
Abstract
The apelin receptor (Aplnr) and its ligands, Apelin and Apela, contribute to metabolic control. The insulin resistance associated with pregnancy is accommodated by an expansion of pancreatic β-cell mass (BCM) and increased insulin secretion, involving the proliferation of insulin-expressing, glucose transporter 2-low (Ins+Glut2LO) progenitor cells. We examined changes in the apelinergic system during normal mouse pregnancy and in pregnancies complicated by glucose intolerance with reduced BCM. Expression of Aplnr, Apelin and Apela was quantified in Ins+Glut2LO cells isolated from mouse pancreata and found to be significantly higher than in mature β-cells by DNA microarray and qPCR. Apelin was localized to most β-cells by immunohistochemistry although Aplnr was predominantly associated with Ins+Glut2LO cells. Aplnr-staining cells increased three- to four-fold during pregnancy being maximal at gestational days (GD) 9–12 but were significantly reduced in glucose intolerant mice. Apelin-13 increased β-cell proliferation in isolated mouse islets and INS1E cells, but not glucose-stimulated insulin secretion. Glucose intolerant pregnant mice had significantly elevated serum Apelin levels at GD 9 associated with an increased presence of placental IL-6. Placental expression of the apelinergic axis remained unaltered, however. Results show that the apelinergic system is highly expressed in pancreatic β-cell progenitors and may contribute to β-cell proliferation in pregnancy.
Similar content being viewed by others


Introduction
The physiology of pregnancy tests the metabolic plasticity of the mother and initiates adaptive responses to metabolic stress. Within the human pancreas, substantial increases in β-cell mass (BCM) normally occur in second and third trimester preceding the appearance of insulin resistance1,2. A failure of β-cells to adaptively expand after the first trimester may place the mother at risk of developing GDM3 associated with elevated levels of pro-inflammatory cytokines4,5,6 which contribute to β-cell dysfunction7. Similarly, β-cell mitogenesis is normally low in adult mice but increases during pregnancy contributing to a two- to three-fold increase in BCM8. In rodents this has been linked to the mitogenic effects of prolactin and placental lactogen (PL) on β-cells8,9,10,11, both of which increase across pregnancy in the maternal circulation9. Targeted over-expression of PL in mouse β-cells resulted in their increased proliferation11, mediated by prolactin receptors. Conversely, targeted deletion of the prolactin receptor prevented a gestational increase in BCM, impaired insulin release and led to glucose intolerance12,13.
An increase in β-cells during pregnancy occurs partly through self-renewal of existing, mature β-cells. In rodents the lifespan of the β-cell in adult life is around 58 days14. An increased rate of proliferation during pregnancy without a change in apoptotic rate results in an accumulation of additional β-cells. However, new β-cells may also derive from a number of progenitor phenotypes during pregnancy. These include insulin-expressing cells that do not express the Fltp gene, a marker of functional β-cells15, which are highly proliferative and which may also express the platelet-derived growth factor (PDGF) receptor-α16. A separate type of multi-lineage progenitor has been identified in mouse and human pancreata throughout life, both within islets and in the small, extra-islet endocrine clusters17. This progenitor cell fraction expresses some insulin, but glucose-stimulated insulin secretion (GSIS) is poor due to low expression of glucose transporter 2 (Ins+Glut2LO cells)18, although they have the capacity to differentiate into functional β-cells in vitro19. Such cells relatively lack β-cell maturity markers such as expression of the transcription factors MafA and Nkx6.1, while over-expressing progenitor cells markers such as neurogenin-3 and MafB18,19. During mouse pregnancy the percentage of Ins+Glut2LO cells that are proliferating increases significantly at gestational day (GD) 9 preceding the increase in BCM20. However, in a mouse model of gestational glucose intolerance characterized by a sub-optimal increase in BCM the number of proliferating Ins+Glut2LO cells was significantly lower21. Neogenesis of new β-cells is also likely during human pregnancy due to the reappearance of C-peptide in women with long-standing type 1 diabetes where residual surviving β-cells are expected to be scarce22.
The local trophic factors contributing to the proliferation and differentiation of β-cell progenitors during pregnancy are not well characterized but may include locally expressed paracrine molecules such as Apelin (Apln) and Apela [Elabela]. Apelin and Apela are endogenous ligands for the G-protein coupled receptors, Aplnr (APJ)23,24 and GPR2525, and both Apelin and the Aplnr are found in multiple tissue types, including pancreas26. The apelinergic system is active in the fetoplacental unit and is thought to promote transplacental glucose transport27. Additionally, Apela is morphogenic for embryonic cardiovascular system formation and early placental development, while Apelin acts in mid or late gestation to mediate fetal angiogenesis and energy homeostasis28. Apelin is released by the placental syncytiotrophoblast into the maternal circulation with concentrations increasing throughout pregnancy in both humans and rodents29,30.
The apelinergic axis may also modulate metabolism since adipose-derived Apelin has been associated with increased glucose uptake and insulin sensitivity28,31,32. Furthermore, Apelin gene-null mice demonstrate a decreased insulin sensitivity and hyperinsulinemia, which could be reversed by Apelin administration, as was similarly reported in the db/db mouse model of type 2 diabetes32. Interestingly, patients who are obese or have type 2 diabetes show increased circulating Apelin levels, which suggests the possibility of Apelin resistance33,34. Similarly, obese and insulin-resistant pregnant rats had increased circulating and placental Apelin levels at term35. However, altered Apelin levels were not associated with a clinical diagnosis of gestational diabetes36.
Within the pancreas, apelin has been localized to, and is released from, β-cells37 and may influence ß-cell number since targeted deletion of the Aplnr from mouse β-cells resulted in a reduced BCM and impaired glucose-stimulated insulin secretion (GSIS)38. Conversely, treatment with apelin protected against cellular stress and promoted β-cell survival in the Akita mouse model of type 1 diabetes39. Additionally, a long-acting depot of apelin reversed insulin resistance and promoted β-cell proliferation in diabetic rats40. Apelin expression has also been associated in other tissues with progenitor cells41, which suggests that it might be involved in the expansion and/or differentiation of Ins+Glut2LO cells. Taken together, these findings suggest that the apelinergic axis could contribute to the increase in BCM during pregnancy, which we have examined in the present studies.
