
Abstract
Using multi-electron–ion coincidence measurements combined with high level calculations, we show that double ionisation of SO2 at 40.81 eV can be state selective. It leads to high energy products, in good yield, via a newly identified mechanism, which is likely to apply widely to multiple ionisation by almost all impact processes.
Similar content being viewed by others
Introduction
Since their detection almost a century ago by mass spectrometry, doubly charged molecular ions have attracted both experimentalists and theoreticians to investigate their formation, stability and reactivity. Their stability is a balance between Coulombic repulsion and chemical bonding, and small polyatomic dications (AB2+) are all unstable or metastable since the charge separation channel (A+ + B+) is located in energy well below the charge retaining channels (AB2+ or A + B2+)1,2. Upon fragmentation, such dications are expected to produce the low energy fragments (A+ + B+) predominantly. All fragmentations tend to produce the lowest energy accessible products, but recent experimental and theoretical studies have shown that state-to-state fragmentations of small neutral molecules may lead to unexpected results. For instance, in some VUV photodissociation of CO2 there is no formation of ground state CO + O species. Instead the O photoproduct is formed in the 1D or the 1S excited states3, whose dipole forbidden emissions are prominent features in the visible spectra of terrestrial atmospheres4. Similarly, the VUV photodissociation of N2 by ns or fs lasers leads to at least one excited atomic nitrogen instead of two 4S ground state nitrogen atoms5,6. Very recently, Zhou et al. showed that the UV photolysis of H2S produces excited S(1D) atoms (+ H2) instead of ground state S(3P)7. Fragmentation of even quite large singly charged molecular ions can also be state-selective rather than statistical, of which a classical example is the dissociation of C2F6+8. These state specific processes are important to planetary atmospheres, plasmas and photochemistry at the fs and shorter time scales. By contrast, no such unexpected behavior has been reported hitherto for molecular dications.
As prototypes of bond-breaking process in molecular dications, the SO22+ spectra and dissociations induced by photoionization or electron impact have been studied for many years accompanied by molecular structure calculations at different levels of theory. These works are reviewed in Ref.9, which presents a resolved spectrum of the doubly-charged ions as obtained by the TOF-PEPECO method, and interpreted using high-level molecular structure theory. Also, double ionization of the SO2 molecule has continued to attract attention10,11, partly because of the great importance of the molecule in atmospheric and astrophysical contexts12,13. Indeed, SO2 is common in volcanic and biological emissions on Earth14,15 and it is a crucial chemical compound in sulphur physical chemistry in Earth13, Venus16, Io17,18 and exoplanet terrestrial atmospheres, where its photochemistry leads to the formation of sulphuric acid or sulphuric acid aerosols with well-known deleterious effects such as acid rains19.
From existing studies, we know that double ionization of SO2 at energies below the triple ionization limit leads to dissociation in four principal ways: (1) SO22+ → O+ + SO+ or (2) SO22+ → O+ + S+ + O or (3) SO22+ → O2+ + S+or (4) SO22+ → O + SO2+. A fifth pathway producing two O+ ions with neutral S is detectable but has extremely low intensity. Dissociative double ionization is brought about not only by direct vertical double ionization, but also by an indirect process shown up clearly in PEPECO studies of this and other small molecules20 by the signatures of distinct electron energies from autoionization of super-excited O, N and other atoms21. The well documented indirect process can be represented for SO2 as: SO2 + hv → SO2+* + e1−; SO2+* → SO+ + O*: O* → O+ + e2−.
We here report multi-particle coincidence measurements using the TOF-PEPEPIPICO technique (cf. SI). This work makes the first direct connection between the energies and states of nascent SO22+ ion with fragmentation leading either to charge separation (SO+ + O+) or to the thermodynamically unfavoured charge retaining (SO2+ + O) products of O–SO bond breaking. A vital characteristic of our technique is that both ions and electrons from ionization at a point-like source are collected and detected with high overall efficiency, being uniquely capable to extract complete mass spectra from single and double ionization separately. This is done by requiring the coincident detection of a single photoelectron of appropriate (relatively high) energy in the one case, and coincident detection of an electron pair within the right energy range in the other. As references—single and double ionization mass spectra of SO2 photoionized at 40.8 eV are shown in the SI. These spectra confirm that the most abundant products at this energy are O+ + SO+ and that SO2+ is the only doubly-charged ion formed.
