuberculosis drug-induced liver injury: A randomized controlled trial. J. Gastroenterol. Hepatol. 31(2), 409–416. https://doi.org/10.1111/jgh.13070 (2016).
Hu, X. et al. Antituberculosis drug-induced adverse events in the liver, kidneys, and blood: Clinical profiles and pharmacogenetic predictors. Clin. Pharmacol. Ther. 104(2), 326–334. https://doi.org/10.1002/cpt.924 (2018).
Tostmann, A. et al. Antituberculosis drug-induced hepatotoxicity: Concise up-to-date review. J. Gastroenterol. Hepatol. 23(2), 192–202. https://doi.org/10.1111/j.1440-1746.2007.05207.x (2008).
Jeong, I. et al. Drug-induced hepatotoxicity of anti-tuberculosis drugs and their serum levels. J. Korean Med. Sci. 30(2), 167–172. https://doi.org/10.3346/jkms.2015.30.2.167 (2015).
Yang, M. et al. Possible association of HMOX1 and NQO1 polymorphisms with anti-tuberculosis drug-induced liver injury: A matched case-control study. J. Clin. Pharm. Ther. 44(4), 534–542. https://doi.org/10.1111/jcpt.12818 (2019).
Piras, I. S. et al. Exploring genome-wide DNA methylation patterns in Aicardi syndrome. Epigenomics 9(11), 1373–1386. https://doi.org/10.2217/epi-2017-0060 (2017).
Widschwendter, M. et al. Methylation patterns in serum DNA for early identification of disseminated breast cancer. Genome Med. 9(1), 115. https://doi.org/10.1186/s13073-017-0499-9 (2017).
Niehrs, C. & Schafer, A. Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol. 22(4), 220–227. https://doi.org/10.1016/j.tcb.2012.01.002 (2012).
Cheishvili, D., Boureau, L. & Szyf, M. DNA demethylation and invasive cancer: Implications for therapeutics. Br. J. Pharmacol. 172(11), 2705–2715. https://doi.org/10.1111/bph.12885 (2015).
He, L. et al. Involvement of cytochrome P450 1A1 and glutathione S-transferase P1 polymorphisms and promoter hypermethylation in the progression of anti-tuberculosis drug-induced liver injury: A case-control study. PLoS ONE 10(3), e0119481. https://doi.org/10.1371/journal.pone.0119481 (2015).
Zhang, J. et al. Correlation of CpG island methylation of the cytochrome P450 2E1/2D6 genes with liver injury induced by anti-tuberculosis drugs: A nested case-control study. Int. J. Environ. Res. Public Health 13(8), 776. https://doi.org/10.3390/ijerph13080776 (2016).
Zhou, M. et al. Drug resistance characteristics of Mycobacterium tuberculosis isolates between 2014 and 2017 in Sichuan, China: A retrospective study. PLoS ONE 13(12), e0209902. https://doi.org/10.1371/journal.pone.0209902 (2018).
Chen, G. et al. Association of UGT2B7 polymorphisms with risk of induced liver injury by anti-tuberculosis drugs in Chinese Han. Int. J. Immunopathol. Pharmacol. 30(4), 434–438. https://doi.org/10.1177/0394632017733638 (2017).
Li, Y. H. et al. Combined 5-hydroxymethylcytosine content of human leucocyte antigen-B and human leucocyte antigen-DQB1 as novel biomarker for anti-tuberculosis drug-induced liver injury. Basic Clin. Pharmacol. Toxicol. 127(3), 234–240. https://doi.org/10.1111/bcpt.13401 (2020).
Saukkonen, J. J. et al. An official ATS statement: Hepatotoxicity of antituberculosis therapy. Am. J. Respir. Crit. Care Med. 174(8), 935–952. https://doi.org/10.1164/rccm.200510-1666ST (2006).
Mushiroda, T. et al. Development of a prediction system for anti-tuberculosis drug-induced liver injury in Japanese patients. Hum Genome Var. 3, 16014. https://doi.org/10.1038/hgv.2016.14 (2016).
Moran, S., Arribas, C. & Esteller, M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 8(3), 389–399. https://doi.org/10.2217/epi.15.114 (2016).
Aryee, M. J. et al. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30(10), 1363–1369. https://doi.org/10.1093/bioinformatics/btu049 (2014).
Zhang, C. et al. Differential DNA methylation profiles of human B lymphocytes and Epstein-Barr virus-immortalized B lymphocytes. Chin. J. Cancer Res. 30(1), 104–111. https://doi.org/10.21147/j.issn.1000-9604.2018.01.11 (2018).
Triche, T. J. Jr., Weisenberger, D. J., Van Den Berg, D., Laird, P. W. & Siegmund, K. D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 41(7), e90. https://doi.org/10.1093/nar/gkt090 (2013).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4), 402–408. https://doi.org/10.1006/meth.2001.1262 (2001).