Results
Differential gene expression in pancreatic Ins+Glut2LO cells
We analyzed differential gene expression in Ins+Glut2LO vs. Ins+Glut2HI cells by DNA microarray following separation by FACS from pancreata of 7-day old neonatal mice. A total of 262 genes were identified where the relative levels of expression were higher by at least tenfold in Ins+Glut2LO cells (Supplementary Table 1). Partek GO enrichment software revealed gene clusters known to be associated with cell lineage commitment, cell proliferation, angiogenesis, and extracellular matrix modeling (Table 1). Within the genes with the greatest differential expression in Ins+Glut2LO cells were Apelin and Aplnr.
The findings from DNA microarray with respect to the apelinergic axis were validated using qPCR quantification of mRNA in fractions of Ins+Glut2LO vs. Ins+Glut2HI cells isolated from 7-day old mouse pancreata, relative to the expression of GAPDH and cyclophilin A. Levels of Apelin, Aplnr and Apela, were all expressed at significantly higher levels in Ins+Glut2LO cells (Fig. 1A). Mean insulin-1 expression was lower in the Ins+Glut2LO population compared with Ins+Glut2HI cells, but not significantly so.

(A) Relative expression levels of mRNA for Apelin, Apela, Aplnr and insulin (INS1) quantified by qPCR in Ins+Glut2HI (closed circles) and Ins+Glut2LO (open circles) populations of β-cells isolated from neonatal mouse pancreas; and (B) Apelin and Aplnr expression in non-pregnant (NP) and pregnant mouse pancreas [gestational day (GD) 9–12 and 18]. Results are shown as fold increase compared to the geometric mean of the expression of housekeeping genes. Values represent mean ± SEM (n = 4–6). *p < 0.05 vs. Glut2Hi in A, *p < 0.001 vs. NP in (B).
Anatomical localization of the apelinergic system within the pancreas
Immunohistochemical staining showed that Apelin was localized predominantly to a sub-population of insulin co-expressing β-cells in islets of Langerhans within adult mouse pancreata (Fig. 2A–C). Aplnr was also present and associated with the cell membrane in a sub-population of β-cells within islets that were mostly located towards the periphery of the islets (Fig. 2M–O). The distribution of Aplnr on the cell membranes was strongly punctate with less intense staining being present within the cytoplasm. Extensive co-localization of Apelin and/or Aplnr with insulin was seen in the small, extra-islet endocrine cell clusters (Fig. 2D,H). When glucagon was localized as a marker of α-cells only occasional co-localization was observed with either Apelin or Aplnr within islets (Fig. 2I–K,M–O) or clusters (Fig. 2L,P). A similar distribution of Apelin and Aplnr was also seen in islets within pancreata from neonatal mice (Supplementary Fig. 1). In addition to localization to β-cells Aplnr immunostaing was also observed to be associated with some vascular endothelial cells within the core of the islets. We also examined the presence of Apelin in human pancreas from a range of donor ages between early childhood and adulthood. Apelin was localized to islet endocrine cells with the intensity of staining decreasing with age. Apelin was also located within a sub-population of acinar cells towards the periphery of the growing pancreas at early ages but was less apparent in adulthood (Fig. 2Q–S). Images from an age range of additional donors are shown on Supplementary Fig. 2).

Immunohistochemical co-localization of insulin (A & E, red), glucagon (I & M, red), Apelin (B & J, green) and Aplnr (F & N, green) in adult mouse islets or extra-endocrine islet clusters (D,H,L,P). Merged images are shown for islets in (C), (G), (K) and (O) and for clusters. In merged images nuclei are shown stained with DAPI (blue). Arrows indicate the localization of Aplnr with the β-cell membranes. Immunohistochemical localization of Apelin in human pancreas is shown in panels (Q–S). Tissue donors were aged 4 months in (Q), 18 years in (R) and 41 years in (S). Apelin is localized to islet cells at all ages (Islet) and to acinar tissue (arrows) at 4 months. Bar represents 50 µm.
We further defined the sub-population of β-cells in mouse islets that contained Apelin and Aplnr by co-staining with Glut2. Apelin predominantly co-localized to β-cells that also showed strong Glut2 staining (Fig. 3A,B). In contrast, Aplnr was largely confined to β-cells in the islet mantle that showed no Glut2 staining (Fig. 3C–E).

Immunohistochemical localization of Apelin alone (A), and in a merged image with insulin and Glut2 (B) in adult mouse pancreatic islets. Localization of insulin, Aplnr and Glut2 is shown in (C) with an expanded view of co-localization of Aplnr to Ins+Glut2LO cells in (D) (arrows). The localization of Glut2 alone in the same section is shown in (E). Nuclei are counter-stained with DAPI (blue). Bar represents 10 µm.
Changes in the pancreatic apelinergic system during pregnancy
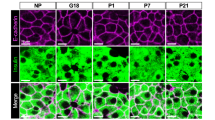
The expression of Aplnr and its ligands were quantified by qPCR in isolated islets from pregnant mice relative to non-pregnant animals. Apelin mRNA levels did not differ between pregnant and non-pregnant mice, but expression of Aplnr significantly declined in late pregnancy (Fig. 1B). The presence of Apela mRNA was not detectable. However, changes in apelinergic gene expression in minority cell populations such as Ins+Glut2LO cells might be difficult to detect within whole islets. Therefore, we examined changes in the number of Aplnr-immunoreactive cells at various gestational ages compared with non-pregnant, age-matched mice. During pregnancy, as in non-pregnant mice, Aplnr was predominantly localized to Ins+Glut2LO cells (Fig. 4A) and the abundance of such cells significantly increased at GD 9 and 12 (p < 0.01) before decreasing at GD 18, when considering whole pancreas (Fig. 4C). When the location of Ins+Glut2LOAplnr+ cells was separated into islet or extra-islet endocrine cluster compartments, a similar ontological profile was seen for islets (Fig. 4E), however, the frequency of these cells was two- to three-fold higher in clusters and did not decline in later gestation (Fig. 4D).