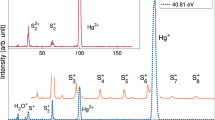
Figure 1 shows the spectra of fragment ion pairs compared to a more highly resolved double ionization spectrum acquired earlier22. The bands in this spectrum due to the direct population of SO22+ states located in the Franck–Condon (F.C.) zone accessed from SO2(X1A1) have been identified in previous work9, and again in the present computations. Whatever the equilibrium geometry of the SO22+ dication, the electronic states are labelled in the C2ν point group since single-photon ionisation occurs from the bent ground neutral SO2 (X 1A1) state. We also mapped the multi-dimensional potentials of the doubly charged ions in the Cs point group (cf. SI for more details). Dissociation following the most abundant ion pair pathway of O+ + SO+ formation sets in at the very onset of double ionization near 34 eV and persists over most of the range of ionization energies. Of the distinct features of the overall spectrum, this charge-separation pathway follows from population of the prominent vibrationally resolved state at 35.3 eV, identified as 11A29, but apparently not from population of the states represented by the bands centered at 36.5 and at 37.5 eV. The onset of the charge retaining pathway forming SO2+ coincides with the peak of the resolved band, and also with the thermodynamic threshold for O + SO2+ at ca. 35.46 eV (Table S3). This channel is populated with substantial quantum yield (about 1/3 of the O+ + SO+ yield) at all energies above threshold, and specifically by decay from a state or states between 37 and 38 eV, seen as a broad peak in the resolved spectrum and apparently correlated with the population of the 31A1 (41A′ in Cs symmetry) state. Contributions of the triplet states lying in the same energy range cannot be ruled out, in particular the {13A1, 23B1, 23B2} set of states. Of the channels not shown in Fig. 1, the one leading to the three-body products O+ + S+ + O (cf. Fig. S2), sets in weakly at ~ 37 eV (thermodynamic threshold at 35.1 eV) and shows no distinctive features. These products are formed by sequential decay of primary SO+, as reflected in Newton diagrams23. Its onset energy agrees with the limit for ground-state products plus the observed total kinetic energy release (KER). Channel (3) will be discussed in a separate paper.

Spectra of electron pairs coincident with O+ + SO+ ion pairs and with specified single ions, are compared with a well-resolved spectrum of all electron pairs, all after photoionization of SO2 at the photon energy hν = 40.81 eV. The SO2+ and O+ curves are displaced vertically on a common scale (with constant off-sets) and show true relative intensities, while the other spectra included for comparison have intensity scales adjusted for display. The combs correspond to the MRCI/aug-cc-pV(Q + d)Z computed vertical double ionization energies of SO2 given in Table S1 and quoted for C2v symmetry.
Insights into the fragmentation mechanisms can be obtained from the kinetic energies released in the fragmentations. The KER magnitudes can be extracted from TOF peak widths and shapes, and for the charge-separating channels this has been done several times before23,24 with generally concordant results, but without initial state selection. The most detailed measurements made possible by use of a position–sensitive ion detector23, gave the full KER distributions for 40.8 eV photoionization. Although we can accurately select ranges of ionization energy (IE), the present experimental data are not ideally suited to the extraction of KERs. In contrast to previous experiments where a continuous light source could be used24, in our set-up there has to be a relatively long delay between the photoionisation event and ion extraction (to ensure that even the slowest electrons leave the source region), ions spread out in the source and the TOF peaks get broadened. Nevertheless, we can estimate the mean KEs released from dications formed in limited ionisation energy ranges. To use the available statistics efficiently we have extracted mass spectra from 1 eV wide IE ranges which correspond quite well to the main features in Fig. 1. Results are given in Table S4.
For the charge separation reaction forming O+ + SO+ we find that the mean KER varies slightly, between 4.5 ± 0.2 to 4.8 ± 0.2 eV (cf. Table S4) over the range of ionization energies (IEs) from 34 to 40 eV. For this two-body dissociation the distributions are quite narrow (FWHM ca. 2 eV) and centered at about 5 eV. When the kinetic energy release is added to the thermodynamic threshold energy, the result (29.6 + 4.5 eV = 34.1 eV) agrees with the observed onset energy. This proves that the products are formed in their ground states as SO+(X2Π) + O+(4Su), at least at threshold.