Kramer, A., Green, J., Pollard, J. Jr. & Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30(4), 523–530. https://doi.org/10.1093/bioinformatics/btt703 (2014).
Crowgey, E. L., Marsh, A. G., Robinson, K. G., Yeager, S. K. & Akins, R. E. Epigenetic machine learning: Utilizing DNA methylation patterns to predict spastic cerebral palsy. BMC Bioinform. 19(1), 225. https://doi.org/10.1186/s12859-018-2224-0 (2018).
Lussier, A. A. et al. DNA methylation as a predictor of fetal alcohol spectrum disorder. Clin. Epigenet. 10, 5. https://doi.org/10.1186/s13148-018-0439-6 (2018).
Hoff, K. et al. DNA methylation profiling allows for characterization of atrial and ventricular cardiac tissues and hiPSC-CMs. Clin. Epigenet. 11(1), 89. https://doi.org/10.1186/s13148-019-0679-0 (2019).
Siggens, L. & Ekwall, K. Epigenetics, chromatin and genome organization: Recent advances from the ENCODE project. J. Intern. Med. 276(3), 201–214. https://doi.org/10.1111/joim.12231 (2014).
Lizio, M. et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 16, 22. https://doi.org/10.1186/s13059-014-0560-6 (2015).
Zhou, S. et al. DNA methylation of METTL7A gene body regulates its transcriptional level in thyroid cancer. Oncotarget 8(21), 34652–34660. https://doi.org/10.18632/oncotarget.16147 (2017).
Arechederra, M. et al. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 9(1), 3164. https://doi.org/10.1038/s41467-018-05550-5 (2018).
Yang, X. et al. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 26(4), 577–590. https://doi.org/10.1016/j.ccr.2014.07.028 (2014).
Wang, Y. W. et al. ITPKA gene body methylation regulates gene expression and serves as an early diagnostic marker in lung and other cancers. J. Thorac. Oncol. 11(9), 1469–1481. https://doi.org/10.1016/j.jtho.2016.05.010 (2016).
Yang, Q., Liu, H., Li, Z., Wang, Y. & Liu, W. Purification and mutagenesis studies of TANC1 ankyrin repeats domain provide clues to understand mis-sense variants from diseases. Biochem. Biophys. Res. Commun. 514(2), 358–364. https://doi.org/10.1016/j.bbrc.2019.04.151 (2019).
Suzuki, T. et al. A novel scaffold protein, TANC, possibly a rat homolog of Drosophila rolling pebbles (rols), forms a multiprotein complex with various postsynaptic density proteins. Eur. J. Neurosci. 21(2), 339–350. https://doi.org/10.1111/j.1460-9568.2005.03856.x (2005).
Wessel, K. et al. 17q23.2q23.3 de novo duplication in association with speech and language disorder, learning difficulties, incoordination, motor skill impairment, and behavioral disturbances: A case report. BMC Med. Genet. 18(1), 119. https://doi.org/10.1186/s12881-017-0479-3 (2017).
Fachal, L. et al. A three-stage genome-wide association study identifies a susceptibility locus for late radiotherapy toxicity at 2q24.1. Nat. Genet. 46(8), 891–894. https://doi.org/10.1038/ng.3020 (2014).
Karouzakis, E. et al. Analysis of early changes in DNA methylation in synovial fibroblasts of RA patients before diagnosis. Sci. Rep. 8(1), 7370. https://doi.org/10.1038/s41598-018-24240-2 (2018).
Fransquet, P. D. et al. Blood DNA methylation as a potential biomarker of dementia: A systematic review. Alzheimers Dement. 14(1), 81–103. https://doi.org/10.1016/j.jalz.2017.10.002 (2018).
Ma, Z. Y. et al. Methylated septin 9 and carcinoembryonic antigen for serological diagnosis and monitoring of patients with colorectal cancer after surgery. Sci. Rep. 9(1), 10326. https://doi.org/10.1038/s41598-019-46876-4 (2019).
Acknowledgements
The authors would like to thank the patients support and participation and Shanghai Biotechnology Corporation for conducting the EPIC array experiments and pyrosequencing assays.
Funding
This work was supported by the National Natural Science Foundation of China (Grant number: 81041096 and 81760609) and Graduate innovation project of North China University of Science and Technology (Grant number: CXZZBS2017128).
Author information
Authors and Affiliations
Contributions
D.X.W. and Y.H.L. designed the research; D.X.W., Y.H.L., Q.R., and S.F.P. performed the experiments; Y.Z.C., L.W. and L.M.Y. analysed data; D.X.W., and Y.H.L. wrote the paper; S.S.F. and F.F.M. supervised the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, D., Li, Y., Ren, Q. et al. TANC1 methylation as a novel biomarker for the diagnosis of patients with anti-tuberculosis drug-induced liver injury. Sci Rep 11, 17423 (2021). https://doi.org/10.1038/s41598-021-96869-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-96869-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
