
Abstract
The microbial diversity and quantitative dynamics during the insect’s development stages constitute recently developed putative tools in forensic and medical studies. Meanwhile, little is known on the role of insects in spreading foodborne pathogenic bacteria and on the impact of these pathogens on the overall insects and feeding substrate microbiome composition. Here, we provide the first characterization of the bacterial communities harbored in adult and immature stages of Lucilia sericata, one of the first colonizers of decomposed human remains, in the presence of the foodborne pathogen Salmonella enterica using 16S rRNA Illumina sequencing and qPCR. The pathogen transmission from the wild adults to the second generation was observed, with a 101.25× quantitative increase. The microbial patterns from both insect and liver samples were not influenced by the artificial introduction of this pathogenic foodborne bacteria, being dominated by Firmicutes and Proteobacteria. Overall, our results provided a first detailed overview of the insect and decomposed substrate microbiome in the presence of a human pathogen, advancing the knowledge on the role of microbes as postmortem interval estimators and the transmission of pathogenic bacteria.
Similar content being viewed by others
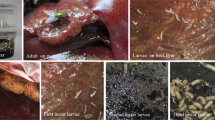
Introduction
The postmortem interval (PMI) estimation represents a key-step in forensic investigations, in the case of a suspicious death or a homicide by providing the time window in which the death occurred1. Most of the time forensic entomology comes to help identify this window, as necrophagous insect species have been used for many years in the court of law as scientific evidence for the PMI estimation2, arriving on carcasses minutes after death to feed and breed on the decaying substrate3,4,5. Frequently, the estimation of the time elapsed since death is based on the immature stages rate of development, though, information is also provided by the chronological succession of species/families6. Blowflies, including Lucilia sericata (Meigen, 1826) (Diptera: Calliphoridae) require this decomposed organic matter substrate for oogenesis and larvae development, as the best survival rates were recorded from such unsterilized mixed bacterial environments7. The importance that the study of insect-microbiome interactions has for the PMI estimation relies in the possibility of identifying common and constant insect-associated bacteria and quantifying their transfer in the colonized tissues (such as a human cadaver). In cases where the body is no longer associated with insects, the presence of insect-specific bacteria can suggest that the body had been colonized by insects, and by quantifying these bacteria through the insect life stages, a link with the development stages can be specified, thus, narrowing the estimation of PMI.
Meanwhile, as flies feed and breed, they acquire a part of the microbiome of the living environment8. As such, blowflies that feed on decomposed human or animal remains are responsible for transmitting pathogens, most species being vectors for important bacteria, such as Streptococcus, Staphylococcus, and Clostridium9. Several studies emphasized the high dispersion ability of blowflies of spreading foodborne pathogens such as various Salmonella serotypes10, Escherichia coli11, Campylobacter species12, and Helicobacter pylori13. The probability of spreading pathogenic bacteria in a human living environment is very high, given the blowflies synanthropic character7. Flies also transfer a part of their own microbiome to their associated environments via regurgitation and defecation14. Thus, the human environment can become indirectly contaminated through the activity of flies15. While the defecation involves a smaller risk than regurgitation, mainly because the gastro-intestinal tract is a hostile environment for non-specific bacteria, the microbial content from the saliva once eliminated, represents a continuous source of pathogens16. However, blowflies can also modify the living substrate microbiome by eliminating substances that have antimicrobial properties, such as defensins and DNase17,18.
Earlier studies demonstrated that bacteria are regulating the blowfly attraction and colonization activities19,20, as substrates associated with Proteus mirabilis were more attractive to flies4. At the opposite pole, several bacterial taxa such us Staphylococcus or Bacillus species21 can inhibit the larval growth. In spite of a wealth of information on the vector capacity of flies9, very little is known about their role in the environmental bacterial spread, as well as in the bacterial transfer throughout the development cycle of various insect species. As microbes rely on insects for spatial and temporal dispersion, all pathogenic bacteria have many more opportunities to be transferred into the environment given the adults capacity of traveling long distances7. Therefore, the microbiome pattern along insect-living environment interactions could lead to a better understanding of the role of flies as main colonizers of decomposed carcasses and as vectors of pathogenic bacteria.
The decomposition process is dependent on many factors, from the cause of death to the environmental factors’ variation, involving both vertebrate and invertebrate scavengers, hosting a poorly understood microbial successional assembly. The decomposing bacterial communities are highly diverse and competitive in terms of acquiring substrate resources22. During the last decade, forensic microbiology studies were initiated in search of microbial patterns useful for the PMI estimation23,24,25. These studies targeted the microbiome associated with the decomposition of human remains26,27, swine and mouse carcasses28,29,30, different decayed organs31,32, and different necrophagous insect species life stages11,33,34. Investigations of both cultured and uncultured bacterial communities showed similar dynamics patterns in animal and humans27, revealing the importance of using animal models in this field. Several molecular methods of identification were used for the microbiome investigation of different Diptera species, starting with MiSeq sequencing of V4 region33, 16S rRNA sequencing11,35, 16S rRNA 454 pyrosequencing36, and whole shot gun sequencing13. All these experimental models helped strengthen the current knowledge on microbes and decomposition. However, these studies focused either on the decomposed tissues necrobiome22 or the insects microbiome33.
Among blowflies, L. sericata is the most common species used in both forensic and medicinal studies, being one of the earliest colonizers of decomposed remains, and a common species used in maggot debridement therapy (MDT)36. This blowfly has a cosmopolitan distribution, from North, Central and South America, Africa, Europe, to Australia37. Considering these aspects, this species was used as the experimental model in our current research, because the resulted data could be used as reference mean worldwide.
In the current experimental research, L. sericata feeding substrate was inoculated with the human pathogen Salmonella enterica subsp. enterica serovar Typhimurium ATCC 14028. This facultative anaerobic mesophilic bacterium, was reported as highly stable to various stress factors including low pH, osmotic stress, low and high temperatures (2–54 °C), and low water activity, being one of the main foodborne pathogens worldwide38. According to a previous study39, Salmonella caused the highest number of deaths in the USA, as the main human foodborne pathogen costing $2.71 billion for more than one million people. In Europe, over 91,000 cases are reported each year, with involved medical costs of over €3 billion a year, being the second most commonly reported gastrointestinal infection40. Most of Salmonella serovars were found in the gastrointestinal tract of humans and animals, being eliminated by feces and further transported by insects or other animals to other environments41.
In this context, our data aimed to provide novel information on pathogen transmission and bacteria as putative indicators of body colonization by insects, in their absence, by answering specific questions: (a) is L. sericata microbiome affected by the presence of a foodborne pathogen in the feeding environment? (b) is the foodborne pathogen transmitted to the new blowfly generation, and if yes, can it be held responsible for future surface/food contamination in a human living environment? (c) what bacteria are associated with the developmental stages of the insect and decomposed liver? and (d) can the fly-associated microbiome provide data for the PMI estimation?
