
Abstract
The Yi is one of fifty-six ethnic populations and one of the most ancient ethnic groups in China. The Liangshan Yi Autonomous Prefecture (LYAP) in Sichuan Province has the single largest Yi community in China. To establish a Yi population database in the LYAP of Sichuan in China, a Goldeneye™ DNA Identification System 20A Kit with 19 autosomal STRs (short tandem repeats) was used. As a result, the total discrimination power (TDP) and the cumulative probability of exclusion (CPE) for these STRs in 1016 unrelated individuals were 0.999999999999999999999897 and 0.9999999597, respectively. Totals of 273 alleles for 19 STRs and 8–22 alleles for each locus were found. The allelic frequencies ranged from 0.0005 to 0.5084. The forensic parameter averages of these STRs were as follows: observed heterozygosity (Hobs) of 78.44%, expected heterozygosity (Hexp) of 79.89%, discrimination power (DP) of 92.66%, and probability of exclusion (PE) of 57.68%. Penta E presented the highest levels of Hobs and DP, whereas TPOX showed the lowest Hobs and DP values. Nei’s standard genetic distance matrix among 31 populations found that the nearest genetic distance to the Yi population was the Sichuan Han (0.0056). Altogether, we first reported the forensic parameters and allele frequencies of 19 autosomal STRs of the Yi group in Liangshan. These 19 STR makers could provide highly informative polymorphisms for individual identification, paternity testing and genetic population analyses.
Similar content being viewed by others

Introduction
The Yi is one of fifty-six ethnic populations and one of the most ancient ethnic groups in China. According to the report of Sixth National Census data in 2010, the Yi is the sixth-largest ethnic minority in China, with a population of 8,714,393. The Liangshan Yi Autonomous Prefecture (LYAP) in Sichuan Province has the single largest Yi community in China with nearly 50% of the 4.5 million inhabitants1. The Honghe and Chuxiong regions in Yunnan Province are the second-largest Yi communities in China. Yi individuals from both Sichuan and Yunnan live in Southwest China. In addition, the Yi ethic group has their own culture and language which belongs to the Loloish language and Sino-Tibetan language close to Burmese2.
Allele frequencies and population data were reported for 9, 15 and 20 autosomal short tandem repeat (STR) loci in the Yi ethnic minority from Yunnan Province of China in 2006 and 2017, respectively3,4,5. Genetic diversity and phylogenetic characteristics obtained from non-combined DNA index system (CODIS) STR markers in the Yi ethnic minority with 95 individuals from Sichuan Province were reported in 20186. Genetic polymorphism studies of the Yi minority in this LYAP area using the Y-STR amplification system have been intensively investigated1,6,7. Moreover, rapidly mutating study for Y-STRs in this Yi population from Sichuan was also reported8. In addition, our group evaluated 23 Y-STR locus mutation rates in Chinese Han father-son pairs from Southwest China9. Forensic characteristics and phylogenetic analyses for 19 X-chromosomal STR loci in the LYAP Yi minority was also reported2. By using insertion/deletion markers, forensic features and population genetic structure of Dong, Yi, Han, and Chuanqing populations in Guizhou of Southwest China were recently determined10. Due to the rich history and complex ethnic makeups in China, it is necessary to study allele frequency for autosomal STRs in different geographic areas and different ethnic groups including Yi Chinese in the LYAP. However, the genetic polymorphisms for autosomal STR loci of the Yi minority in the LYAP are unclear, and the genetic relationships of populations between the Yi minority and other populations are unknown. It is necessary to use autosomal STRs to fill the gap among Y-STRs and X-STRs of this Yi minority. Thus, in this study we focus on establishing a Yi population database from the LYAP in China by using the Goldeneye™ DNA Identification System 20A Kit, which includes 19 autosomal STRs.
Results
Linkage disequilibrium, forensic parameters and allele frequencies
We first performed a linkage disequilibrium (LD) analysis of 19 autosomal STR loci in the Yi group in LYAP. For the LD test, only 3 pairs (Supplementary Table S1) of loci showed significant LD after Bonferroni correction (p = 0.05/171 ≈ 0.0003), indicating that most loci were statistically independent. These few pairs in linkage disequilibrium may be due to random sampling errors because the pairs were located on different autosomal chromosomes or chromosome arms.