Immunohistochemical localization of Aplnr (white), insulin (red), Glut 2 (green) and cell nuclei (DAPI, blue) in islets from pregnant mice at GD 12 exposed in early life to control (A) or LP (B) diet. Co-localization of Aplnr to Ins+ Glut2LO cells is indicated by arrows. Bar represents 80 µm in (A) and 50 µm in (B). The percent Ins+ Glut2LO Aplnr+ cells relative to all Ins+ cells is shown for total pancreas (C), extra-islet clusters (D) or within islets (E) for control (closed circles, black bars) or LP pregnancies (open circles, grey bars). Values represent mean ± SEM (n = 4–6) in non-pregnant females (NP) or at gestational day (GD) 9, 12 or 18. *p < 0.05, **p < 0.001 vs. control.
We utilized a mouse model of glucose intolerance in pregnancy where female offspring of dams exposed to a low protein (LP) diet between conception and weaning have a lower BCM when pregnant, as compared to offspring of control-fed dams21. We examined the abundance of Ins+Glut2LOAplnr+ cells in pregnant mice exposed to the maternal LP diet in early life. The abundance of such cells was significantly reduced in pregnant mouse pancreata from LP-exposed mice at GD 12 and 18 compared to control-fed animals, although a pregnancy-associated increase in their number still occurred (Fig. 4B,C). A similar pattern was seen when data was separated into islet and extra-islet cluster compartments (Fig. 4D,E). Of note, these differences may originate prior to pregnancy as the abundance of Ins+Glut2LOAplnr+ cells was significantly lower in the pancreas of non-pregnant mice that previously received the LP diet.
To determine if this decrease in abundance of Ins+Glut2LOAplnr+ cells in pancreata from glucose intolerant pregnant mice reflected a general decrease of Ins+Glut2LO cells related to LP diet we compared the percentage of Ins+Glut2LO cells relative to all Ins+ cells at each gestational day. For both control and LP pregnancies, Ins+Glut2LO cell presence significantly deceased after GD 9 in whole pancreas and when considering clusters alone but did not differ with prior diet (Table 2). Therefore, the reduced presence of Aplnr immunoreactivity in Ins+Glut2LO cells in LP vs. control pregnancies was not due to an associated change in Ins+Glut2LO cell abundance.
Biological actions of Apelin and Apela on β-cells
A 4,6-diamidino-2 phenylindole, dihydrochloride (MTT) cell proliferation assay was used to examine the actions of Apelin and Apela on INS1E β-cells. Apelin significantly increased cell proliferation at 10 nM (Fig. 5A), but not at lower or higher concentrations. Apela did not significantly increase cell proliferation at 4 nM or greater concentrations relative to controls. Co-incubation with the specific Aplnr antagonist, ML221 significantly negated the proliferative effects of both Apelin and Apela (Fig. 5A) while having no detrimental effect alone. This suggests that any endogenous release of Apelin or Apela from INS1E cells was minimal. We also examined the ability of Apelin to increase β-cell DNA synthesis within islets isolated from neonatal mice. Immunohistochemistry was performed on whole islets to co-localize insulin and Ki67 as a measure of β-cells in G1 and S phase of cell replication (Fig. 5B,C). Apelin significantly increased DNA synthesis in β-cells, as measured by co-localization of insulin and Ki67, at 100 nM but not at greater concentrations (Fig. 5D).

(A) Proliferation of INS1E cells measured by MTT assay in the presence or absence of Apelin (10 nM) or Apela (4 nM) with or without the addition of the Aplnr inhibitor ML221 (10 µM). Values represent mean ± SEM (n = 4–6). *p < 0.05 vs. control, **p < 0.01, ***p < 0.001 vs. Apelin or Apela alone. Cell proliferation of β-cells visualized by fluorescence immunohistochemistry for insulin (green) and Ki67 (red) in islets isolated from neonatal mice incubated without (B) or with Apelin (100 nM, C). Bar represents 50 µm. The percentage of Ki67-positive β-cells was quantified (D) in control culture and in the presence of 100 nM or 1 μM apelin. Values represent mean ± SEM (n = 5–6 replicates per experiment). *p < 0.05 vs. control.
Similarly, the actions of Apelin on GSIS were investigated in both isolated islets from neonatal mice and in INS1E cells (Fig. 6). Incubation in the presence of 16.7 mM glucose (islets) or 28.8 mM glucose (INS1E cells) resulted in a significant increase in insulin secretion (expressed as log values) relative to control cultures containing 2.8 mM glucose. However, the addition of Apelin at 0.1 or 1 mM did not modify insulin release at either basal or stimulating glucose concentrations. When expressed as a fold increase in insulin release between the lower and higher glucose concentrations for islets mean values for control cultures were 6.4 ± 2.1, 7.2 ± 1.7 for Apelin at 0.1 mM and 2.6 ± 0.4 at 1 mM Apelin. Fold increase values for INS1E cell cultures were 10.2 ± 1.4, 8.8 ± 0.3 for Apelin at 0.1 mM and 9.2 ± 0.2 at 1 mM Apelin. Thus, the delta changes in glucose-stimulated insulin release were not significantly altered by Apelin.

Insulin release (log pg/ml) from (A) isolated neonatal mouse islets and (B) INS1E cells in the presence of basal glucose (2.8 mM, black bars) or a stimulating concentration (16.7 mM glucose for islets or 28.8 mM for INS1E cells; grey bars), with or without Apelin at 0.1 mM or 1 mM concentrations. Values represent mean ± SEM (n = 4). *p < 0.05, ***p < 0.001 vs. 2.8 mM glucose.
The placental apelinergic axis
The mitogenic effects of Apelin on β-cells coupled with the increased BCM that occurs during pregnancy could be linked to a placental production of Apelin or Apela. We found no significant change in maternal serum levels of Apelin through gestation during normal pregnancy (Fig. 7A). Maternal Apelin levels in dams who were exposed to the LP diet in early life were significantly greater than those in control-fed animals at GD 9, but not at other times. We also quantified mRNA levels for Apelin, Apela and Aplnr in placental tissues from mice at GD12 and 18 (Fig. 7B). All three proteins were expressed, but levels did not change between GD 12 and 18 in control pregnancies. In glucose intolerant pregnancies the levels of placental Aplnr expression were higher at GD 12 than at GD 18, but did not differ with diet. Expression levels of Apelin and Apela also did not differ with diet.