Results
Quantitative data on Salmonella enterica dynamics in Lucilia sericata and liver samples
L. sericata adults were collected and fed on swine liver inoculated with low (102 CFU/ml) and high (104 CFU/ml) concentrations of S. enterica in order to monitor the pathogen transmission throughout the insect life stages. No S. enterica could be identified from the wild L. sericata adults collected, nor from the reference specimens that were fed with non-inoculated liver, during the assigned experimental time frame.
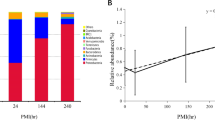
After being fed for three hours with the inoculated liver, L. sericata adults presented similar values for the low (102.59) and high (102.81) S. enterica concentrations (Fig. 1a). Following, the abundance of S. enterica increased for the first instar larvae (103.79, 104.60), being relatively constant during the second instar larvae stage. The maximum bacterial abundance was recorded for the third instar larvae (105.15), exhibiting 102.34× higher values than the initial adult concentrations. The pupal stage was marked by a slightly descending trend, with maximum and minimum recorded values of 104.33 and 102.85, respectively. S. enterica was also identified in the newly emerged adults, with values ranging between 103.60 and 103.84. The bacterial concentration in the new generation was 101.03× higher than the initially assimilated one.

Relative abundance of Salmonella enterica throughout (a) Lucilia sericata life stages (A adult, LI first instar larvae, LII second instar larvae, LIII third instar larvae, P pupa, T teneral specimen) and from (b) decomposed liver. Standard deviations are indicated by error bars (n = 3).
Regarding the differences between the specimens fed with low and high S. enterica inoculated liver, the most noticeable difference was recorded for the third instar larvae, with 10–1.21× lower concentration in the larvae fed with the low inoculated liver. S. enterica content from the low and high initial inoculations was very similar in value between several samples, such as the adult (102.59, 102.81), third instar larvae (103.59, 103.74), pupa (103.53, 103.55) and teneral specimens (103.60, 103.84) (Fig. 1a). Moreover, the specimens fed with both concentrations showed a similar dynamic, namely an ascending trend up to the third larval stage and then a descending slope to the new generation.
No S. enterica could be identified in the fresh liver samples, as in the case of the wild L. sericata adults. The pathogen content after the first day of inoculation increased up to 103.79 and 104.38 (Fig. 1b). Subsequently the trend was slightly ascendent, reaching a maximum value of 105.33 in the last experimental day. The increase in bacterial abundance of 104.65 (Fig. 1b) corresponded to an increased value in the third larvae stage of 105.15. The liver samples were collected and analyzed as long as the larvae fed on it. Therefore, there are fewer liver sampling points than insect sampling points. Nevertheless, the liver samples may have undergone changes in S. enterica concentration due to the actively feeding larvae and the progress of decomposition.
Bacterial community diversity and profile throughout Lucilia sericata life stages and from the liver associated-samples
The bacterial diversity and composition were investigated and found to be specific and clearly delimited for L. sericata life stages, represented by the adult, larvae stages, pupa and teneral specimens. Overall, there were 14 phyla, 117 orders, and 274 genera identified for the insect samples, while the liver tissue bacterial diversity contained 28 phyla, 113 orders and 183 genera.
Firmicutes constituted the dominant phyla present in the adult (91.08 ± 5.08%) and larvae (93.47 ± 8.69%) stages, presenting lower relative abundance in the pupa (11.33 ± 10.18%) and teneral (6.17 ± 7.04%) specimens (Fig. 2a). On the other hand, the insect samples presented a greater bacterial diversity during the pupae and teneral stages, with Proteobacteria (67.13 ± 11.29%) prevailing in the pupa, and Actinobacteria (67.51 ± 16.59%) in the teneral specimens, respectively. Bacteroidetes exhibited lower values, being the most abundant in the pupa stage (16.32 ± 7.19%) (Fig. 2a).

Bacterial diversity composition of Lucilia sericata life stages: (a) Phylum; (b) Class (sample symbols, except control: “–” specimens fed with 102 S. enterica concentration; “P” specimens fed with 104 S. enterica concentration).
Among the most representative classes, Bacilli dominated in the adult and larvae stages (92.81 ± 7.92%), with Lactobacillales as the prevalent order (72.54 ± 14.71%) (Figs. 2b, 3a). The pupa stage corresponded with an increase in the Gammaproteobacteria representatives up to 63.16 ± 9.73% (Fig. 2b), while presenting the highest diversity among the insect samples at the order level (Fig. 3a). Actinobacteria (67.51 ± 16.59%) represented by Corynebacteriales (66 ± 16.04%) prevailed in the teneral specimens (Figs. 2b, 3a).

Bacterial diversity composition of Lucilia sericata life stages: (a) Order; (b) Family (sample symbols, except control: “–” specimens fed with 102 S. enterica concentration; “P” specimens fed with 104 S. enterica concentration).
Out of a total of 155 identified families, Lactobacillaceae and Corynebacteriaceae had the highest relative abundance values (Fig. 3b), while Lactobacillus (72.54 ± 14.71%), Acinetobacter (20.05 ± 13.96%), Proteus (18.63 ± 10.69%) and Myroides (16.19 ± 14.05%) were among the most abundant genera detected in all analyzed insect samples.
No significant differences in the bacterial communities’ diversity were observed in the untreated and treated liver samples with S. enterica, while variable community structure was visible during the decomposition process being time dependent (Fig. 4a). The fresh liver samples used as control showed a completely different bacterial community as compared to the decomposed liver tissues (Fig. 4a). While in most samples, Firmicutes constitute the dominant phyla, in the control tissues Actinobacteria had the highest relative abundance (52.81 ± 24.11%), followed by Proteobacteria (30.69 ± 4.75%) (Fig. 4a). During liver decomposition, Firmicutes relative content increased from 83.72 ± 14.70% to 91.32 ± 9.63%, while Proteobacteria registered lower abundances in the first 3 days of decay, recording an average value of 29.09 ± 12.40% (Fig. 4a). A similar pattern was registered at the Order level, Bacilli being the most representative in most samples (86.61 ± 13.45%), except for the control liver tissue (Fig. 4b). Corynebacteriales represented by Corynebacteriaceae (50.86 ± 26.37%) were dominant in the untreated liver, while Lactobacillales represented by Lactobacillaceae (66.84 ± 21.32%) showed the highest relative content in all treated samples (Fig. 5a, b). At the family level, the highest diversity registered during the first experimental days comprised Lactobacillaceae, Enterobacteriaceae, Streptococcaceae, Hafniacea, Leuconostocaceae, i.eg. (Fig. 5b), with Lactobacillus (60.94 ± 27.75%) dominating the bacterial community from decomposed liver samples, while Lawsonella (50.55 ± 26%) was dominant in the untreated liver.