The forensic parameters, allelic frequencies and p-values of exact tests for Hardy–Weinberg equilibrium (HWE) of 19 autosomal STR loci in the Yi group in Liangshan are presented in Table 1 and Supplementary Table S2. In the present study, eighteen of the 19 STR loci were observed to show HWE after Bonferroni correction (p = 0.05/19 ≈ 0.0026), and only the D21S11 locus was a significant HWE departure. Individual migration or genetic exchange may be a possible explanation for this deviation. The total discrimination power (TDP) and the cumulative probability of exclusion (CPE) of the 19 STRs in 1016 unrelated individuals were 0.999999999999999999999897 and 0.9999999597, respectively.
A total of 273 alleles for 19 STRs with corresponding allelic frequencies from 0.0005 to 0.5084 were noticed, and 8–22 alleles for each locus were found (Supplementary Table S2). The forensic parameter averages of these STRs were as follows: observed heterozygosity (Hobs) of 78.44%, expected heterozygosity (Hexp) of 79.89%, discrimination power (DP) of 92.66%, and probability of exclusion (PE) of 57.68%. Locus Penta E presented the highest levels of Hobs (0.8917) and DP (0.9839), whereas TPOX showed the lowest Hobs of 0.6230 and DP of 0.8154.
Population comparisons and population differentiation
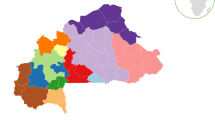
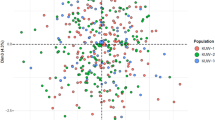
Nei’s standard genetic distance matrix among 31 populations (Fig. 1, Supplementary Table S3) is presented in Table 2. From Table 2, we found that the nearest genetic distance to the Yi population was the Sichuan Han (0.0056), followed by the Anhui Han (0.0058) and the Guangdong Han (0.0059). The Euclidean distance model of a multidimensional scale (MDS) and a representation plot are indicated in Fig. 2. Eighteen Han origin groups (except Guizhou Han), Chengde Manchu and Liaoning Hui are closely located together and stand far apart from Yunnan Miao, Xinjiang Kazakh, Xinjiang Uyghur, Gansu Hui, Xinjiang Mongolian, Inner Mongolia Mongolian, Dongbei Korean, Yunnan Yi and Hainan Li ethnic populations. The neighbour joining (NJ) phylogenetic tree (Fig. 3) of Nei’s standard distance matrix showed that Yunnan Han, Sichuan Han, Guizhou Han, Chongqing Han, Hunan Han, Fujian Xiamen Han, Guangdong Han, Zhejiang Han, Ningbo Han, Hubei Han, Shanghai Han, Anhui Han, and Jiangsu Han, are formed as a southern China Han population. Hebei Han, Heilongjiang Han, Shanxi Han, Shandong Han, Liaoning Han, and Beijing Han are formed as a northern China Han population. Xinjiang Uyghur, Xinjiang Mongolian, Xinjiang Kazakh and Inner Mongolia Mongolian ethnic minorities presented a relatively close but distant relationship to the Han groups. Chengde Manchu, Dongbei Korean, Gansu Hui, and Liaoning Hui ethnic minorities were relatively close to the Northern China Han population. The Yunnan Han and Yunnan Miao ethnic minorities clustered a unique cluster and were relatively obviously distant from the Southwest China Han population. The Yunnan Yi and Hainan Li ethnic minorities clustered a unique cluster. Yunnan Miao showed an obvious distant relationship from the other groups. Thus, the NJ phylogenetic tree and MDS corresponded with the cultural, historical, and geographical distribution of the studied majority groups.

Map showing the geographic positions of the studied Yi ethnic minority in Liangshan Yi Autonomous Prefecture of Sichuan and the 30 reference populations. The sample numbers are presented at the bottom left of the figure. The longitude and latitude of all 31 reference populations are presented in Supplementary Table S3. The R project software (version 4.0.4) (https://www.r-project.org/) and DATAV GeoAtlas (areas_v3) (https://datav.aliyun.com/tools/atlas/index.html) were used to create this map.

Multidimensional scaling (MDS) plots displaying the genetic relationships between the Yi ethnic minority and 30 reference populations.

Phylogenetic tree displaying the genetic relationships between the population and 30 reference populations. The phylogenetic tree was constructed using the neighbour-joining method based on 19 overlapping STR loci with MEGA-X software.