(A) Serum levels of Apelin detectable in non-pregnant female mouse serum (NP) and at gestational days (GD) 9, 12 and 18 in animal receiving control (closed circles, black bars) or LP diet (open circles, grey bars) in early life. (B) Expression levels of mRNA for Apelin, Aplnr and Apela in placenta from (C) (black bars) or LP-exposed (grey bars) pregnant mice on GD 12 and 18; and (C) expression levels of TNFα, IL-1β and IL-6 in placenta at the same gestational ages. Values represent mean ± SEM (n = 4–6). *p < 0.05, **p < 0.01, ***p < 0.001 vs. control or between days.
Lastly, since GDM is characterized by an enhanced pro-inflammatory environment with elevated levels of pro-inflammatory cytokines that might precipitate β-cell dysfunction, we also quantified mRNA levels of TNFα, IL-1β and IL-6 in placentae from control and LP pregnancies. IL-6 was more highly expressed in LP pregnancies when compared to controls at GD 12, but not at GD 18, whilst TNFα and IL-1β levels did not differ with prior diet (Fig. 7C).
Discussion
Our findings confirm that Apelin, Apela and Aplnr are preferentially expressed within the mouse pancreatic β-cell population from neonatal life until adulthood, and that Aplnr is predominantly expressed within Ins+Glut2LO β-cells most abundant in the periphery of the islets of Langerhans. The abundance of Aplnr-stained Ins+Glut2LO β-cells increased three- to four-fold during pregnancy being maximal at gestational days 9–12 and were significantly reduced in glucose intolerant mice. The likelihood that increased signaling through the apelinergic axis in pancreas contributes to the increased β-cell proliferation seen during pregnancy is supported by the mitogenic effects of Apelin in isolated mouse islets and INS1E cells.
These findings reinforce previous evidence19,20,42 that Ins+Glut2LO pancreatic endocrine cells have a different distribution and gene expression profile compared to Ins+Glut2HI cells. Specifically, the Ins+Glut2LO cells more highly express a wide range of genes associated with β-cell lineage commitment (such as delta like non-canonical notch ligand 143 and fatty acid binding protein-444), proliferation, extracellular matrix remodeling, and angiogenesis. This further supports the hypothesis that Ins+Glut2LO cells are a source of new β-cells given the intimate relationship between the islet microvasculature and BCM, and the role of the extracellular matrix in defining the paracrine signaling that supports the proliferation of both β-cells and endothelial cells45,46. Many of the genes differentially expressed in Ins+Glut2LO cells have also been reported as being selectively up-regulated in mouse islets during pregnancy47, supporting the likelihood that Ins+Glut2LO cells contribute to the increase in BCM observed during mouse pregnancy.
The relatively high expression of apelinergic system genes, Apelin, Apela, and Aplnr, within Ins+Glut2LO cells was confirmed by qPCR for isolated islets relative to Ins+Glut2HI cells while immunohistochemistry showed that Apelin was preferentially co-localized within most β-cells in mouse and human pancreas. The semi-quantitative nature of immunohistochemistry may explain why staining for Apelin was not noticeably different between Ins+Glut2HI and Ins+Glut2LO β-cells despite mRNA expression being significantly greater in the latter. The Ins+Glut2LO cells were preferentially localized in the periphery of the islets, as we described previously48 which may represent a ‘niche’ for new β-cell development from progenitor cells49. Aplnr was more abundantly expressed in Ins+Glut2LO than Glut2HI cells and the peptide was similarly preferentially localized by immunohistochemistry, although localization was also seen in a minority of α-cells as described before37. Additionally, Aplnr was localized to some small cells in the core of the islet with the morphology of endothelial cells. This is consistent with the reported ability of Apelin to promote endothelial cell differentiation50 Apelin was also present in the acinar cells around the periphery of the human pancreas in neonatal subjects, but not adults. In rodent species new pancreatic lobes continue to develop in early postnatal life with proliferation of acinar cells51. If pancreatic lobes continue to be formed postnatally in human then Apelin expression could possibly contribute to this process.
Aplnr has been previously linked to the β-cell generation38. However, this action could be indirect due to the ability of Apelin to promote angiogenesis through the maturation of endothelial cell progenitor cells52. Our findings suggest that Apelin directly promotes β-cell DNA synthesis as seen in both isolated islets and INS1E cells, and the use of a selective Aplnr antagonist demonstrated that the actions were mediated by the Aplnr receptor. Both Apelin and Apela have been shown to activate the PI3K/AKT/mTORC1 signaling pathways, which are potent regulators of proliferation and facilitate a reduction in apoptosis53.
During mouse pregnancy pancreatic Ins+Glut2LO cells are highly proliferative at mid-gestation but this declines in late gestation, possibly through their maturation into functional β-cells20. A similar pattern was seen here during pregnancy for the number of Ins+Glut2LO cells expressing Aplnr, suggesting that the apelinergic system may contribute to the increased BCM. In support of this hypothesis a long-acting Apelin analogue was shown to increase β-cell area within islets following administration of streptozotocin, or following a high fat diet, in mice54. The relative abundance of Ins+Glut2LOAplnr+ cells was significantly reduced in a mouse model of gestational hyperglycemia characterized by a lower BCM further suggesting a causal relationship.
We found no effect of Apelin on GSIS in vitro from INS1E cells or from isolated mouse islets. Previous reports using the same cell line, isolated islets or administration in vivo have been inconsistent37,55,56. However, Apelin has several metabolic actions including the inhibition of lipolysis, regulation of glucose uptake and fatty acid oxidation, and increased mitochondrial bioactivity57. Thus, glucose homeostatic actions in vivo may be a combination of both direct and indirect effects on metabolic tissues. The biological actions of Apelin might also differ between molecular forms. Apelin is synthesized as a 77 amino acid prepropeptide that can be differentially cleaved in a tissue-specific manner at the C-terminal to yield peptides of 35, 17 or 13 amino acids, each with different potencies with respect to Aplnr signaling58. In our studies we utilized the shorter, Apelin-13 form.