Bacterial diversity composition for the decomposed liver samples: (a) Phylum; (b) Class (sample symbols, except control: “–” liver inoculated with 102 S. enterica concentration; “P” liver inoculated with 104 S. enterica concentration).

Bacterial diversity composition for the decomposed liver samples: (a) Order; (b) Family (sample symbols, except control: “–” liver inoculated with 102 S. enterica concentration; “P” liver inoculated with 104 S. enterica concentration).
Regarding the genera consistence within the investigated samples, Lactobacillus was prevalent in both insect (95.24%) and liver (100%) samples (Fig. 6). Among the other common genera identified (Fig. 7), Weissella had different prevalence values, being 29% higher in the liver tissues, representing the second most prevalent genus of this sample type (Fig. 6). Furthermore, Citrobacter, Acinetobacter, and Vagococcus were the among the prevalent genera detected in both insect and liver samples.

Prevalence of main bacterial genera in insect and liver associated samples. Prevalence was calculated by counting the relative abundance > 0.001% of each individual genus from the total number of samples.

Venn diagram of the bacterial taxa of L. sericata and liver tissues. The number of distinct and shared bacterial genera between adult, larva, pupa and teneral insect and liver samples, regardless of S. enterica treatment were indicated.
Among the total of 457 identified genera, eight were shared by all sample types, from which the most abundant taxa were represented by Myroides, Kurthia, Lawsonella, Acinetobacter, and Lactobacillus, while the pupa and the liver samples presented the highest number of distinct genera (Fig. 7). When testing for a differential prevalence of bacterial genera, 54 genera were differentially represented between liver and insect samples. Seven genera prevailed in the liver samples, including Enterobacter, Aeromonas, Lelliotia, Serratia, Citrobacter, Hafnia Obesumbacterium and an Enterobacteriaceae, while 47 genera prevailed in insect samples, including Comamonas, Lawsonella, Stenotrophomonas, Pseudochrobactrum, Providencia and Morganella among others.
The Shannon alpha diversity index showed no significant variation among the insect samples subject to Salmonella feeding treatment and the samples of insects fed with non-inoculated liver (Fig. 8a). The pupa stage displayed significantly higher values in terms of richness, Shannon index and evenness, while the teneral stage displayed the lowest values of richness and Shannon index (Fig. 8a). Moreover, the presence of S. enterica in the feeding substrate did not affect the overall microbiome diversity of the insect specimens, as the two-way ANOVA revealed that the pathogen concentration accounts for < 0.1% of the total variance, while time represented by the development stages (adult, larva, pupa and teneral specimens) accounts for 66.97% (p < 0.0001) of the total variance. On the other hand, for the liver samples collected immediately after being purchased the microbiome diversity was the lowest, according to the Shannon index, while the overall diversity observed between treatments showed slight differences (Fig. 8b). The concentration interactions given by the Salmonella treatment accounted for 30.08% (p = 0.0156), while time accounted for 31.67% (p = 0.0127) of the total variance, suggesting that the diversity differences recorded are likely due in part to the pathogens’ presence but also to the decomposition process over time. Furthermore, the alpha diversity for the insect samples had a slightly higher range compared to the liver samples (Fig. 8a,b).

Alpha diversity represented by the Shannon’s diversity index for: (a) insect associate samples; and (b) liver associated samples. PCoA based on weighted UniFrac distances for (c) insect; and (d) liver groups.
The PCoA based on weighted UniFrac distances was used to further investigate and explain the grouping of samples according to Salmonella treatment. In the insect case, the PCoA explained only 88.3% of the variance on the two axes (A1 and A2), where R was − 0.026 and p = 0.966 (Fig. 8c). The PCoA results were similar for the liver associated samples, where the analysis explained 89.4% of the variance, at an R value of 0.256 and p = 0.002 (Fig. 8d).
Discussion
Salmonella enterica was introduced in the experimental model to observe whether or not this foodborne pathogenic bacterium can cause changes in the general structure of the microbiome during L. sericata development, as well as in the inoculated substrate, and whether or not these quantitative and qualitative changes would affect a possible estimation of the insect development stage. At the same time, the pathogen transfer during fly development was investigated. Given the interaction of blowflies with a large number of bacterial species they are ideal candidates for the study of fly-bacteria interactions. Our study involving L. sericata and S. enterica helps to understand these interactions in the wild, and their forensic and medical repercussions, by providing data on the bacterial specificity for certain insect life stages that can be used to track their transfer in the colonized tissues. By identifying bacteria-life stages specificity the development stage of the insect along with the body decomposition can be estimated.
For humans, the infectious dose of Salmonella strains depends on age, physical condition, health status, as well as the bacterial strains involved in the infection. In general, 105–106 CFU (colony forming unit) bacterial cells are necessary to induce acute salmonellosis42. This large inoculum is needed due to gastro-intestinal tract conditions, such as low acidity, presence of bile salts and enzymes, and normal intestinal microbiome which generates nutrient competition. A low gastric acidity related to a variety of medical conditions or in the elderly population can decrease the infectious dose to 103 cells43. Consequently, for the current study the feeding environment of L. sericata was inoculated with 102 CFU/ml and 104 CFU/ml. An important aspect from a forensic perspective is that there was no registered delay in the colonization of the Salmonella inoculated liver by L. sericata, indicating that the presence of this pathogen did not prevent the feeding of the adult specimens. A previous study44, where pigs were exposed to contamination with Salmonella enterica subsp. enterica serovar Typhimurium by direct intranasal inoculation, or indirect, by environmental contamination required more than 103 Salmonella to induce an acute infection. Moreover, the market-exposed pigs during routine resting or holding could be infected by Salmonella when subjected to a bacterial concentration of 103 CFU45. As resulted from these previous studies44,45, a Salmonella concentration of 104× can cause an infection in a weakened animal or human, by food/surface contamination.
Regarding the overall microbiome characterization, studies investigating the fly’s bacterial content showed that Proteobacteria, Firmicutes, Bacteroidetes and Actinobacteria prevailed in both blowfly and housefly life stages9,13,46, while another study36 revealed that Flavobacteria dominated the pupa stage. Singh and collaborators research36 showed that the microbiome pattern was specific and similar for the adult and larva stages, being characterized by Firmicutes with Lactobacillales, for the pupa being dominated by Proteobacteria with the greatest order and family diversity, and for the teneral stage prevailing Actinobacteria with Corynebacteriales. In a recent study, Actinobacteria was not that consistent among L. sericata and L. cuprina dissected organs, however, were more abundant46. Interestingly, during the present research Actinobacteria was present in high abundances in the teneral specimens, suggesting that this phylum can play a role within the species metabolic activities. In contrast, another study revealed that the microbiome between the adult and eggs, and between the pupa and larvae stages was similar33. Although a bacterial diversity delimitation, characteristic for certain stages of development could be emphasized, this was not reflected in the diversity dynamics of the liver samples, which were dominated by Firmicutes. Most studies revealed Gammaproteobacteria as the main class identified from the blowfly specimens9,11,33,35, with a relative abundance of up to 81%11. Furthermore, in Maleki-Ravasan et al.11 Morganellaceae represented the most abundant family detected both by culture-dependent and independent methods, while in the current survey Lactobacillaceae dominated, along with Corynebacteriaceae. At genera level, Citrobacter was identified among the most prevalent taxa, being also identified by both methods mentioned above from L. sericata life stages11. This genus plays a role in the production of putrefaction products, such as indole, by converting tryptophan, but also has the ability to ferment lactose47, being among the most prevalent genera detected in our samples.