For the reaction(s) producing SO2+ the KER is much smaller and produces roughly triangular peak shapes, as is known from the work of Field and Eland24 (their Fig. 5), where a full list of SO2+ KERs as a function of ionization energy range (Table 2 in Ref.24) is also presented. The KER values given in the present work agree satisfactorily with the more complete values given in the past24. In the 34–40 eV range, this two-body channel SO2+ + O shows two distinct energy distributions: (i) a close to zero KER (0.0–0.2 eV) for 35 < IE < 37 eV range and ~ 0.5 eV for 37 < EI < 40 eV (cf. Table S4). These KERs are derived from the peak widths (FWHM) by first subtracting the thermal width assuming that it and the release width add quadratically. There is a clear increase in energy release as the IE increases, but as the absolute magnitudes are uncertain, we give no error limits. It is apparent from Fig. 1 that production of the SO2+ ion starts at the estimated thermodynamic limit for its formation, 35.3 eV. The estimate depends on a theoretical value for the double ionization ionisation energy of SO25, but this is sufficiently accurate to confirm that ground-state products, SO2+(X1Σ+) + O(3P) must be formed at threshold. The rise in KER as a function of IE seems continuous, with no special excursion at the energy of the state(s) near 37.5 eV, which decay specifically to these products.
To investigate the fragmentation dynamics of the SO22+ ions more deeply we have performed ab initio computations on the potential energy surfaces (PESs) of the singlet and triplet mainly two-hole (2 h) electronic states located in the whole 33–40 eV range above SO2(X1A1). These computations are done at the CASSCF/MRCI/aug-cc-pV(Q + d)Z level for a wide range of nuclear configurations (cf. Figs. S3–S6), which show a high density of states, consistent with the expected number (20 to 30) of 2 h orbital combinations. Also, we anticipate the existence of a larger number of three-hole-one particle (3h-1p) states accessible in the same regions. Such a high density of electronic states normally implies multiple mutual interactions by vibronic and spin–orbit interactions. In the main dissociation channel, for example, no singlet states correlate to the lowest asymptote, SO+(X2Π) + O+(4Su), so intersystem crossings (from singlets) and internal conversions (from higher triplets) are needed to populate the lowest triplet that leads to these products (Fig. 2). The formation of the other products, in particular SO2+ + O, apparently from specific states is intriguing since the surfaces leading to these products are embedded in a dense manifold of surfaces, mostly leading to the lowest dissociation channel (cf. Table S3 of the SI).

Minimal energy paths (MEPs) along the SO distance (R) for the 1A′, 1A″, 3A′ and 3A″ PESs. These MEPs are deduced from the SO22+ PESs (Figs. S3–S6) by looking for the minimal energy for each SO distance along the different OSO angles. We show also the 41A′ potential for RSO = 2.7 Bohr and Θ = 120°. The asymptotes are located using the data from Table S3. The dashed dotted vertical line corresponds to the middle of the Franck–Condon (F.C.) region for the SO2(X1A1) → SO22+ + 2e- transition. The arrows illustrate the fragmentations discussed in the text.
To identify the SO22+ electronic states decaying to SO+ + O+ and to O + SO2+ and to understand their dissociation pathways we take inspiration from the earlier calculations9, and also make use of the present theoretical data. For the O+ + SO+ channel, the KERs are between 4.5 and 4.8 eV whatever the initial SO22+ state. These experimental values are close to the computed KER where the products are formed in their electronic ground states O+(4S) + SO+(X2Π) via the following mechanisms (Fig. 2):
-
(i)
Internal conversions within the singlets manifold of states populating the lowest 1A′ state.
-
(ii)
Spin–orbit conversion from this 1A′ to the lowest 3A″ potential. This triplet can be also reached via internal conversion within the triplets.
-
(iii)
Afterwards, the lowest triplet 3A″ leads to the O+(4Su) + SO+(X2Π) products. The spin–orbit integrals presented in Fig. S7 have magnitudes of several cm-1, which are large enough for such internal conversions to be rapid.