The short biological half-life of Apelin means that circulating levels are low (0.02–0.05 pmol/mL in rats)59, implying that locally produced Apelin is likely of most relevance to the control of BCM. However, this may differ during pregnancy when maternal levels increase due to the release of Apelin from the placental syncytiotrophoblast, as reported in humans28. We could not confirm an increasing gestational presence of Apelin in mice, although circulating levels were higher in both non-pregnant and pregnant mice (approximately 1 nM) than those described in women. However, mRNAs for Apelin, Apela and Aplnr were each expressed in mouse placenta. In hyperglycemic mouse pregnancies Apelin levels only differed from values in control pregnancies in mid-gestation and the placental expression of Apelin, Apela, and Aplnr did not differ. However, cellular stress may have been occurring in placentae from glucose intolerant pregnant mice related to a selective increase in IL-6 expression, as was also observed in human gestational diabetes60. Interestingly, incubation of human syncytiotrophoblast cells with increasing concentrations of human Apelin decreased the release of human placental lactogen61, a major trophic factor for the expansion of BCM during pregnancy8,9,10,11. Notably, in human pregnancies with GDM, maternal levels of Apelin were relatively increased in the second trimester, as was observed in the present studies for hyperglycemic mouse pregnancies, whilst levels of Apela were decreased62. The relationship between placental expression of Apelin and BCM during pregnancy is therefore likely to be complex.
In summary, our studies demonstrate the presence of Apelin in pancreatic β-cells throughout mouse pregnancy and show that Apelin exerts mitogenic effects on β-cells through the Aplnr receptor. Aplnr was preferentially localized to pancreatic Ins+Glut2LO cells during pregnancy, and the proportion of such cells immunopositive for Aplnr was decreased in glucose intolerant pregnancy. Thus, we speculate that the apelinergic axis contributes to the increased BCM of pregnancy.
Materials and methods
Animals
A total of 180 C57B6/6J mice (Charles River Laboratories, Wilmington, MA, USA) were used in the studies that generated the data reported. Animals received standard mouse chow and water ad libitum unless otherwise indicated. The studies were compliant with the ARRIVE guidelines both in the design and reporting of the findings. All animal procedures received animal ethics committee approval from Western University, Canada and were undertaken with adherence with the standard operating practices established by Western University and in agreement with published guidelines of the Canadian Council for Animal Care.
Time-mated pregnant mice aged 42–50 days were allowed to deliver and neonatal animals were euthanized by decapitation at 7 days of age before removal of the pancreas for enzymatic dispersal to single cell suspensions prior to fluorescence-activated cell sorting (FACS). The rationale for using neonatal mice was that the number of Ins+Glut2LO cells is greatest in early life42. Pancreata were also collected from pregnant mice at gestational days (GD) 9 and 12 and age-matched non-pregnant females following euthanasia by CO2 asphyxia prior to isolation of the islets of Langerhans.
In separate experiments female mice were time-mated and randomly allocated when pregnant to either a C diet (20% protein, Bioserv, NJ, USA) or a low protein (LP) isocaloric diet (8% protein with a balance of calories from sucrose, Bioserv)21. The respective diets were maintained throughout gestation until weaning (post-natal day 21), at which point the offspring (F1) were transferred to the C-diet. At age 42–50 days, F1 female mice previously exposed to LP or C diets were randomized into pregnant or non-pregnant groups. Those in the pregnancy group were time-mated with C diet-fed C57BL/6J males. LP- and C-exposed pregnant F1 mice were euthanized on either GD 9, 12 or 18. Pancreata and placentae were then removed, weighed, and either fixed in 4% paraformaldehyde for histology, or placed into RNA later (QIAGEN, Hilden, Germany) prior to storage at – 20 °C for future RNA isolation. Blood was collected via cardiac puncture after death and serum separated to quantify circulating apelin.
Additional details of animal care and analytical methods are provided as Supplementary Information.
Fluorescence activated cell sorting (FACS) and DNA microarray analysis
Dispersed cells from whole pancreata of 7-day-old mice were subjected to FACS as described previously19. Cells fractions were separated based on the binding of antibodies against GPm6a (a cell surface marker specific for mouse β-cells63) and Glut 2 to create Ins+Glut2HI or Ins+Glut2LO fractions. Using the RNeasy Plus Mini kit (QIAGEN), total RNA was extracted and purified from each cell pool and DNA microarray analysis performed at the London Regional Genomics Centre, Western University, London, ON, Canada (Mouse Genome 430 2.0 (MOE430 2.0) array, Affymetrix, Santa Clara, CA, USA). All procedures, including cRNA synthesis, labelling, and hybridization were performed as described in the Affymetrix Technical Analysis Manual. The GeneChips were scanned with the Affymetrix GeneChip Scanner 3000 and probe level data from the .CEL files were analysed using Partek Genomics Suite v6.5 (Partek, St. Louis, MO, USA). Probes were imported and summarized using multi-array averaging and ANOVA was used to determine fold changes. Only those genes with a tenfold or greater difference in expression between Ins+Glut2HI or Ins+Glut2LO cell fractions were considered further.
Quantitative polymerase chain reaction (qPCR)
RNA was extracted from Ins+Glut2HI or Ins+Glut2LO cell fractions from 7 day-old neonatal mouse pancreata, isolated islets of Langerhans from pregnant mouse pancreata, and from mouse placentae and stored at – 80 °C. Quantitative PCR was performed on a QuantStudio5 Real-time PCR System (Applied Biosystems, Waltham, MA, USA) using TaqMan primers for Apelin, Apela, Aplnr, insulin, TNF-α, IL-1ß, IL-6 and for the control genes, cyclophilin A (cycloA) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to quantify relative gene expression using the ∆∆ cycle threshold (CT) method. Relative gene expression was calculated as fold change compared to the geometric mean of the housekeeping genes GAPDH and cyclophilin A.