The same recent studies on L. sericata bacterial content11,35 revealed Proteus as the most abundant genus detected, being identified from the digestive tract, salivary glands and the adult stage48,49. However, the study of Gasz et al.46 revealed the dominant presence of Pseudomonas and Corynebacterium in all L. sericata organs, while Proteus was observed in low abundances. During our investigation, this taxon was the 7th most abundant genera, Lactobacillus being the most abundant in all investigated samples. The presence in high number of Lactobacillus can be explained by this genus fermentative abilities, being also well adapted to the insect hosts50. This genus was identified as being more abundant in the insect salivary gland36, and was able to inhibit other bacterial growth by producing bacteriocins51. However, in the recent study of Gasz et al.46, Lactobacillus was identified in lower quantities, compared to our study, where this taxon prevailed. The presence of Lactobacillus in low abundance46 can be a consequence of the investigated organs and feeding substrate that differed from the initial bait (dog dung), the later one could also explain the variation between insect and bait samples. Furthermore, Proteus was previously identified as less abundant as 0.4% in L. sericata specimens33, however, it was demonstrated that this bacterium can transmit swarming signals to attract blowflies52, so its presence in insects may be explained by its acquisition from the surrounding environment. Even though in our study Proteus was one of the top ten most abundant genera, it was ranked 16th in prevalence in the insect samples, and 26th in the liver; the highest values being recorded in the last experimental day; 18.86% and 28.75%, respectively. In the insect samples, the higher relative abundance of 37.02% was recorded for the pupa stage, not being detected in the adult, first and second larvae life stages. In addition, Providencia, Vagococcus, Lactococcus, and Leuconostoc prevailed as main genera detected in L. sericata life stages33. Lactococcus was previously reported as being associated with L. sericata11,36, while in our study it was among the top ten genera in the insect and liver samples, constituting a shared taxon for the adult, larva, pupa and liver samples. Lactococcus has a homofermentative metabolism53, while Leuconostoc is a heterofermentative bacteria54, their presence being associated with the fermentative phase of decomposition. Furthermore, Providencia was previously associated with insects55, being considered as an opportunistic human pathogen, similar to Vagococcus56.
Regarding the investigated life stages, several authors researched the eggs from the first and second generation, third instar larvae, pupa and first-generation adult33. Others studied the first and second instars, male and female specimens, and the digestive tract of the third instar larvae11, as well as the internal and external contents of the third instar larvae. These type of studies revealed via culture-based bacteriological method that more bacterial species were present in the third instar larvae after feeding, opposite to the unfed specimens, with a three to seven ratio, respectively11, but also that the larvae external content presented significant higher Enterobacteria abundances. Simultaneously, the same study showed that several bacteria genera, such as Enterococcus faecalis and Proteus spp., were present in most of L. sericata life stages, having transstadial and transovarial transmission routes11. Our data revealed Lactobacillus dominated in the adult and larvae stages being present in all investigated life stages.
In terms of pathogen introduction in an experimental model, a study of bacterial transmigration emphasized that the inoculation of mouse bodies with Staphylococcus aureus did not affect the overall microbial diversity pattern22, not even in organisms that have been sterilized on the surface. However, even if our data showed a similar trend, it is of note that both the inoculated substrate and L. sericata life stages that fed on it were investigated, unlike the previous research that limited the access of invertebrate decomposers22. This aspect is very important, since it is known that the insect presence can have an influence on the microbial composition of the feeding substrate. Concerning the blowfly’s ability to perform the microbiome exchange in and from the living environment, carrying and transmitting pathogens, was demonstrated by the same microbial patterns identified both in flies and the feces of sympatric animals9. Additionally, during the decomposed tissue pre-digestion, blowflies secrete certain enzymes that have antimicrobial actions, thus having the potential to alter the microbial pattern of the substrate36,57. These excretions/secretions (ES) with antimicrobial properties can be one of the reasons why many of the bacteria identified in the liver samples were not transferred to the insect specimens. However, the interaction between S. enterica and L. sericata life stages and liver samples must be considered, as these interactions can play a role in the bacterial acquisition and transmission58.
As revealed from the present and past studies, flies can contain numerous pathogens of human and veterinary importance, such as Enterococcus, Streptococcus, Pseudomonas, Proteus, and Serratia9. All of these genera comprise numerous species, that can cause different infections in animals and humans, from minor illnesses to deadly diseases, such as the streptococcal toxic shock syndrome59. Some species are present in the gastrointestinal tract of healthy people, such as Enterococcus species, but they are also the cause of severe nosocomial infections in hospitalized patients, being inherently resistant to various antibiotics60. These infections often target people who are hospitalized or have a compromised immune system, but the infection in healthy individuals can also occur through the consumption of contaminated food or water61. Staphylococcus, another bacterium with pathological significance, was detected only in the wild adults and teneral specimens with relative abundance values ranging from 9.6 up to 16.18%, data observed in other research where the values were higher in adults than in the larval stage33. Among all identified pathogens, Salmonella spp. exhibited low abundances of 0.01% in the investigated blowflies and in the initial feeding substrate, assuming that the presence of larvae caused an increase in the substrate abundance of up to 2.01%36. Our data could support this statement, given that an increase in the liver bacterial abundance corresponded to an increased S. enterica value in the third larvae stage.
Considering all these aspects, the identification of these possible bacterial pathogens in different insect species is very important, in order to monitor their transfer in both invertebrates and humans’ living environments. However, the living environment must be also examined, as most studies investigated only the fly bacterial content, and as has been demonstrated so far36, L. sericata microbiome is thoroughly connected to the feeding substrate represented by the decomposed organic matter. As such, an increase in abundance of several pathogenic bacteria in the liver samples, identified at the same time in lower abundance in the insect samples, could suggest that the feeding substrate abundances were increased by the feeding larvae36. In addition, the experimental introduction of a potential “disturbing” element, as the inoculation of the feeding substrate with S. enterica, could bring new and interesting information about the overall microbiome structure. Moreover, by studying the transmission of these pathogens and the environment of the serovars, that can be represented by the gastro-intestinal tract of humans, farm animals, reptiles, birds, and insects, we can have a better understanding on the dispersal of these microorganisms.