The Fst values and corresponding p values of the population comparisons between the Liangshan Yi population and the other 30 compared populations at 19 STR loci are shown in Supplementary Table S4. Yunnan Miao showed significant genetic differences with Liangshan Yi at seventeen STR loci and no significant differences were observed with the exception of Yunnan Miao at the locus of D5S818 and Penta D. The calculation revealed that there were statistically significant differences between Liangshan Yi and Xinjiang Mongolian at twelve STR loci, followed by Xinjiang Kazakh at nine STR loci, Yunnan Yi at six STR loci, Hainan Li at five STR loci, Zhejiang Han, Yunnan Han and Fujian Xiamen Han at two STR loci, and among the Dongbei Korean, Xinjiang Uyghur, Sichuan Han, Guizhou Han, Guangdong Han, Ningbo Han, Jiangsu Han, Anhui Han, Hubei Han, Hebei Han, Shanxi Han and Heilongjiang Han at one locus after Bonferroni correction (p < 0.0003). However, no statistically significant differentiation was obtained for the Gansu Hui, Liaoning Hui, Chengde Manchu, Inner Mongolia Mongolian, Chongqing Han, Shanghai Han, Hunan Han, Beijing Han, Shandong Han or Liaoning Han at any of the 19 STR loci.
Discussion
China is currently populated by 1,443,497,378 people who belong to at least 56 officially recognized linguistically and ethnically different Chinese groups, according to China’s Seventh National Census Bulletin (http://www.stats.gov.cn/tjsj/tjgb/rkpcgb/) published on May 11, 2021. Compared with the China’s Sixth National Census in 2010, the Han population increased by 60,378,693 (4.93%) and the ethnic minorities increased by 11,675,179 (10.26%) (http://www.stats.gov.cn/tjsj/tjgb/rkpcgb/qgrkpcgb/202106/t20210628_1818821.html), showing a more than twofold increase for ethnic minorities. Genetic studies of Chinese populations from ethnic minorities are of great interest due to China’s complex demographics, complex geographical characteristics and large population sizes. The Liangshan Yi Autonomous Prefecture (LYAP) in Sichuan Province has the single largest Yi community. However, the genetic polymorphisms responsible for the autosomal STR loci of the Yi minority in LYAP are unclear, and the genetic relationships between the Yi minority population and other ethnic populations are unknown.
In this study, by recruiting 1016 unrelated individuals, the total discrimination power (TDP) and the cumulative probability of exclusion (CPE) for 19 STRs were found to be 0.999999999999999999999897 and 0.9999999597, respectively. Totals of 273 alleles for 19 STRs and 8–22 alleles for each locus were found. The observed heterozygosity (Hobs) was 78.44%, the expected heterozygosity (Hexp) was 79.89%, the discrimination power (DP) was 92.66%, and the probability of exclusion (PE) was 57.68%. Penta E presented the highest levels of Hobs (0.8917) and DP (0.9839), whereas TPOX showed the lowest Hobs of 0.6230 and DP of 0.8154. Nei’s standard genetic distance matrix among 31 populations found that the nearest genetic distance to the Yi population was the Sichuan Han (0.0056), followed by the Anhui Han (0.0058) and Guangdong Han (0.0059). Nei’s standard genetic distance of the Yi population in Sichuan Province to the Yi population in Yunnan Province was 0.0189, showing the 24th nearest genetic distance. Surprisingly, Yunnan Miao showed an obvious distant relationship from other groups, which was consistent with other analyses11, showing far distance to Filipinos from the Philippines. Altogether, the NJ phylogenetic tree and MDS corresponded with the cultural, historical, and geographical distribution of the studied majority groups.
The Fst values and corresponding p values of the population comparisons between the Liangshan Yi population and the other 30 compared populations indicated that no statistically significant differentiation was obtained with Gansu Hui, Liaoning Hui, Chengde Manchu, Inner Mongolia Mongolian, Chongqing Han, Shanghai Han, Hunan Han, Beijing Han, Shandong Han or Liaoning Han at any of the 19 STR loci. The exploration of the origin and diversification of ethnic populations may be affected by the natural environment, cultural background, population migration and national policies.