Immunohistochemistry
At least two longitudinal cryosections (7 μm) were examined from each mouse pancreas with an interval greater than 100 μm between each. Immunofluorescence histochemistry was performed to localize Apelin, Aplnr, insulin, glucagon, somatostatin and Glut2 as described previously20. Full details of antibody sources and dilutions are provided in the Supplementary Methods. Formalin-fixed, paraffin embedded sections of non-diabetic human pancreas were obtained from the Department of Pathology and Laboratory Medicine, Western University with institutional approval from the Western University Human Research Ethics Board. All methods were performed in accordance with the guidelines and regulations governing the use of human pathological samples by Western University through the research ethics board.
Immunohistochemical staining for Apelin was performed using diaminobenzidine (DAB) as the chromogen. Tissue sections were de-identified and the histology quantified using a Nikon Eclipse TS2R inverted microscope (Nikon, Minato, Tokyo, Japan) with the program NIS elements (Nikon, Minato). Images were captured and analyzed using cell counter on ImageJ software. Every insulin, Aplnr, or Glut2-expressing cell was imaged for each section and for each animal. In this study, an “islet” was considered to contain six or more β-cells, and an extra-islet endocrine “cluster” containing 1–5 β-cells19.
Isolated islet and INS1E cell culture
Pancreata from neonatal or pregnant mice were digested with collagenase V and islets separated using a Dextran density gradient consisting of 27, 23 and 11% concentrations and collected from the 23/11% interface. Islets were incubated for 24 h and allocated the following day into 6-well ultra-low attachment multiwell plate (Falcon, VWR International) in RPMI medium for 48 h, with and without Pyr-Apelin 13 (100 nM, 1 μM; Sigma. Following exposure to Apelin, islets (approximately 20 islets/treatment) were hand-picked and allowed to affix to glass-bottom dishes (MatTek Life Sciences, Ashland, MA, USA) pre-adsorbed with diluted Cell-Tak adhesive (BD Biosciences), fixed in 4% paraformaldehyde for 30 min at room temperature and stored at 4 °C in phosphate buffered saline (PBS). Immunofluorescent staining for insulin and Ki67 was performed on whole islets to assess the percentage of β-cells undergoing DNA synthesis.
Z-stack images were collected from control or Apelin-treated islets using confocal microscopy (Nikon A1R, Nikon Canada, Mississauga, ON, Canada) with an average of 26 images per stack. Four to six randomly selected images per islet (20–25 islets/treatment) were analyzed using the cell counter on ImageJ software and the percentage of Ki67+ cells relative to insulin+ cells was calculated.
An MTT assay was used to determine the effects of Apelin or Apela on the proliferation of INS1E cells (gifted by Dr. D. Kilkenny, University of Toronto). Culture medium contained Apelin (10 nM) or Apela (4 nM) with or without the specific Aplnr antagonist, ML221 (10 µM; BioTechne, Minneapolis, MN, USA). Spectrophotometric absorbance of the purple formazan crystal product was measured using a microplate reader at an absorbance wavelength of 590 nm.
Isolated islets (approximately 20 islets/well) and INS1E cell cultures (approximately 70% confluency) were also incubated in the presence or absence of Apelin (100 nM or 1 μM) for 48 h prior to incubation with either low (2.8 mM) or high (islets 16.7 mM, INS1E cells 28.8 mM) glucose for 90 min at 37 °C. Conditioned culture medium was removed and insulin release determined using an Ultra-Sensitive Mouse Insulin ELISA kit (Crystal Chem, Elk Grove Village, IL, USA).
Apelin measurement
A competitive enzyme-linked immunosorbent assay (ELISA) was used to quantify Apelin in pregnant and non-pregnant mouse serum using the manufacturer’s instructions (NBP2-68235, Novus Biologicals, Centennial, CO, USA). Data was then collected using an iMark Plate Reader (Bio-Rad) and analyzed using Microplate Manager Software (Bio-Rad).
Statistical analysis
A sample size of 4–6 animals per variable was utilized based on achieving a statistically significant difference (p < 0.05) with an expected standard deviation around mean values of 15% or less for pancreas immunohistochemistry, based on previous studies19,20. Where duplicate tissue sections from the same pancreas gave discordant values two further sections were examined. DNA microarray analysis was repeated twice. Statistics were conducted using GraphPad Prism software and data are presented as mean ± SEM. The data were analyzed using either a Student’s unpaired t-test, a one-way ANOVA or a two-way ANOVA and a Tukey post-hoc test was conducted to compare study groups at different experimental time points. Insulin values were log transformed before analysis.
Data availability
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
References
Van Assche, F. A., Aerts, L. & De Prins, F. A morphological study of the endocrine pancreas in human pregnancy. Br. J. Obstet. Gynaecol. 85, 818–820 (1978).
Butler, A. E. et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 53, 2167–2176 (2010).
Xiang, A. et al. Longitudinal changes in insulin sensitivity and beta-cell function between women with and without a history of gestational diabetes mellitus. Diabetologia 56, 2753–2760 (2013).
Lorenzo, P. I., Martín-Montalvo, A., Cobo Vuilleumier, N. & Gauthier, B. R. Molecular modelling of islet β-cell adaptation to inflammation in pregnancy and gestational diabetes mellitus. Int. J. Mol. Sci. 20, 6171. https://doi.org/10.3390/ijms20246171 (2019).
Yang, Y. et al. Functional defects of regulatory T cell through interleukin 10 mediated mechanism in the induction of gestational diabetes mellitus. DNA Cell. Biol. 37, 278–285 (2018).
Vitoratos, N. et al. Pre- and early post-partum adiponectin and Interleukin-1beta levels in women with and without gestational diabetes. Hormones 7, 230–236 (2018).
Chen, L., Chen, R., Wang, H. & Liang, F. Mechanisms linking inflammation to insulin resistance. Int. J. Endocrinol. 2015, 1–9 (2015).
Sorenson, R. L. & Brelje, T. C. Adaptation of islets of Langerhans to pregnancy: Beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm. Metab. Res. 29, 301–307 (1997).
Parsons, J. A., Brelje, T. C. & Sorenson, R. L. Adaptation of islets of Langerhans to pregnancy: Increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology 130, 1459–1466 (1992).
Newbern, D. & Freemark, M. Placental hormones and the control of maternal metabolism and fetal growth. Curr. Opin. Endocrinol. Diabetes Obes. 18, 409–416 (2011).
