
Abstract
Early detection improves survival and increases curative probability in hepatocellular carcinoma (HCC). Peripheral blood mononuclear cells (PBMCs) can provide an inexpensive, less-invasive and highly accurate method. The objective of this study is to find the potential marker for HCC screening, utilizing gene expression of the PBMCs. Data from the NCBI GEO database of gene expression in HCC patients and healthy donor's PBMCs was collected. As a result, GSE 49515 and GSE 58208 were found. Using both, a statistical significance test was conducted in each gene expression of each data set which resulted in 187 genes. We randomized three selected genes (FLNA, CAP1, and CLU) from the significant p-value group (p-values < 0.001). Then, a total of 76 healthy donors, 153 HCC, 20 hepatic fibrosis, 20 non-alcoholic fatty liver were collected. Quantitative RT-PCR (qRT-PCR) was performed in cDNA from all blood samples from the qRT-PCR, The Cycle threshold (Ct) value of FLNA, CLU, CAP1 of HCC group (28.47 ± 4.43, 28.01 ± 3.75, 29.64 ± 3.90) were lower than healthy group (34.23 ± 3.54, 32.90 ± 4.15, 32.18 ± 5.02) (p-values < 0.0001). The accuracy, sensitivity and specificity of these genes as a screening tool were: FLNA (80.8%, 88.0%, 65.8%), CLU (63.4%, 93.3%, 31.3%), CAP1 (67.2%, 83.3%, 39.1%). The tests were performed in two and three gene combinations. Results demonstrated high accuracy of 86.2%, sensitivity of 85% and specificity of 88.4% in the FLNA and CLU combination. Furthermore, after analyzed using hepatic fibrosis and non-alcoholic fatty liver as a control, the FLNA and CLU combination is shown to have accuracy of 76.9%, sensitivity of 77.6% and specificity of 75%. Also, we founded that our gene combination performs better than the current gold standard for HCC screening. We concluded that FLNA and CLU combination have high potential for being HCC novel markers. Combined with current tumor markers, further research of the gene’s expression might help identify more potential markers and improve diagnosis methods.
Similar content being viewed by others
Introduction
Hepatocellular carcinoma (HCC) is the fourth most common cause of cancer-related mortality worldwide1. Annually, liver cancer accounts for approximately 840,000 new cases and 780,000 death cases2. In Thailand, HCC tends to be diagnosed late and until it becomes worse, for example, 75% of patients diagnosed as HCC have already reached stage B or C in Barcelona Clinic Liver Cancer staging, and 25% of the patients were asymptomatic3.
Primary screening for HCC is mainly conducted either by Alpha-Fetoprotein (AFP) level from blood sample or by ultrasound imaging. If an abnormality is found, a contrast-enhanced multiphase Computerized Tomography or Magnetic Resonance Imaging study would be done4. However, the AFP blood test produces a wide variation of results with sensitivity ranging from 32 to 79.5% and specificity ranging from 29.4 to 98.5%5,6,7. Ultrasound is also problematic because it is operator dependent. The results may vary from each operator thus, reducing its capability. Ultrasound test sensitivity for the detection of HCC ranges from 29 to 100%, whereas its specificity ranges from 94 to 100%. This means both AFP and ultrasound performance as screening/diagnosis markers are not very satisfactory8.
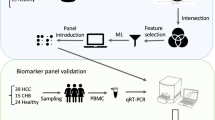
Recently, there are multiple studies regarding the change in gene expression in circulating white blood cells of the cancer patient9,10,11. Also, there are experiments utilizing the gene expression on peripheral white blood cells to detect these cancer’s influences12,13,14,15,16. Therefore, an invention of markers that are inexpensive, simple, less-invasive and highly accurate can be expected. We focused on the usability of peripheral white blood cells (WBCs), especially peripheral blood mononuclear cells (PBMCs) as a biomarker for liver cancers17. The objective of this study is to find the high-performance novel markers for HCC screening, utilizing gene expression of PBMCs (Fig. 1b).

Summary of the experiment. (a) Venn diagram based on the overlapping number of upregulated genes between two microarray data extracted from Gene Expression Omnibus (GEO) data repository. (b) A schematic design flow of the experiment.
Results
Data summary
Bioinformatic data of the samples
Results of the CU-DREAM program showed a significant p-value in the comparison between GSE4951518 and GSE5820819 (p-value = 9.91 × 10−19, Odd ratio = 2.08, upper 95%CI = 2.46 and lower 95%CI = 1.76). All 187 upregulated genes were classified to identify the highest significant p-value. Then, three genes with high significant p-values including CLU (p-value = 8.16 × 10−5), FLNA (p-value = 3.35 × 10−5), and CAP1 (p-value = 2.84 × 10−7) were selected and applied to observe the gene expression in this study (Fig. 1a, Table 1) .