The fact that O+ + SO+ continues to be formed over the whole range of ionisation energies up to 40 eV with little change in kinetic energy release means that the products must be created with increasingly large amounts of internal energy. Only a limited amount of energy can be accommodated as vibration in undissociated SO+26, so much of the excess energy must be electronic excitation of the product ions. This is consistent with the observation of the sequential formation of O+ + S+ + O and of optical emission in coincidence with the O+ + SO+ ion pair with a significant quantum yield27, attributed particularly to SO+(A2Π → X2Π). It seems to indicate that internal conversions and intersystem crossings within the SO22+ state manifold, although dominant, are not quite complete.
For the SO2+ + O channel, we refer to Fig. 2, which presents the minimal energy paths (MEPs) of the 1A′, 1A″, 3A′ and 3A″ PESs along the SO distance, and to the KERs listed in Table S4. For IE < 37 eV, computations suggest the production of these fragments occurs on the 1A′ and/or the 1A″ MEPs. For 1A′, we find a Morse-like potential that leads to a plateau at intermediate bond extension (ca. 4 Bohr) very close in energy to the SO2+(X1Σ+) + O(3Pg) asymptote. We also found a 1A″ potential with a small potential barrier (of 0.04 eV) before a level at almost exactly the same energy. We suggest that production of ground-state SO2+ may occur by intersystem crossing from these surfaces to one of the triplet surfaces correlated to SO2+(X1Σ+) + O(3Pg), which are expected to be relatively flat at these distances in the presence of charge-induced dipole attraction and the absence of Coulomb repulsion. At such distances other competitive curve crossings, which could lead to eventual SO+ + O+ production are less frequent (cf. Figs. S3–S6). With this interpretation, the computed KERs and the measured ones are the same within the error bars. But in the F.C. region the MEPs concerned are iso-energetic with high vibronic levels of SO22+(X1Σg+/X1A1) and levels of the vibrationally resolved 11A2 state in regions where there are multiple curve crossings. There is also a weak peak at 35.7 eV on the SO2+ yield curve in Fig. 1 which suggests that some vertically accessed levels of the resolved 11A2 state at 35.3 eV, decay into this channel rather than to the lowest energy products. It follows that to support the proposed mechanisms, we still need an explanation of how the long O–SO bond extension required can be reached in significant yield, without undergoing prior conversion to lower energy states.
For production of SO2+ in the range of IE above 37 eV, we measure a KER of ~ 0.5 eV. This value is sufficiently different from the previous case to suggest that a different potential surface is involved. Figure 2 shows that the formation of this product coincides with the occurrence of the SO2+(X1Σ+) + O(1Dg) asymptote where there is no difficulty over the spin. Computations indicate that the SO22+(41A′/31A1) state which leads directly to these products can be populated by direct ionization in the F.C. zone. Figure 2 shows that the SO22+(41A′/31A1) surface exhibits a potential barrier toward dissociation of ~ 0.4 eV, which is in good agreement with the measured KER at these IEs (cf. Table S4). Again, for this route to be followed in the high observed yield after direct vertical double ionisation in the F.C. zone, the expected competitive internal conversions would need to be somehow turned-off to allow the necessary bond extension. Because this seems both ad hoc and inherently unlikely, we looked for another explanation.
The clue to an alternative mechanism is given by the observation in double ionisation of autoionising O* over the range of ionisation energy from near threshold right up to the photon energy, as visible in the electron pair coincidence map9 and shown in more detail in Fig. S8. The observation of O* autoionization over the whole energy range proves that super-excited states of SO2+* are also populated over the same wide energy range to act as precursors. There is indeed no lack of suitable states, because below each of the 20 to 30 2-h states of SO22+ (Figs. S3–S6) there is a quasi-infinity of singly-charged 2h-1p Rydberg states in series converging on those dication states. The high Rydbergs have long lifetimes (proportional roughly to n*3) towards both optical emission and autoionisation28. The cores of the higher Rydberg states, being doubly ionised, excited and hardly influenced by the attached electron, will start to dissociate in the same way as bare SO22+ ions. Many will follow the most favorable path of O+–SO+ bond extension leading to breakage. A departing O+ may then capture the Rydberg electron on its way out, producing the autoionisating O* states observed. The same or other Rydbergs, some with shorter but still relatively long lifetimes to autoionisation, will undergo significant O–SO bond extension before production of nascent SO22+. The new geometry at the moment of autoionisation can bring the molecular dications into regions of parameter space where curve-crossings and the consequent state conversions are rare, allowing the formation of energetically disfavored products with apparent state selectivity, exactly as is seen in the case of SO2+. At the same time, the molecular autoionisation will contribute to the whole double ionisation spectrum, adding to the direct vertical process. This mechanism should be operative in all molecular double ionisation by photon or electron impact not just in SO2.