In conclusion, our work was the first to report the forensic parameters and allele frequencies of 19 autosomal STR markers of the Yi group in Liangshan, China, and the population genetic relationships between the Yi minority and 30 other neighbouring populations. These 19 STR makers could provide highly informative polymorphisms for individual identification, paternity testing and genetic population analyses.
Methods
Sample collections and genomic DNA isolation
Bloods from 1016 unrelated healthy Yi individuals were recruited from Xichang City and its surrounding areas of the LYAP, Sichuan Province, P. R. China. Chelex-100 protocol was used to extract DNA12,13. The study was approved by the institutional review boards of Southwest Medical University. Informed consent from participants and ethical approval from the Committee of Southwest Medical University were obtained. All experiments were performed in accordance with relevant guidelines and regulations.
PCR amplification and STR genotyping
In this study, the Goldeneye™ DNA Identification System 20A Kit from Beijing in China was used, which included 19 autosomal STRs, namely D2S1338, D3S1358, D5S818, D6S1043, D7S820, D8S1179, D12S391, D13S317, D16S539, D18S51, D19S433, D21S11, CSF1PO, Penta D, vWA, TPOX, Penta E, TH01, and FGA.
PCRs were generated according to the manufacturer's instructions. For details, a 10 μL PCR volume was used for each sample, including 2.5 × PCR Buffer III 4.0 μL, 5 × 20A Primer Mix 2.0 μL, Taq DNA Polymerase III 0.16 μL and ddH2O 3.84 μL. PCR amplification was conducted using the Goldeneye™ DNA ID System 20A Kit from Peoplespot (Cat#: 20AMC401, Beijing, China) in an Applied Biosystems Veriti® 96-Well Thermal Cycler (Applied Biosystems, Life Technology, USA) with the following steps: pre-denatured 96 °C for 2 min, followed by 30 cycles of 94 °C for 5 s and 60 °C for 70 s, then a step of 60 °C for 30 min, and finally held at 15 °C until for capillary electrophoresis. The PCR products were genotyped by using the 3500 Dx Genetic Analyzer (Applied Biosystems, Life Technology, USA) and analyzed by GeneMapper ID-X software (Thermo Fisher, USA)14. For quality, DNA as a control was also supplied in this kit. Genotype analysis for STR was performed by reference to the provisions of the ISO/IEC 17025:2005 General Requirements for the Competence of Testing, the Specification for Parentage Testing by China (GB/T 37223-2018).
Data analysis
Modified-PowerStats software was used to evaluate forensic parameters, allele frequencies and Hardy–Weinberg equilibrium (HWE). The Arlequin v3.5 software was used to analyze linkage disequilibrium (LD) and population differentiation between the studied group and previously published data15. Nineteen STR information from 30 Chinese populations, including one Manchu16, two Hui17,18, one Kazakh19, one Uyghur20, one Miao21, one Yi3, one Li11, one Korean22, two Mongolian23,24 and 19 Han25,26,27,28,29,30,31,32,33,34 populations (Fig. 1), were extracted from other studies to evaluate the genetic similarities and divergences. The longitude and latitude of all 31 reference populations are presented in Supplementary Table S3. A modified PHYLIP program was used to calculate Nei’s standard genetic distance between Yi populations and the 30 other populations reported in previous studies35. MEGA-X software was used to develop and visualize the phylogenetic tree36.
Compliance with ethical standards
This study was approved by Southwest Medical University (KY2021168).
Data availability
All data used for the analyses in this report are available from the corresponding author on reasonable request.
References
He, G. et al. Genetic polymorphism investigation of the Chinese Yi minority using PowerPlex(R) Y23 STR amplification system. Int. J. Legal Med. 131, 663–666. https://doi.org/10.1007/s00414-017-1537-2 (2017).
He, G. et al. Forensic characteristics and phylogenetic analyses of the Chinese Yi population via 19 X-chromosomal STR loci. Int. J. Legal Med. 131, 1243–1246. https://doi.org/10.1007/s00414-017-1563-0 (2017).
Zhang, X. et al. Population data for 20 autosomal STR loci in the Yi ethnic minority from Yunnan Province, Southwest China. Forensic Sci. Int. Genet. 28, e43–e44. https://doi.org/10.1016/j.fsigen.2017.02.017 (2017).
Li, J. et al. Allele frequencies of 15 autosomal STR loci in the Yi ethnic minority from Yunnan Province in the far southwest of China. Forensic Sci. Int. Genet. 26, e23–e24. https://doi.org/10.1016/j.fsigen.2016.10.013 (2017).