Characteristics of participants
Age (Mean ± SD) of HCC group was 58.93 ± 9.99, Healthy donor group was 48.32 ± 5.16, hepatic fibrosis (HF) group was 57.85 ± 8.92, and non-alcoholic fatty liver (NAFL) group was 57.50 ± 8.31. Gender data shows that our samples had more males than females (Table 2); The HCC group has 124 males to 29 females, the Healthy group has 45 males to 31 females, the HF group has 16 males to 4 females, and the NAFL group has 18 males to 2 females. The staging of HCC from the cancer group showed 12 samples in stage 0, 54 samples in stage A, 61 samples in stage B, 26 samples in stage C, and no samples in stage D according to the BCLC staging system.
The laboratory result of The HCC group show that more than half of the HCC patient has Child–Pugh class A (80 from 153) while class C are lesser in number (57 from 153) and class B has the least number (16 from 153). Also, the majority of the HCC samples have cirrhosis (120 from 153). The Blood cell count is notably within the normal range. Hemoglobin is 12.36 ± 1.95 (g/dL), Hematocrit (%) is 37.60 ± 5.02, Platelet count (109/L) is 164.08 ± 91.58, and WBC count (109/L) is 6.21 ± 3.59. The liver function test is predictively increased to that of chronic liver disease. Aspartate aminotransferase (IU/L) level is 67.68 ± 44.95, Alanine aminotransferase (IU/L) level is 53.73 ± 38.62, Alkaline phosphatase level (IU/L) is 133.41 ± 68.56. The serum alpha fetoprotein level is also increased (13,251.54 ± 69,742.37 ng/mL). Additionally, the HF and the NAFL groups’ laboratory result shown that both are within normal range. Sample from both HF and NAFL group are belong only to class A from Child–Pugh classification system (Table 2).
Quantitative Real-time PCR analysis with 2−ΔΔCt calculation
In the HCC group, The Ct value was 28.47 ± 4.43 for FLNA, 28.01 ± 3.75 for CLU and 29.64 ± 3.90 for CAP1. The level of Ct value in the healthy group was 34.23 ± 3.54 for FLNA, 32.90 ± 4.15 for CLU and 32.18 ± 5.02 for CAP1. The Ct value of HF group was 30.75 ± 3.90 for FLNA, 32.14 ± 0.78 for CLU and 30.01 ± 4.94 for CAP1. The Ct value of NAFL group was 30.46 ± 3.45 for FLNA, 32.13 ± 0.80 for CLU and 28.98 ± 3.30 for CAP1 (Table 3). These results demonstrated that the Ct values in HCC group were significantly lower than the healthy group, (p-value < 0.0001 in FLNA, CLU and p-value = 0.0003 in CAP1) (Fig. 2).

Ct values and expression (2−ΔΔCt) of each gene compare to the housekeeping gene (GAPDH) within Healthy Donor group and HCC cancer group, shown in boxplot (Mean ± SD); (a) Ct value of FLNA gene, (b) Ct value of CLU gene, (c) Ct value of CAP1 gene, (d) Expression of FLNA gene, (e) Expression of CLU gene, (f) Expression of CAP1 gene. The Ct values in HCC group were significantly lower than the healthy group (p-value < 0.0001 in FLNA, CLU and p-value = 0.0003 in CAP1). The expression of FLNA, CLU, CAP1 was increase in the HCC group when compare with the healthy group (p-value < 0.0001 in FLNA and CLU and p-value = 0.4663).
Moreover, when we utilized either HF group or NAFL group or the combined HF and NAFL group as the control and compare each gene expression against HCC group, we found that only CLU gene is shown to express significantly difference from other genes. The significant different of each gene expression when pairing between HCC group and HF group are p-value = 0.08 for FLNA gene, p-value < 0.0001 for CLU gene, p-value = 0.64 for CAP1 gene. The significant different of each gene expression when pairing between HCC group and NAFL group are p-value = 0.06 for FLNA gene, p-value < 0.0001 for CLU gene, p-value = 0.32 for CAP1 gene. The significant different of each gene expression when pairing between HCC group and HF group with NAFL group are p-value = 0.013 for FLNA gene, p-value < 0.0001 for CLU gene, p-value = 0.34 for CAP1 gene. Also, there is no statistically significant difference of each gene expression between HF group and NAFL group (Table 4).