The present technique is the only existing means of acquiring such detailed information on dicationic decays from the states of ions formed by photoionisation. We note that similar information for the different set of mainly singlet states accessed by the Auger effect could be acquired by Auger-electron–ion coincidence techniques. There are only six other molecules whose fragmentation after double ionisation can be compared with that of SO2. In BrCN2+, ICN2+29 and CF3I2+30 decays no state-specific behavior can be seen. The N2O2+ ion decays radiatively from specific states, but it shows no other state-specific decay behavior. The CS22+ ion both emits fluorescence from two specific excited states and also decays to C+ + S2+ from states in a limited energy range31, and is the closest to SO22+ in respect of this behavior. The electron–electron coincidence map for CS2 at 40.81 eV shows evidence of autoionisation29 akin to that in SO2, further confirming the validity of the proposed mechanism.
We have here identified a new non-conventional mechanism involved in the unimolecular decomposition of doubly charged molecular dications which accounts for the observation of energy-disfavored dissociation channels with substantial quantum yield in the decay of doubly ionized SO2. The mechanism almost certainly operates more widely. In the case of SO2, we have identified particular electronic states as probable surfaces upon which the dissociations to high energy products occur. The mechanism is one of indirect ionisation, akin to the usual Auger effect, providing a non-vertical overall pathway to double ionisation of molecules, outside the F.C. zone. In many cases the outcome will be indistinguishable from vertical double ionization in the Franck–Condon region, but in some cases it can permit energetically disfavored pathways to be followed to a much greater extent than would be expected on the basis of free internal energy flow.
Experimental method
Multi-electron multi-ion coincidence experiments were done at 304 Å (40.8 eV), well above the estimated adiabatic single-photon double ionization onset 33.5 eV9. The TOF-PEPEPIPICO apparatus which has been described before27 allows continuous (simultaneous) detection of two electrons with energy measurement and two positive ions with mass/charge measurement. In brief, ionisation occurs where molecules in an effusive jet from a hollow needle intersect wavelength-selected light from a pulsed discharge in He. At the moment of ionisation there is a weak electric field in the source region which accelerates any near-zero energy photoelectrons towards a 2 m distant detector, giving them sufficient velocity to arrive there in less than 10 μs. A continuous, divergent strong magnetic field in the same region guides almost all photoelectrons, irrespective of kinetic energies of less than a few hundred eV, into a solenoidal field which carries them all to the detector. After about 150 ns, when all relevant electrons have left the source region, a strong electric field pulse extracts ions in the opposite direction through a two-field time-of-flight mass spectrometer to a separate detector. The strength of the ion drawout pulse and the associated acceleration field, as set for time-focus conditions, determine the mass resolution, and the time-width of signals for fragment ions formed with more than thermal kinetic energies. The drawout pulse field strength affects the detectability of ion pairs of equal mass-to-charge ratio (m/z) such as O+ + O+ or O2+ + S+ because the ion detector and associated electronics used in these experiments imposed a deadtime of 50 ns.
At the time of these experiments the efficiency was about 30% for electrons and 10% for ions at m/z = 100. Because the final ion energy at the detector was less than 2 keV, the efficiency was somewhat higher for lighter ions. Electron resolution (E/ΔE) was about 20 throughout, while mass resolution ranged from 30 to 100 (FWHM) according to the strength of the drawout pulse; all ions of interest from SO2 except equal mass pairs were fully resolved under all conditions, but the 34S isotopic variants were only partially resolved.