Gao, Y., Jin, T. B., Lai, J. H., Zheng, H. B. & Li, S. B. [The genetic polymorphism of 9 short tandem repeat loci in Yi ethnic group of Yunnan in China]. Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics 23, 216–218 (2006).
He, G. et al. Genetic diversity and phylogenetic characteristics of Chinese Tibetan and Yi minority ethnic groups revealed by non-CODIS STR markers. Sci. Rep. 8, 5895. https://doi.org/10.1038/s41598-018-24291-5 (2018).
Wang, F. et al. Genetic reconstruction and phylogenetic analysis by 193 Y-SNPs and 27 Y-STRs in a Chinese Yi ethnic group. Electrophoresis https://doi.org/10.1002/elps.202100003 (2021).
Wang, Q. et al. Rapidly mutating Y-STRs study in Chinese Yi population. Int. J. Legal Med. 133, 45–50. https://doi.org/10.1007/s00414-018-1894-5 (2019).
Fu, J. et al. Assessing 23 Y-STR loci mutation rates in Chinese Han father-son pairs from southwestern China. Mol. Biol. Rep. 47, 7755–7760. https://doi.org/10.1007/s11033-020-05851-3 (2020).
Liu, Y. et al. Forensic features and population genetic structure of Dong, Yi, Han, and Chuanqing human populations in Southwest China inferred from insertion/deletion markers. Front. Genet. 11, 360. https://doi.org/10.3389/fgene.2020.00360 (2020).
Fan, H. et al. Population data of 19 autosomal STR loci in the Li population from Hainan province in southernmost China. Int. J. Legal Med. 133, 429–431. https://doi.org/10.1007/s00414-018-1828-2 (2019).
Walsh, P. S., Metzger, D. A. & Higuchi, R. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10, 506–513 (1991).
Fu, J. et al. Technical note: Multi-alleles at the DYS385ab locus with high frequency in a Han Chinese population from southwestern China. Int. J. Legal Med. https://doi.org/10.1007/s00414-021-02592-1 (2021).
Fu, J. et al. Evaluation genotypes of cancer cell lines HCC1954 and SiHa by short tandem repeat (STR) analysis and DNA sequencing. Mol. Biol. Rep. 45, 2689–2695. https://doi.org/10.1007/s11033-018-4438-7 (2018).
Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resourc. 10, 564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x (2010).
Zhao, L. et al. Genetic polymorphisms of 19 STR loci in Chengde Manchu Population from Hebei Province. Chin. J. Forensic Med. 31, 72–73 (2016).
Hu, S., Zhao, W. & Wang, T. Genetic polymorphisms of 19 STR loci in Pingliang Hui population from Gansu Province. Chin. J. Forensic Med. 30, 620–621 (2015).
Shen, H. et al. Genetic polymorphisms of 19 STR loci in Hui population in Liaoning province. Chin. J. Forensic Med. 28, 504–505 (2013).
Zhang, L. et al. Genetic polymorphism of 19 STR loci in Xinjiang Barkol Kazakh population. J. Central South Univ. Med. Sci. 37, 934–938. https://doi.org/10.3969/j.issn.1672-7347.2012.09.014 (2012).
Tursun, N., Gulinar, K. & Zhang, L. Genetic polymorphisms of 20 STR loci in the Xinjiang Uyghur population. Chin. J. Forensic Med. 27, 138–140 (2012).
Zhang, X. et al. Genetic analysis of 20 autosomal STR loci in the Miao ethnic group from Yunnan Province, Southwest China. Forensic Sci. Int. Genet. 28, e28–e29. https://doi.org/10.1016/j.fsigen.2017.02.003 (2017).
Liu, L., Song, H. & Jiang, X. Genetic polymorphism of 23 autosomal STR loci in Korean population from Dongbei. Chin. J. Forensic Med. 30, 612–614 (2015).
Wei, Y. Y. et al. Genetic distribution and forensic evaluation of multiplex autosomal short tandem repeats in the Chinese Xinjiang Mongolian group. J. Zhejiang Univ. Sci. B 20, 287–290 (2019).
Si, Y. et al. Genetic polymorphisms of 20 STR loci in Mongolian population from InnerMongolia. Chin. J. Forensic Med. 33, 299–301 (2018).