Furthermore, we used the 2−ΔΔCt method to calculate expression power compared to the house keeping gene from both HCC and healthy groups. We found that within the HCC group FLNA, CLU, CAP1 gene expressed (Median) 112.7 folds, 134.2 folds, 11.3 folds to the house keeping gene expression, respectively while the Healthy group expressed 1.9 folds, 0.1-fold, 17.1 folds to the house keeping gene, respectively with p-values of < 0.0001, < 0.0001, 0.4663, respectively (Fig. 2).
The performance of genes as a screening test
The performance of results (Fig. 3) are reported hereafter. When using the cut-off value of Ct value < 33, the results of accuracy, sensitivity and specificity were FLNA (80.8% accuracy, 88.0% sensitivity, 65.8% specificity) (Fig. 3a), CLU (63.4% accuracy, 93.3% sensitivity, 31.3% specificity) (Fig. 3b) and CAP1 (67.2% accuracy, 83.3% sensitivity, 39.1% specificity) (Fig. 3c). Then, the two and three-gene combinations were performed. The results showed that the combination of FLNA & CLU (86.2% accuracy, 85.0% sensitivity, 88.4% specificity) (Fig. 3d) demonstrated higher proficiency than the combination of FLNA & CAP1 (74.1% accuracy, 77.5% sensitivity, 68.1% specificity) (Fig. 3e), CLU & CAP1 (76.4% accuracy, 80.8% sensitivity, 67.7% specificity) (Fig. 3f). However, the three-gene combination could not affect the efficiency of the test (80.2% accuracy, 75.8% sensitivity, 88.7% specificity) (Fig. 3g). From all the ROC graphs (Fig. 3) we showed that the combination of FLNA & CLU has the greatest discriminate capacity than the other tests.

The ROC graph and the result of accuracy, sensitivity, specificity in each combination including; (a) FLNA, (b) CLU, (c) CAP1, (d) FLNA and CLU, (e) FLNA and CAP1, (f) CLU and CAP1, (g) FLNA and CLU and CAP1. The combination of FLNA and CLU has the greatest discriminate capacity than other tests (86.2%accuracy, 85.0%sensitivity, 88.4%specificity).
The performance of the genes when using sample from HF group with NAFL group are as follow. When using the cut-off value of Ct value < 31 the results of accuracy, sensitivity and specificity were FLNA (64.8% accuracy, 71.9% sensitivity, 37.5% specificity) (Fig. 4a), CLU (88.5% accuracy, 86.2% sensitivity, 95% specificity) (Fig. 4b) and CAP1 (52.9% accuracy, 63.2% sensitivity, 22.5% specificity) (Fig. 4c), FLNA & CLU (76.9% accuracy, 77.6% sensitivity, 75% specificity) (Fig. 4d), FLNA & CAP1 (51% accuracy, 55.6% sensitivity, 37.5% specificity) (Fig. 4e), CLU & CAP1 (71.8% accuracy, 70.7% sensitivity, 75% specificity) (Fig. 4f), and the three-gene combination (68.6% accuracy, 66.4% sensitivity, 75% specificity) (Fig. 4g). From all the ROC graphs (Fig. 4).

The ROC graph and the result of accuracy, sensitivity, specificity in each combination including; (a) FLNA, (b) CLU, (c) CAP1, (d) FLNA and CLU, (e) FLNA and CAP1, (f) CLU and CAP1, (g) FLNA and CLU and CAP1.This analysis use samples from the hepatic fibrosis group and the non-alcoholic fatty liver group as a control.
In addition, the performance of the genes to detect the early liver cancer stage is also analyzed. We found that all three genes can distinguish between the healthy group and the early HCC group (Table 3). However, all three genes show no statistically significant distinction between liver cancer stages (Table 3).
Furthermore, we test the early HCC group (stage 0, A of BCLC staging system) (66 subjects) with both the current gold standard for HCC screening (AFP and ultrasonography) and our gene combination (FLNA and CLU). We found that our gene combination is superior in identifying the early HCC than the gold standard. While the gold standard has an accuracy of 30.3% (20 of 66), our gene combination has accuracy of 69.7% (46 of 66) with p-value < 0.0001. This reinforces the capability of our gene combination as a novel screening marker for HCC.