Regardless of the acquired pathogens throughout their lifetime, the fly’s microbiome can provide useful data during forensic studies regarding the presence or even the development stage of the insects present on the body, since it was not influenced by the artificial introduction of a specific bacterial taxa. Nevertheless, other pathogens (with increased antibiotic resistance) must be tested to see if their presence alters the microbiome structure during the different development stages of various necrophagous insect species.
Material and methods
Experimental setup
The hypothesis of this study relied on the blowfly’s potential of transmitting pathogens and on their role as main colonizers of decomposed human remains. Thus, the quantitative transmission of Salmonella enterica subsp. enterica serovar Typhimurium ATCC 14028 through one generation of L. sericata life stages (adult to teneral specimens) was investigated, as well as the dynamics within the inoculated feeding substrate (swine liver) via qPCR. Moreover, for an in-depth investigation of the pathogen impact and bacterial putative role as PMI estimators, both insect and liver microbiome were investigated via 16S rRNA gene Illumina MiSeq sequencing (Fig. 9).

Visual representation of the experimental setup, methods and expected outcomes.
Lucilia sericata sampling and laboratory rearing
Lucilia sericata adults were collected from an urban area of Bucharest (Romania) (N 44° 26′ 49.349′′ E 26° 2′ 45.352′′) during August 2019. Liver tissue was used as bait and the adult sampling was performed via entomological net. Further, the specimens were immediately transferred to a 46 cm rearing cage (BioQuip Products, Rancho Dominguez, CA, USA), transported to the laboratory and reared under constant laboratory conditions (25 °C ± 1 and 60% relative humidity, LD 13:11) (Fig. 9). The species has been taxonomically identified and genetically confirmed.
Adult specimens were subsequently transferred to nine sterilized rearing jars, 21 × 12 cm (BioQuip Products, Rancho Dominguez, CA, USA). Each rearing jar contained 300 g of swine liver. Three jars contained liver without S. enterica, three contained liver inoculated with 102 CFU/ml S. enterica, while the last three contained liver inoculated with 104 CFU/ml S. enterica. Under these conditions the adult specimens were allowed to feed for three hours and to lay egg clusters. After feeding, they were immediately collected and preserved in 200 µl Tris–EDTA pH 8 (TE) buffer at − 20 °C until further investigations. Under aseptic conditions, using a microbiological safety cabinet, and sterilized rearing jars, the egg clusters (one egg cluster/rearing jar) were transferred on the liver containing the same S. enterica concentrations, and monitored under constant laboratory conditions (25 °C ± 1 and 60% relative humidity, LD 13:11) (Fig. 9).
All experimental steps (rearing, transfer, isolation, inoculation, molecular assays) were performed under aseptic conditions to avoid any microbial cross contamination.
Bacterial cultivation and inoculation
Salmonella enterica subsp. enterica serovar Typhimurium ATCC 14028 was cultivated in BHI broth (Brain Heart Infusion—Merck, Darmstadt, Germany) at 37 °C in aerobic conditions. Fresh over-night culture was washed two times and resuspended in sterile saline buffer. Dilutions of 102 CFU/ml and 104 CFU/ml were obtained. The bacterial concentrations were obtained by serial dilutions (1/10) of the over-night culture (washed and resuspended in sterile saline buffer) with sterile saline buffer.
Samples of 1 ml of the dilutions were inoculated on the entire surface of each liver and the feeding substrates were incubated for two hours at room temperature for colonization. The liver surface was inoculated by five consecutive times with 200 µl bacterial culture of appropriate concentration (102 or 104) using a sterile cotton swab. The bacterial concentration was measured by CFU counting per ml. From previous experiments and determinations, the concentration of the fresh, over-night culture of Salmonella enterica subsp. enterica serovar Typhimurium ATCC 14028 was predictable, consequently, the desired inoculants were obtained by appropriate dilutions. To confirm the prospective results, the CFU/ml was determined for each inoculum by spreading 100 µl of serial dilutions onto solid BHI medium (BHI supplemented with 1.5% (w/v) agar), followed by incubation at 37 °C until colonies were observed. To calculate the number of CFU/ml, the number of colony forming units on the countable plate was multiplied by 10 (100 µl to ml) × 10the dilution of the sample. The results confirmed the expected bacterial concentrations.
Alternatively, liver treated with saline buffer was used as control. The inoculum dilutant was sterile saline buffer, the same used for the bacterial culture inoculations and final resuspension.
Sample collection and DNA extraction
Insect and liver samples were collected daily, in triplicate, from all nine jars, and preserved in 200 µl Tris–EDTA pH 8 (TE) buffer at − 20 °C. Prior to DNA extraction, the insect specimens were surface sterilized in 70% ethanol and sterile water for 30 s each. The liver was first sampled 1 h after purchase.
Total DNA was extracted from the whole fly homogenates and liver samples, according to the manufacturer protocol from DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA). The DNA extraction comprised an additional step of cell disruption with 2 mm ZR Bashing Beads (Zymo research, Irvine, CA, USA). The samples were vigorously disrupted at 20 °C, 50 Hz-power, for 12 min via a SpeedMill PLUS Cell Homogenizer (Analitik Jena, Jena, Germany). The DNA purity and concentration were measured with a Biodrop spectrophotometer (Denville Scientific Inc, Holliston, MA, USA).
Salmonella enterica quantitative PCR assays
Control adults and liver samples were tested for the presence of Salmonella via PCR amplification and no bacteria could be identified. Salmonella enterica quantification from the insect specimens and liver tissues was carried out using Sal_enterF/Sal_enterR (5′-AGCGTACTGGAAAGGGAAAG-3′; 5′-ATACCGCCAATAAAGTTCACAAAG-3′) specific primers62. The primers were selected given their capability to detect up to 329 Salmonella isolates, belonging to all subspecies of S. enterica62. For the qPCR assays the total DNA was extracted from the liver and insect samples as previously described.
Thermo Scientific Maxima SYBR Green (1×) Master Mix (ThermoFisher Scientific) reagents were used for the investigation in a total volume of 10 µl, containing 10 µM of each forward and reverse primer, 200 ng DNA template and nuclease free water. All reaction components were thawed and kept on ice, while the entire procedure was performed under a microbiological safety cabinet.
The qPCR amplifications were carried out using a Mastercycler ep gradient S thermocycler PCR (Eppendorf, Wien, Austria) and protocol: initial incubation step of 10 min at 95 °C followed by 40 cycles of 15 s at 95 °C, 30 s at 47 °C, and 30 s at 72 °C. A melting curve analysis was included at the end of each program, being used as amplification control.
Quantification was performed by interpolation in a standard regression curve of cycle threshold (Ct) values generated from samples of known concentration of DNA and plotted as Ct versus log10 (ng DNA/reaction) and considered when R2 > 0.99. Standard curve was generated using corresponding PCR amplified DNA fragments from the known pure culture of S. enterica.