Vasavada, R. C. et al. Targeted expression of placental lactogen in the beta cells of transgenic mice results in beta cell proliferation, islet mass augmentation, and hypoglycemia. J. Biol. Chem. 275, 15399–153406 (2000).
Huang, C., Snider, F. & Cross, J. C. Prolactin receptor is required for normal glucose homeostasis and modulation of β-cell mass during pregnancy. Endocrinology 150, 1618–1626 (2009).
Brelje, T. C., Stout, L. E., Bhagroo, N. V. & Sorenson, R. L. Distinctive roles for prolactin and growth hormone in the activation of signal transducer and activator of transcription 5 in pancreatic islets of Langerhans. Endocrinology 145, 4162–4175 (2004).
Finegood, D. T., Scaglia, L. & Bonner-Weir, S. Dynamics of β-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes 44, 249–256 (1995).
Bader, E. et al. Identification of proliferative and mature beta-cells in the islets of Langerhans. Nature 535, 430–434 (2016).
Chen, H. et al. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature 478, 349–355 (2011).
Seaberg, R. M. et al. Clonal identification of multipotent precursors from adult mouse pancreas that generate neural and pancreatic lineages. Nat. Biotechnol. 22, 1115–1124 (2004).
Smukler, S. R. et al. The adult mouse and human pancreas contain rare multipotent stem cells that express insulin. Cell Stem Cell 8, 281–293 (2011).
Beamish, C. A., Strutt, B. J., Arany, E. J. & Hill, D. J. Insulin-positive, Glut2-low cells present within mouse pancreas exhibit lineage plasticity and are enriched within extra-islet endocrine cell clusters. Islets 8, 65–82 (2016).
Beamish, C. A., Zhang, L., Szlapinski, S. K., Strutt, B. S. & Hill, D. J. An increase in immature β-cells lacking Glut2 precedes the expansion of β-cell mass in the pregnant mouse. PLoS One 12, e0182256. https://doi.org/10.1371/journal.pone.0182256 (2017).
Szlapinski, S. et al. A mouse model of gestational glucose intolerance through exposure to a low protein diet during fetal and neonatal development. J. Physiol. 597, 4237–4250 (2019).
Nielsen, L. R., Rehfeld, J. F., Pedersen-Bjergaard, U., Damm, P. & Mathiesen, E. R. Pregnancy-induced rise in serum C-peptide concentration in women with type 1 diabetes. Diabetes Care 32, 1052–1057 (2009).
Cantley, J. The control of insulin secretion by adipokines: Current evidence for adipocyte-beta cell endocrine signalling in metabolic homeostasis. Mamm. Genome 25, 442–454 (2014).
Deng, C., Chen, H., Yang, N., Feng, Y. & Hsueh, A. J. Apela regulates fluid homeostasis by binding to the APJ receptor to activate Gi signaling. J. Biol. Chem. 290, 18261–18268 (2015).
Zhang, J. et al. The orphan G protein-coupled receptor 25 (GPR25) is activated by Apelin and Apela in non-mammalian vertebrates. Biochem. Biophys. Res. Commun. 501, 408–414 (2018).
Ladeiras-Lopes, R., Ferreira-Martins, J. & Leite-Moreira, A. The apelinergic system: The role played in human physiology and pathology and potential therapeutic applications. Arq. Bras. Cardiol. 90, 343–349 (2008).
Mayeur, S. et al. Apelin controls fetal and neonatal glucose homeostasis and is altered by maternal undernutrition. Diabetes 65, 54–560 (2015).
Eberlé, D. et al. Elabela and apelin actions in healthy and pathological pregnancies. Cytokine Growth Factor Rev. 46, 45–53 (2019).
Van Mieghem, T. et al. Apelin in normal pregnancy and pregnancies complicated by placental insufficiency. Reprod. Sci. 23, 1037–1043 (2016).
Van Mieghem, T. et al. Maternal apelin physiology during rat pregnancy: The role of the placenta. Placenta 31, 725–730 (2010).
O’Carroll, A., Lolait, S. J., Harris, L. E. & Pope, G. R. The apelin receptor APJ: Journey from an orphan to a multifaceted regulator of homeostasis. J. Endocrinol. 219, R13-35 (2013).
Yue, P. et al. Apelin is necessary for the maintenance of insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 298, E59-67 (2010).
Krist, J. et al. Effects of weight loss and exercise on apelin serum concentrations and adipose tissue expression in human obesity. Obes. Facts 6, 57–69 (2013).
Noori-Zadeh, A., Bakhtiyari, S., Khanjari, S., Haghani, K. & Darabi, S. Elevated blood apelin levels in type 2 diabetes mellitus: A systematic review and meta-analysis. Diabetes Res. Clin. Pract. 14, 43–53 (2018).
Hanssens, S. et al. Maternal obesity alters the apelinergic system at the feto-maternal interface. Placenta 39, 41–44 (2016).
Sun, J. et al. Circulating apelin, chemerin and omentin levels in patients with gestational diabetes mellitus: A systematic review and meta-analysis. Lipids Health Dis. 19, 26. https://doi.org/10.1186/s12944-020-01209-7 (2020).
Ringström, C. et al. Apelin is a novel peptide. Regul. Pept. 162, 44–51 (2010).
Han, S. et al. Pancreatic islet APJ deletion reduces islet density and glucose tolerance in mice. Endocrinology 156, 2451–2460 (2015).
Chen, H. et al. Apelin alleviates diabetes-associated endoplasmic reticulum stress in the pancreas of Akita mice. Peptides 32, 1634–1639 (2011).
Gao, L. R. et al. Overexpression of apelin in Wharton’ jelly mesenchymal stem cell reverses insulin resistance and promotes pancreatic β cell proliferation in type 2 diabetic rats. Stem Cell Res. Ther. 9, 339. https://doi.org/10.1186/s13287-018-1084-x (2018).
Hou, J. et al. Hypoxia preconditioning promotes cardiac stem cell survival and cardiogenic differentiation in vitro involving activation of the HIF-1α/apelin/APJ axis. Stem Cell Res. Ther. 8, 215. https://doi.org/10.1186/s13287-017-0673-4 (2017).