Theoretical methods
All the ab-initio electronic calculations with different levels of theory were performed using the MOLPRO program suite32. In the present study, we mapped the SO22+ six lowest A′ and six lowest A′′ three-dimensional potential energy surfaces (PESs) of singlet and triplet spin multiplicities along the SO distance and the in-plane angle, θ = OSO varying from 40° to 180°. The other SO distance is kept fixed at 2.7 Bohr i.e. its value in SO2(X1A1) at equilibrium.
The PESs have been generated using the Complete Active Space Self Consistent Field (CASSCF)33,34 approach followed by the internally contracted multi reference configuration interaction method (MRCI)35,36,37. The CASSCF active space is constituted by the whole set of all configurations which is allowing all the possible excitations of all valence electrons in valence orbitals. The electronic states having the same spin multiplicity are computed according to the state-average procedure, as implemented in MOLPRO. For MRCI calculations, all configurations having a coefficient greater than 0.005 in the CI expansion of the CASSCF wave functions were taken into account as a reference. The computations were done in the Cs point group with wave functions leading to more than 7 × 108 uncontracted configurations for the singlet electronic states. For the triplet electronic states, we considered more than 26 × 108 uncontracted configurations. For these calculations, the aug-cc-pV(Q + d)Z and the aug-cc-pVQZ basis sets of Dunning et al. were used to describe the S and O atoms, respectively38,39,40. In fact, adding the tight-d functions to describe the sulfur atom improve the accuracy of the results as shown in different works41,42,43. Furthermore, to investigate the spin-obit coupling at the crossings between some electronic SO22+ states, the spin orbit integrals where computed using the CASSCF wave functions and the uncontracted spd cc-pVTZ basis set. We used the Breit–Pauli spin–orbit Hamiltonian as implemented in MOLPRO. Indeed, the spin–orbit integrals were evaluated, in Cartesian coordinates, over the CASSCF wavefunctions, where the effective Breit–Pauli SO operator, HSO, as implemented in MOLPRO was used44.
To evaluate the energies of the dissociation limits of SO22+ forming the [SO + O]2+, the partially spin restricted coupled cluster method including perturbative treatment of triple excitation (RCCSD(T))45,46,47,48 was used. For the RCCSD(T) computations we used the aug-cc-pV(5 + d)Z basis set for sulfur and the aug-cc-pV5Z for oxygen.
Data availability
The experimental and theoretical datasets generated during and/or analysed during the current study are available from the corresponding authors on reasonable request.
References
Schröder, D. & Schwarz, H. Generation, stability, and reactivity of small, multiply charged ions in the gas phase. J. Phys. Chem. A 103, 7385–7394 (1999).
Falcinelli, S. & Rosi, M. Production and characterization of molecular dications: Experimental and theoretical efforts. Molecules 25, 4157 (2020).
Lu, Z. et al. State-to-state vacuum ultraviolet photodissociation study of CO2 on the formation of state-correlated CO(X1Σ+; v) with O(1D) and O(1S) photoproducts at 11.95–12.22 eV. Phys. Chem. Chem. Phys. 17, 11752–11762 (2015).
Raghuram, S., Jain, S. K. & Bhardwaj, A. Forbidden atomic oxygen emissions in the martian dayside upper atmosphere. Icarus 359, 114330 (2021).
Song, Y. et al. Quantum-state dependence of product branching ratios in vacuum ultraviolet photodissciation of N2. ApJ 819, 23 (2016).
Trabattoni, A. et al. Mapping the dissociative ionization dynamics of molecular nitrogen with attosecond time resolution. Phys. Rev. X 5, 041053 (2015).
Zhou, J. et al. Ultraviolet photolysis of H2S and its implications for SH radical production in the interstellar medium. Nat. Commun. 11, 1547 (2020).
Simm, I. G., Danby, C. J. & Eland, J. H. D. Direct observation of an isolated state in the ion C2F6+: A violation of the quasi-equilibrium theory of mass spectra. J. Chem. Soc. Chem. Comm. 21, 832–833 (1973).
Hochlaf, M. & Eland, J. H. D. A theoretical and experimental study of the SO22+ dication. J. Chem. Phys. 120, 6449–6460 (2004).