The DNA decimal dilutions series used for the standard curve ranged between 5.8 × 10–2 and 5.8 × 10–9 ng/μl. Samples were run in triplicate, with a Ct threshold value over 35 not considered, while no-template controls were included in all PCR runs.
The gene copy number per reaction was calculated according to Staroscik 63: Copy number = (ng DNA × 6.023 × 1023)/(length × 1 × 109 × 650), and the plot was performed between the logarithmic scale (base 10) of copy gene number and the sampling points.
16S rRNA gene Illumina sequencing and data analysis
The concentration of the total genomic DNA from the liver tissue and insects was measured with Qubit fluorometer (ThermoFisher Scientific). The primer pair 341F/805R64 was used for the PCR amplification of the 16S rRNA gene fragments (V3–V4 variable region) (McGill University and Génome Québec Innovation Centre, Canada). The amplicons pooled in equal concentrations were sequenced at the McGill University and Génome Québec Innovation Centre via Illumina MiSeq PE300 sequencing platform.
The samples were analysed using dada2 package (v1.11.3) implemented in R (v3.4.4). Forward and reverse primers were removed using cutadapt (v1.2.1)65, then sequences were trimmed to 250 bases and filtered (truncLen = c(250, 250), maxEE = 1, maxN = 0, truncQ = 11, rm.phix = TRUE). Amplicon Sequence Variants (ASVs) were inferred from de-replicated sequences. Chimeras were removed using the “consensus” removal method.
Taxonomic assignment of the ASVs was performed using the Silva v138 16S rRNA database. The community analysis was performed in R environment with the phyloseq and MicrobiomeSeq packages66. The sequencing depth varied between 601 and 729.269 sequences per sample with a median of 120,703 sequences. The final dataset consisted of a total of 2798 ASVs from 96 samples. The raw data were deposited in the NCBI SRA Sequence Read Archive under the BioProject: PRJNA646601.
Alpha-diversity was used to evaluate the diversity differences at the ASVs level using Shannon’s diversity index, and significant differences were evaluated using t-tests67,68. Principal coordinate analysis (PCoA) based on Bray–Curtis dissimilarities69 was used to assess the beta diversity. PERMANOVA and ANOSIM tests were performed in MicrobiomeAnalyst platform67,68 to determine statistical differences between a priori groups. Microbial community profiles of the variable V3/V4 region of the 16S rRNA gene sequences and graphical visualization were conducted using MicrobiomeAnalyst67 with the parameters for counter filters = 4, prevalence in samples = 20%, and inter-quartile variance filter percentage to remove = 10%. Data were normalized using the Total Sum Scaling (TSS) method67. Additional statistical analysis was performed using R version 3.6.370, to investigate statistical differences among treatments using a two-way ANOVA, followed by the Tukey multiple comparison test a posteriori using the package dplyr71.
Venn diagram of all identified genera was generated using the Bioinformatics and Evolutionary Genomics online calculator72.
Data availability
The datasets generated during and/or analyzed during the current study were deposited in the NCBI SRA Sequence Read Archive under the BioProject: PRJNA646601.
References
Dash, H. R. & Das, S. Thanatomicrobiome and epinecrotic community signatures for estimation of post-mortem time interval in human cadaver. Appl. Microbiol. Biotechnol. 104, 9497–9512. https://doi.org/10.1007/s00253-020-10922-3 (2020).
Smith, K. G. A Manual of Forensic Entomology (The Trustees of British Museum, 1986).
Grassberger, M. & Frank, C. Initial study of arthropod succession on pig carrion in a central European urban habitat. J. Med. Entomol. 41, 511–523. https://doi.org/10.1603/0022-2585-41.3.511 (2004).
Chaudhury, M. F., Zhu, J. J. & Skoda, S. R. Bacterial volatiles attract gravid secondary screwworms (Diptera: Calliphoridae). J. Econ. Entomol. 109, 947–951. https://doi.org/10.1093/jee/tov390 (2016).
Thompson, C. R., Brogan, R. S., Scheifele, L. Z. & Rivers, D. B. Bacterial interactions with necrophagous flies. Ann. Entomol. Soc. Am. 106, 799–809. https://doi.org/10.1603/an12057 (2013).
Catts, E. P. & Haskell, N. H. Entomology and Death: A Procedural Guide (Joyce’s Print Shop Inc., 1990).
Tomberlin, J. K. et al. A review of bacterial interactions with blow flies (Diptera: Calliphoridae) of medical, veterinary, and forensic importance. Ann. Entomol. Soc. Am. 110, 19–36. https://doi.org/10.1093/aesa/saw086 (2017).
Jordan, H. R. & Tomberlin, J. K. Abiotic and biotic factors regulating inter-kingdom engagement between insects and microbe activity on vertebrate remains. Insects. https://doi.org/10.3390/insects8020054 (2017).
Poudel, A. et al. Comparison of microbiota, antimicrobial resistance genes and mobile genetic elements in flies and the feces of sympatric animals. FEMS Microbiol. Ecol. https://doi.org/10.1093/femsec/fiaa027 (2020).
Mian, L. S., Maag, H. & Tacal, J. V. Isolation of Salmonella from muscoid flies at commercial animal establishments in San Bernardino County, California. J. Vector Ecol. 27, 82–85 (2002).
Maleki-Ravasan, N. et al. New insights into culturable and unculturable bacteria across the life history of medicinal maggots Lucilia sericata (Meigen) (Diptera: Calliphoridae). Front. Microbiol. 11, 505. https://doi.org/10.3389/fmicb.2020.00505 (2020).
Royden, A. et al. A role for flies (Diptera) in the transmission of Campylobacter to broilers?. Epidemiol. Infect. 144, 3326–3334. https://doi.org/10.1017/S0950268816001539 (2016).
Junqueira, A. C. M. et al. The microbiomes of blowflies and houseflies as bacterial transmission reservoirs. Sci. Rep. 7, 16324. https://doi.org/10.1038/s41598-017-16353-x (2017).
Pava-Ripoll, M., Pearson, R. E., Miller, A. K. & Ziobro, G. C. Prevalence and relative risk of Cronobacter spp., Salmonella spp., and Listeria monocytogenes associated with the body surfaces and guts of individual filth flies. Appl. Environ. Microbiol. 78, 7891–7902. https://doi.org/10.1128/AEM.02195-12 (2012).
Daeschlein, G. et al. Maggots as potential vector for pathogen transmission and consequences for infection control in waste management. GMS Hyg. Infect. Control. https://doi.org/10.3205/dgkh000250 (2015).
Onwugamba, F. C. et al. The role of ‘filth flies’ in the spread of antimicrobial resistance. Travel Med. Infect Dis. 22, 8–17. https://doi.org/10.1016/j.tmaid.2018.02.007 (2018).
Cerovsky, V. et al. Lucifensin, the long-sought antimicrobial factor of medicinal maggots of the blowfly Lucilia sericata. Cell Mol. Life Sci. 67, 455–466. https://doi.org/10.1007/s00018-009-0194-0 (2010).