Beamish, C. A. et al. Decrease in Ins+GLUTLO β-cells with advancing age in mouse and human pancreas. J. Endocrinol. 233, 229–241 (2017).
Friedrichsen, B. N. et al. Expression, biosynthesis and release of preadipocyte factor-1/delta-like protein/fetal antigen-1 in pancreatic β-cells: Possible physiological implications. J. Endocrinol. 176, 257–266 (2003).
Wu, L. E. et al. Identification of fatty acid binding protein 4 as an adipokine that regulates insulin secretion during obesity. Mol. Metab. 3, 465–473 (2014).
Lammert, E. & Thorn, P. The role of the islet niche on beta cell structure and function. J. Mol. Biol. 432, 1407–1418 (2020).
Aamodt, K. I. & Powers, A. C. Signals in the pancreatic islet microenvironment influence β cell proliferation. Diabetes Obes. Metab. 19(Suppl 1), 124–136 (2017).
Horn, S. et al. Research resource: A dual proteomic approach identifies regulated islet proteins during β-cell mass expansion in vivo. Mol. Endocrinol. 30, 133–143 (2016).
Szlapinski, S. K., Bennett, J., Strutt, B. J. & Hill, D. J. Increased alpha and beta cell mass during mouse pregnancy is not dependent on transdifferentiation. Exp. Biol. Med. 246, 617–628 (2021).
Huising, M. O., Lee, S. & van der Meulen, T. Evidence for a neogenic niche at the periphery of pancreatic islets. BioEssays 40, e1800119. https://doi.org/10.1002/bies.201800119 (2018).
Masoud, A. G. et al. Apelin directs endothelial cell differentiation and vascular repair following immune-mediated injury. J. Clin. Investig. 130, 94–107 (2020).
Bonner-Weir, S., Aguayo-Mazzucato, C. & Weir, G. C. Dynamic development of the pancreas from birth to adulthood. Upsala J. Med. Sci. 121, 155–158 (2016).
Yu, M., Feng, H. J., Abdalla, A. M. E., Teng, Y. F. & Li, Q. Apelin-13 promotes late endothelial progenitor cells differentiation by regulating Krüppel-like factor 4. Eur. Rev. Med. Pharmacol. Sci. 23, 7098–7109 (2019).
Kinjo, T., Higashi, H., Uno, K. & Kuramoto, N. Apelin/apelin receptor system: Molecular characteristics, physiological Roles, and prospects as a target for disease prevention and pharmacotherapy. Curr. Mol. Pharmacol. https://doi.org/10.2174/1874467213666200602133032 (2020).
Tanday, N., Irwin, N., Moffett, R. C., Flatt, P. R. & O’Harte, F. P. M. Beneficial actions of a long-acting apelin analogue in diabetes are related to positive effects on islet cell turnover and transdifferentiation. Diabetes Obes. Metab. 22, 2468–2478 (2020).
Guo, L. et al. Apelin inhibits insulin secretion in pancreatic beta-cells by activation of PI3-kinase-phosphodiesterase 3B. Endocr. Res. 34, 142–154 (2009).
Sörhede Winzell, M., Magnusson, C. & Ahrén, B. The APJ receptor is expressed in pancreatic islets and its ligand, apelin, inhibits insulin secretion in mice. Regul. Pept. 131, 12–17 (2005).
Feng, J., Zhao, H., Du, M. & Wu, X. The effect of apelin-13 on pancreatic islet beta cell mass and myocardial fatty acid and glucose metabolism of experimental type 2 diabetic rats. Peptides 114, 1–7 (2019).
Carpéné, C. et al. Expanding role for the apelin/APJ system in physiopathology. J. Physiol. Biochem. 63, 359–373 (2007).
Kawamata, Y. et al. Molecular properties of apelin: Tissue distribution and receptor binding. Biochim. Biophys. Acta 1538, 162–171 (2001).
Barbour, L. A. et al. Cellular mechanisms for insulin resistance in normal pregnancy and gestational diabetes. Diabetes Care 30(Suppl. 2), 112–119 (2007).
Dawid, M., Mlyczynska, E., Kurowska, P., Sierpowski, M. & Rak, A. Apelin decreased placental hormone secretion by human trophoblast BeWo cells via apelin receptor, protein kinase A and extracellular signal-regulated kinases 1/2 activation. J. Physiol. Pharmacol. https://doi.org/10.26402/jpp.2019.6.08 (2019).
Guo, Y. Y. et al. Circulating levels of Elabela and Apelin in the second and third trimesters of pregnancies with gestational diabetes mellitus. Gynecol. Endocrinol. 2, 1–5 (2020).
Dorrell, C. et al. Isolation of mouse pancreatic alpha, beta, duct and acinar populations with cell surface markers. Mol. Cell. Endocrinol. 339, 144–150 (2011).
Acknowledgements
SS was a recipient of the Ontario Graduate Scholarship. We thank Houshang Azimi for technical support and Kristin Chadwick at the Cell Sorting Core facility, Western University for expert advice. This work was supported by grants from the Canadian Institute of Health Research (MOP-15263), the Alan Thicke Centre for Juvenile Diabetes Research, and the Lawson Foundation, Toronto, ON, Canada.
Author information
Authors and Affiliations
Contributions
B.S., S.S., T.G., S.D., J.H. and J.B. were responsible for experimental procedures generating data included in the report. D.H., B.S., S.S. and J.H. each contributed to the experimental plan. All authors contributed to the writing and editing of the report. The guarantor is D.H. who accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
Corresponding author
Ethics declarations
Competing interests
The study funders were not involved in the design of the study; the collection, analysis, and interpretation of data; writing the report; and did not impose any restrictions regarding the publication of the report. The authors had no financial or professional conflicts of interest with the funding agencies.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Strutt, B., Szlapinski, S., Gnaneswaran, T. et al. Ontology of the apelinergic system in mouse pancreas during pregnancy and relationship with β-cell mass. Sci Rep 11, 15475 (2021). https://doi.org/10.1038/s41598-021-94725-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-94725-0
This article is cited by
-
ID1 marks the tumorigenesis of pancreatic ductal adenocarcinoma in mouse and human
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