McConkey, J. W. et al. Electron impact dissociation of oxygen-containing molecules: A critical review. Phys. Rep. 466, 1 (2008).
Fletcher, J. D., Parkes, M. A. & Price, S. D. Electron ionisation of sulfur dioxide. J. Chem. Phys. 138, 184309 (2013).
Barbu, A. L., Segers, A. J., Schaap, M., Heemink, A. W. & Builtjes, P. J. H. A multi-component data assimilation experiment directed to sulphur dioxide and sulphate over Europe. Atmos. Environ. 43, 1622–1631 (2009).
Broadfoot, A. L. et al. Extreme ultraviolet observations from voyager 1 encounter with jupiter. Science 204, 979 (1979).
Holland, H. D. The oxygenation of the atmosphere and oceans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 903–915 (2006).
Farquhar, J., Bao, H. & Thiemens, M. Atmospheric influence of earth’s earliest sulfur cycle. Science 289, 756 (2000).
Zhang, X. et al. Photolysis of sulphuric acid as the source of sulphur oxides in the mesosphere of venus. Nat. Geosci. 3, 834–837 (2010).
Pearl, J. et al. Identification of gaseous SO2 and new upper limits for other gases on Io. Nature 280, 755–758 (1979).
Lellouch, E., McGrath, M. A. & Jessup, K. L. Io’s atmosphere. In Io After Galileo (eds Lopes, R. M. C. & Spencer, J. R.) 231–264 (Springer, 2007).
Hu, R., Seager, S. & Bains, W. Photochemistry in terrestrial exoplanet atmospheres II: H2S and SO2 photochemistry in anoxic atmospheres. ApJ 769, 14 (2013).
Eland, J. H. D. & Feifel, R. Double Photoionisation Spectra of Molecules (Oxford University Press, 2017).
Eland, J. H. D. Dynamics of double photoionization in molecules and atoms. Adv. Chem. Phys. 307, 103–151 (2009).
Eland, J. H. D. Studies of double photoionisation using a fast pulsed VUV source. J. Elec. Spectr. Relat. Phen. 144–147, 1145–1150 (2005).
Hsieh, S. & Eland, J. H. D. Reaction dynamics of three-body dissociations in triatomic molecules from single-photon double ionization studied by a time- and position-sensitive coincidence method. J. Phys. B At. Mol. Opt. Phys. 30, 4515 (1997).
Field, T. & Eland, J. H. D. Fragmentation dynamics of SO22+. Int. J. Mass Spectr. 192, 281 (1999).
Ben Houria, A., Ben Lakhdar, Z., Hochlaf, M., Kemp, F. & McNab, I. R. Theoretical investigation of the SO2+ dication and the photo-double ionization spectrum of SO. J. Chem. Phys. 122, 054303 (2005).
Ben Houria, A., Ben Lakhdar, Z. & Hochlaf, M. Spectroscopic and spin-orbit calculations on the SO+ radical cation. J. Chem. Phys. 124, 054313 (2006).
Taylor, S. & Eland, J. H. D. Light emissions accompanying molecular ionization found by a new triple coincidence technique. Chem. Phys. 315, 8–16 (2005).
Lefebvre-Brion, H. Lifetimes of Rydberg states in small molecules: Fluorescence, predissociation and autoionization. In The Role of Rydberg States in Spectroscopy and Photochemistry. Understanding Chemical Reactivity Vol. 20 (ed. Sándorfy, C.) (Springer, 1999).
Eland, J. H. D. & Feifel, R. Double ionisation of ICN and BrCN studied by a new photoelectron–photoion coincidence technique. Chem. Phys. 327, 85–90 (2006).
Eland, J. H. D., Feifel, R. & Hochlaf, M. Double photoionization and dication fragmentation of CF3I: Experiment and theory. J. Chem. Phys. 128, 234303 (2008).
Taylor, S., Eland, J. H. D. & Hochlaf, M. Fluorescence from the CS22+ dication: Theory and experiment. Chem. Phys. 330, 16–25 (2006).
Werner, H. et al. MOLPRO, Version 2015.1, a Package of Ab Initio Programs (University of Cardiff Chemistry Consultants, 2015).