Brown, A. et al. Blow fly Lucilia sericata nuclease digests DNA associated with wound slough/eschar and with Pseudomonas aeruginosa biofilm. Med. Vet. Entomol. 26, 432–439. https://doi.org/10.1111/j.1365-2915.2012.01029.x (2012).
Hobson, R. P. Sheep blowfly investigations. II. Substances which induce Lucilia sericata Mg to oviposit on Sheep. Ann. Appl. Biol. 22, 294–300 (1935).
DeVaney, J. A., Eddy, G. W., Ellis, E. M. & Harrington, R. Jr. Attractancy of inoculated and incubated bovine blood fractions to screwworm flies (Diptera: Calliphoridae): Role of bacteria. J. Med. Entomol. 10, 591–595. https://doi.org/10.1093/jmedent/10.6.591 (1973).
Zurek, L., Schal, C. & Watson, D. W. Diversity and contribution of the intestinal bacterial community to the development of Musca domestica (Diptera: Muscidae) larvae. J. Med. Entomol. 37, 924–928. https://doi.org/10.1603/0022-2585-37.6.924 (2000).
Burcham, Z. M. et al. Bacterial community succession, transmigration, and differential gene transcription in a controlled vertebrate decomposition model. Front. Microbiol. 10, 745. https://doi.org/10.3389/fmicb.2019.00745 (2019).
Pechal, J. L. et al. The potential use of bacterial community succession in forensics as described by high throughput metagenomic sequencing. Int. J. Legal Med. 128, 193–205. https://doi.org/10.1007/s00414-013-0872-1 (2014).
Iancu, L., Carter, D. O., Junkins, E. N. & Purcarea, C. Using bacterial and necrophagous insect dynamics for post-mortem interval estimation during cold season: Novel case study in Romania. Forensic Sci. Int. 254, 106–117. https://doi.org/10.1016/j.forsciint.2015.07.024 (2015).
Guo, J. et al. Potential use of bacterial community succession for estimating post-mortem interval as revealed by high-throughput sequencing. Sci. Rep. 6, 24197. https://doi.org/10.1038/srep24197 (2016).
DeBruyn, J. M. & Hauther, K. A. Postmortem succession of gut microbial communities in deceased human subjects. PeerJ 5, e3437. https://doi.org/10.7717/peerj.3437 (2017).
Hauther, K. A., Cobaugh, K. L., Jantz, L. M., Sparer, T. E. & DeBruyn, J. M. Estimating time since death from postmortem human gut microbial communities. J. Forensic Sci. 60, 1234–1240. https://doi.org/10.1111/1556-4029.12828 (2015).
Metcalf, J. L. et al. A microbial clock provides an accurate estimate of the postmortem interval in a mouse model system. Elife 2, e01104. https://doi.org/10.7554/eLife.01104 (2013).
Metcalf, J. L. et al. Microbial community assembly and metabolic function during mammalian corpse decomposition. Science 351, 158–162. https://doi.org/10.1126/science.aad2646 (2016).
Burcham, Z. M. et al. Fluorescently labeled bacteria provide insight on post-mortem microbial transmigration. Forensic Sci. Int. 264, 63–69. https://doi.org/10.1016/j.forsciint.2016.03.019 (2016).
Javan, G. T. et al. Human thanatomicrobiome succession and time since death. Sci. Rep. 6, 29598. https://doi.org/10.1038/srep29598 (2016).
Lawrence, K. E., Lam, K. C., Morgun, A., Shulzhenko, N. & Lohr, C. V. Effect of temperature and time on the thanatomicrobiome of the cecum, ileum, kidney, and lung of domestic rabbits. J. Vet. Diagn. Investig. 31, 155–163. https://doi.org/10.1177/1040638719828412 (2019).
Wohlfahrt, D., Woolf, M. S. & Singh, B. A survey of bacteria associated with various life stages of primary colonizers: Lucilia sericata and Phormia regina. Sci. Justice 60, 173–179. https://doi.org/10.1016/j.scijus.2019.11.001 (2020).
Iancu, L., Necula-Petrareanu, G. & Purcarea, C. Potential bacterial biomarkers for insect colonization in forensic cases: Preliminary quantitative data on Wohlfahrtiimonas chitiniclastica and Ignatzschineria indica dynamics. Sci. Rep. 10, 8497. https://doi.org/10.1038/s41598-020-65471-6 (2020).
Ulanova, R. V. T. & Kravchenko, I. K. Bacteria associated with Lucilia sericata larvae reared on fish wastes. Entomol. Exp. et Appl. 168, 573–581. https://doi.org/10.1111/eea.12918 (2020).
Singh, B. et al. A metagenomic assessment of the bacteria associated with Lucilia sericata and Lucilia cuprina (Diptera: Calliphoridae). Appl. Microbiol. Biotechnol. 99, 869–883. https://doi.org/10.1007/s00253-014-6115-7 (2015).
Rueda, L. C., Ortega, L. G., Segura, N. A., Acero, V. M. & Bello, F. Lucilia sericata strain from Colombia: Experimental colonization, life tables and evaluation of two artificial diets of the blowfly Lucilia sericata (Meigen) (Diptera: Calliphoridae), Bogota, Colombia strain. Biol. Res. 43, 197–203 (2010).
Andino, A. & Hanning, I. Salmonella enterica: Survival, colonization, and virulence differences among serovars. ScientificWorldJournal 2015, 520179. https://doi.org/10.1155/2015/520179 (2015).
Batz, M. B., Hoffmann, S. & Morris, J. G. Jr. Ranking the disease burden of 14 pathogens in food sources in the United States using attribution data from outbreak investigations and expert elicitation. J. Food Prot. 75, 1278–1291. https://doi.org/10.4315/0362-028X.JFP-11-418 (2012).
European Centre for Disease Prevention and Control. Salmonellosis. In ECDC. Annual Epidemiological Report for 2017. https://www.ecdc.europa.eu/. Accessed 10 April 2020.
Sanderson, K. E. & Roth, J. R. Linkage map of Salmonella typhimurium, edition VII. Microbiol. Rev. 52, 485–532 (1988).
Kothary, M. H. & Babu, U. S. Infective dose of foodborne pathogens in volunteers: A review. J. Food Saf. 21, 49–73. https://doi.org/10.1111/j.1745-4565.2001.tb00307.x (2001).
WHO.Vaccines and Biologicals. Background Document. https://www.who.int/immunization/monitoring_surveillance/burden/vpd/WHO_SurveillanceVaccinePreventable_21_Typhoid_R1.pdf?ua=1. Accessed 15 January 2021.
Loynachan, A. T. & Harris, D. L. Dose determination for acute Salmonella infection in pigs. Appl. Environ. Microbiol. 71, 2753–2755. https://doi.org/10.1128/AEM.71.5.2753-2755.2005 (2005).