Zou, X. et al. Genetic polymorphisms for 19 autosomal STR loci of Chongqing Han ethnicity and phylogenetic structure exploration among 28 Chinese populations. Int. J. Legal Med. 131, 1539–1542. https://doi.org/10.1007/s00414-017-1577-7 (2017).
Ruan, X. et al. Genetic variability and phylogenetic analysis of 39 short tandem repeat loci in Beijing Han population. Hereditas 37, 683–691. https://doi.org/10.16288/j.yczz.15-025 (2015).
Tong, D. et al. Polymorphism analysis and evaluation of 19 STR loci in the Han population of Southern China. Ann. Hum. Biol. 40, 191–196. https://doi.org/10.3109/03014460.2012.750685 (2013).
Sun, L. et al. Analysis of genetic polymorphisms and mutations at 19 STR loci in Hebei Han population. Forensic Sci. Int. Genet. 31, e50–e51. https://doi.org/10.1016/j.fsigen.2017.07.016 (2017).
Xiao, C., Zhang, W., Wei, T., Pan, C. & Huang, D. Population data of 21 autosomal STR loci in Chinese Han population from Hubei province in Central China. Forensic Sci. Int. Genet. 20, e13–e14. https://doi.org/10.1016/j.fsigen.2015.11.002 (2016).
Yin, C. et al. Analysis of 19 STR loci reveals genetic characteristic of eastern Chinese Han population. Forensic Sci. Int. Genet. 14, 108–109. https://doi.org/10.1016/j.fsigen.2014.09.015 (2015).
Zhang, M. X. et al. Genetic polymorphisms of 19 STR loci in Shandong Han population. Fa Yi Xue Za Zhi 29, 440–443 (2013).
Xie, J. et al. Genetic distribution on 20 STR loci from the Han population in Shanghai, China. Forensic Sci. Int. Genet. 9, e30-31. https://doi.org/10.1016/j.fsigen.2013.08.007 (2014).
Xiang, C. et al. Genetic variation of the 20 STRs in Han population from Yunnan-Kweichow plateau of China. Forensic Sci. Int. Genet. 23, e10–e11. https://doi.org/10.1016/j.fsigen.2016.03.003 (2016).
He, G. et al. Genetic diversity of 21 autosomal STR loci in the Han population from Sichuan province, Southwest China. Forensic Sci. Int. Genet. 31, e33–e35. https://doi.org/10.1016/j.fsigen.2017.07.006 (2017).
Shimada, M. K. & Nishida, T. A modification of the PHYLIP program: A solution for the redundant cluster problem, and an implementation of an automatic bootstrapping on trees inferred from original data. Mol. Phylogenet. Evol. 109, 409–414. https://doi.org/10.1016/j.ympev.2017.02.012 (2017).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. https://doi.org/10.1093/molbev/msw054 (2016).
Acknowledgements
This work was funded by the Special Training Program for Young Science and Technology Talents from Southwest Medical University (No. 00031726), the Joint Research Foundation of Luzhou City and Southwest Medical University (No. 2018LZXNYD-YL01), and the Translational Medicine Foundation of Southwest Medical University (Nos. 00031476 and 00031477), and was partially funded by the National Natural Science Foundation of China (Nos. 81672887, 31701087, and 30371493).
Author information
Authors and Affiliations
Contributions
J.F. conceptualized and designed the project. Ji.F., J.C., B.S. and X.Z. performed the experiments, interpreted the results. B.S. and J.C. analysed the data. J.F. wrote the manuscript. J.F. and T.H. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cheng, J., Song, B., Fu, J. et al. Genetic polymorphism of 19 autosomal STR loci in the Yi ethnic minority of Liangshan Yi autonomous prefecture from Sichuan province in China. Sci Rep 11, 16327 (2021). https://doi.org/10.1038/s41598-021-95883-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-95883-x
This article is cited by
-
Comorbidity and drug resistance of smear-positive pulmonary tuberculosis patients in the yi autonomous prefecture of China: a cross-sectional study
BMC Infectious Diseases (2023)
-
Novel, heterozygous, de novo pathogenic variant (c.4963delA: p.Thr1656Glnfs*42) of the NF1 gene in a Chinese family with neurofibromatosis type 1
BMC Medical Genomics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