Knowles, P. J. & Werner, H.-J. An efficient second-order MC SCF method for long configuration expansions. Chem. Phys. Lett. 115, 259–267 (1985).
Werner, H. J. & Knowles, P. J. A second order multiconfiguration SCF procedure with optimum convergence. J. Chem. Phys. 82, 5053–5063 (1985).
Werner, H. J. & Knowles, P. J. An efficient internally contracted multiconfiguration–reference configuration interaction method. J. Chem. Phys. 89, 5803–5814 (1988).
Knowles, P. J. & Werner, H.-J. An efficient method for the evaluation of coupling coefficients in configuration interaction calculations. Chem. Phys. Lett. 145, 514–522 (1988).
Shamasundar, K. R., Knizia, G. & Werner, H.-J. A new internally contracted multi-reference configuration interaction method. J. Chem. Phys. 135, 054101 (2011).
Kendall, R. A., Dunning, T. H. Jr. & Harrison, R. J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 96, 6796–6806 (1992).
Woon, D. E. Jr. & Dunning, T. H. Jr. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 98, 1358–1371 (1993).
Dunning, T. H. Jr., Peterson, K. A. & Wilson, A. K. Gaussian basis sets for use in correlated molecular calculations. X. The atoms aluminum through argon revisited. J. Chem. Phys. 114, 9244–9253 (2001).
Ben Yaghlane, S. et al. Theoretical spectroscopic investigations of HNSq and HSNq (q = 0, +1, −1) in the gas phase. J. Chem. Phys. 140, 244309 (2014).
Ben Yaghlane, S., Cotton, C. E., Francisco, J. S., Linguerri, R. & Hochlaf, M. Ab initio structural and spectroscopic study of HPSx and HSPx (x = 0,+1,−1) in the gas phase. J. Chem. Phys. 139, 174313 (2013).
Trabelsi, T., Mogren Al-Mogren, M., Hochlaf, M. & Francisco, J. S. Mechanistic study of the photoexcitation, photoconversion, and photodissociation of CS2. J. Chem. Phys. 149, 064304 (2018).
Berning, A., Schweizer, M., Werner, H.-J., Knowles, P. J. & Palmieri, P. Spin-orbit matrix elements for internally contracted multireference configuration interaction wave functions. Mol. Phys. 98, 1823–1833 (2000).
Hampel, C., Peterson, K. A. & Werner, H.-J. A comparison of the efficiency and accuracy of the quadratic configuration interaction (QCISD), coupled cluster (CCSD), and Brueckner coupled cluster (BCCD) methods. Chem. Phys. Lett. 190, 1–12 (1992).
Deegan, M. J. O. & Knowles, P. J. Perturbative corrections to account for triple excitations in closed and open shell coupled cluster theories. Chem. Phys. Lett. 227, 321–326 (1994).
Knowles, P. J., Hampel, C. & Werner, H.-J. Erratum: Coupled cluster theory for high spin, open shell reference wave functions. J. Chem. Phys. 99, 5219 (1993).
Knowles, P. J., Hampel, C. & Werner, H.-J. Erratum: Coupled cluster theory for high spin, open shell reference wave functions. J. Chem. Phys. 112, 3106–3107 (2000).
Acknowledgements
We thank the Swedish Research Council and the Knut and Alice Wallenberg Foundation for financial support. This study was carried out while M.H. was Waernska Guest Professor at the University of Gothenburg. The computations involved the Swedish National Infrastructure for Computing (SNIC) at Chalmers Centre for Computational Science and Engineering (C3SE) partially funded by the Swedish Research Council through Grant No. 2018-05973.
Funding
Open access funding provided by University of Gothenburg.
Author information
Authors and Affiliations
Contributions
J.H.D.E. and R.F. conducted the experimental research. J.H.D.E. and M.W. performed the data analysis. M.J., M.W., S.B.Y. and M.H. carried out the theoretical calculations. All authors discussed the results and contributed to the writing of the manuscript at several instances.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jarraya, M., Wallner, M., Nyman, G. et al. State selective fragmentation of doubly ionized sulphur dioxide. Sci Rep 11, 17137 (2021). https://doi.org/10.1038/s41598-021-96405-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-96405-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