Hurd, H. S., Gailey, J. K., McKean, J. D. & Rostagno, M. H. Rapid infection in market-weight swine following exposure to a Salmonella typhimurium-contaminated environment. Am. J. Vet. Res. 62, 1194–1197. https://doi.org/10.2460/ajvr.2001.62.1194 (2001).
Gasz, N. E. G., Doggett, S. L. & Harvey, M. L. Bacterial association observations in Lucilia sericata and Lucilia cuprina organs through 16S rRNA gene sequencing. Appl. Microbiol. Biotechnol. 105, 1091–1106. https://doi.org/10.1007/s00253-020-11026-8 (2021).
Dworkin, M. et al. (eds) The Prokaryotes: Volume 6: Proteobacteria: Gamma Subclass 90–98 (Springer, 2006).
Wei, T., Ishida, R., Miyanaga, K. & Tanji, Y. Seasonal variations in bacterial communities and antibiotic-resistant strains associated with green bottle flies (Diptera: Calliphoridae). Appl. Microbiol. Biotechnol. 98, 4197–4208. https://doi.org/10.1007/s00253-013-5498-1 (2014).
Yuan, Y. et al. Genome sequence of a Proteus mirabilis strain isolated from the salivary glands of larval Lucilia sericata. Genome Announc. https://doi.org/10.1128/genomeA.00672-16 (2016).
Matos, R. C. & Leulier, F. Lactobacilli-Host mutualism: “Learning on the fly”. Microb. Cell Fact 13, S6. https://doi.org/10.1186/1475-2859-13-S1-S6 (2014).
Inglin, R. C., Stevens, M. J., Meile, L., Lacroix, C. & Meile, L. High-throughput screening assays for antibacterial and antifungal activities of Lactobacillus species. J. Microbiol. Methods 114, 26–29. https://doi.org/10.1016/j.mimet.2015.04.011 (2015).
Ma, Q. et al. Proteus mirabilis interkingdom swarming signals attract blow flies. ISME J. 6, 1356–1366. https://doi.org/10.1038/ismej.2011.210 (2012).
Wessels, S. A. et al. The lactic acid bacteria, the food chain, and their regulation. Trends Food Sci. Technol. 15, 498–505. https://doi.org/10.1016/j.tifs.2004.03.003 (2004).
Dworkin, M. (ed.) The Prokaryotes: A Handbook on the Biology of Bacteria: Firmicutes, Cyanobacteria Vol. 4, 267–319 (Springer, 2006).
Juneja, P. & Lazzaro, B. P. Providencia sneebia sp. nov. and Providencia burhodogranariea sp. nov., isolated from wild Drosophila melanogaster. Int. J. Syst. Evol. Microbiol. 59, 1108–1111. https://doi.org/10.1099/ijs.0.000117-0 (2009).
Matsuo, T. et al. Vagococcus fluvialis as a causative pathogen of bloodstream and decubitus ulcer infection: Case report and systematic review of the literature. J. Infect. Chemother. 27, 359–363. https://doi.org/10.1016/j.jiac.2020.09.019 (2021).
Wilson, M. R., Nigam, Y., Jung, W., Knight, J. & Pritchard, D. I. The impacts of larval density and protease inhibition on feeding in medicinal larvae of the greenbottle fly Lucilia sericata. Med. Vet. Entomol. 30, 1–7. https://doi.org/10.1111/mve.12138 (2016).
Pace, R. C. T., Crippen, T. L. & Wayadande, A. C. Filth fly transmission of Escherichia coli O157:H7 and Salmonella enterica to Lettuce, Lactuca sativa. Ann. Entomol. Soc. Am. 110, 83–89 (2017).
Stevens, D. L. Streptococcal toxic-shock syndrome: Spectrum of disease, pathogenesis, and new concepts in treatment. Emerg. Infect. Dis. 1, 69–78. https://doi.org/10.3201/eid0103.950301 (1995).
Pana, M. et al. Enterococcal urinary tract infections analyzed at Cantacuzino National Research Institute. Infectio Ro 51, 21–23. https://doi.org/10.26416/Inf.51.3.2017.1198 (2017).
Hanchi, H., Mottawea, W., Sebei, K. & Hammami, R. The genus Enterococcus: Between probiotic potential and safety concerns—An update. Front. Microbiol. 9, 1791. https://doi.org/10.3389/fmicb.2018.01791 (2018).
Kasturi, K. N. & Drgon, T. Real-time PCR method for detection of Salmonella spp. in environmental samples. Appl. Environ. Microbiol. https://doi.org/10.1128/AEM.00644-17 (2017).
Staroscik, A. (2004). www.cels.uri.edu/gsc/cndna.html. Accessed 20 February 2020.
Takahashi, S., Tomita, J., Nishioka, K., Hisada, T. & Nishijima, M. Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PLoS ONE 9, e105592. https://doi.org/10.1371/journal.pone.0105592 (2014).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17(3), 2011. https://doi.org/10.14806/ej.17.1.200 (2011).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217. https://doi.org/10.1371/journal.pone.0061217 (2013).
Dhariwal, A. et al. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 45, W180–W188. https://doi.org/10.1093/nar/gkx295 (2017).
Chong, J., Liu, P., Zhou, G. & Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 15, 799–821. https://doi.org/10.1038/s41596-019-0264-1 (2020).
Paliy, O. & Shankar, V. Application of multivariate statistical techniques in microbial ecology. Mol. Ecol. 25, 1032–1057. https://doi.org/10.1111/mec.13536 (2016).
R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria (2020). URL: https://www.R-project.org/.
Wickham et al. Welcome to the Tidyverse. J. Open Source Softw 4(43), 1686. https://doi.org/10.21105/joss.01686 (2019).
Bioinformatics & Evolutionary Genomics. http://bioinformatics.psb.ugent.be/webtools/Venn/. Accessed 10 November 2020.
Acknowledgements
This research received partially financial support from Unitatea Executiva pentru Finantarea Invatamantului Superior, a Cercetarii, Dezvoltarii si Inovarii (ERANET-MARTERA-MOBILTOX-2-224/2021) and Academia Româna (RO1567-IBB05/2021) Grants.
Author information
Authors and Affiliations
Contributions
L.I. performed the experimental design, insect identification, Quantitative polymerase chain reaction (qPCR), data interpretation and article writing; L.I., I.R.A. and V.I.P. performed the experimental sampling; C.H.C., P.L. and V.I.P. performed the bioinformatics and statistical analyses; I.R.A. contributed to article writing; C.P. performed data interpretation and contributed to article writing. All authors discussed the results, wrote, review, and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Iancu, L., Angelescu, I.R., Paun, V.I. et al. Microbiome pattern of Lucilia sericata (Meigen) (Diptera: Calliphoridae) and feeding substrate in the presence of the foodborne pathogen Salmonella enterica. Sci Rep 11, 15296 (2021). https://doi.org/10.1038/s41598-021-94761-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-94761-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