Discussion
FLNA and CLU gene combination might be a prospective marker for HCC. We demonstrated that the PBMCs are affected by the HCC and the result contained the upregulated FLNA and CLU gene (86.2% accuracy, 85.0% sensitivity, 88.4% specificity) which possess high performance as novel screening markers for discriminating the presence or absence of HCC in patients. Furthermore, both genes shown to be able to separate early cancer patient and healthy people. Moreover, this test only requires peripheral blood for testing e.g., AFP and is considered one of the least invasive types of testing available. Furthermore, this test is operator independent, unlike ultrasound which require years of training and hours of procedures to be able to produce reliable results.
From the result of our initial analysis of the microarray data, we found that there are differences in gene expressions of the PBMCs between HCC patients and normal people (Fig. 1a, Table 1). The genes that significantly upregulate are involved in the immunotolerance process, anti-apoptosis, and pro-proliferative pathways. The mechanisms behind these modifications are still unknown but could be theoretically divided into three processes.
The first process is by various cytokines released from cancer cells or the immune cell responding to the cancer cells. The cytokines and chemokines are known to be involved in the interaction between malignant cells and immune cells20,21,22,23. Mostly, in the roles of chemotaxis, pro-angiogenesis, and inflammatory response. Recent studies show that the cancer cells are responsible for the changes in the release of the cytokines from PBMCs and those cytokines are also associated with tumor growth and invasion24.
The second process is by direct cell to cell interaction between the cancer cells and immune cells. Cancer cells and immune cells have a predator–prey-like relationship. If the immune cells recognize the cancer cell, the immune cell will initiate the apoptotic process of the recognized cancer cell25. But, if the immune cells fail to recognize the cancer cell, it will continue to grow unimpeded. This process is governed by multiple receptors and ligands such as major histocompatibility complex (MHC), program death ligand 1 or 2 (PD-L1 or PD-L2), CD80, CD86, CD28, cytotoxic T-lymphocytes-associated protein 4 (CTLA-4), T-cell receptor (TCR), and many others26,27. The malignant cell could disrupt these processes by genetic mutations. The result of the mutation could lead to the transmutations or the removal of the receptor/ligand/key protein in the processes interrupting the immune cell response. Furthermore, the alteration in these cell–cell signals might lead to the change in WBCs itself. A recent study shows that there is a dynamic relationship between the cancer cells and peripheral immune cell phenotype28. If the cancer cell has over-expressed PD-L1 it will lead immune cells’ dysfunction and also lead to the reduction in the number of circulating immune cells because the proliferation of the immune cells is prohibited29. This suggests a connection between cell–cell interaction and immune cells gene expressions.
The third process is due to the shift in the body's physiology that causes the changes in gene expression of PBMCs. the third process is targeting the macro-environment of the cancer cells. This third process involves multiple origins such as the inherited genetic susceptibility, the socio-economic issues which lead to different lifestyles (e.g., Alcohol consumption, smoking, profession-related environment, etc.), and mental health issue (e.g., stress from both personal or from the society at large). These will result in changes in the internal mechanism of the body and could potentially cause the change in PBMCs30,31,32,33,34. Multiple studies report on the correlation between stress and immune responds35. Some studies report on alcohol consumption and immunosuppression. All of this supports the approach of the immune cell gene expression is tied to whole-body physiology status. However, the precise mechanisms responsible for these three processes are still eluding the scientists. Thus, further research into the component of these pathways may benefit both the laboratory and clinical fields. This not only includes many possible laboratory investigations but also a potential treatment for multiple cancer as well.
The relationship between HCC cells and each candidate gene does exist. There are reports on FLNA gene that it has a complex role on the cytoskeleton18. FLNA gene encodes Filamin A protein. This protein is a non-muscle actin filament cross-linking protein36. The most common role of this protein is for being a scaffolding molecule to regulate various cell functions. More than 90 types of protein are reported to bind to this protein, and many of those genes are responsible for cytoskeleton reorganizing37. Thus, making it involved in cell migration by prompting transcription factor SRF along with MLK138. This act of triggering both genes result in the tumor undergoes local invasion and metastasis. It is also reported that elevated FLNA gene expression in the harvested HCC tissue could be a predictor of recurrence of HCC39 and could also be a marker for the progression of HCC40. Moreover, there is a report on the importance of Filamin A protein within the immune cell. Mainly, Filamin A is necessary for the activation of human T-cells41. Protein kinase C-θ (PKC-θ) which plays a major role in the early activation of T-cell is reported to require Filamin A protein to translocate itself within the cell. There is also an association between PKC-θ and Interleukin-2 (IL-2) production. IL-2 is a cytokine essential in the maturation and differentiation of the regulatory T-cell which responsible for immunotolerance. The PKC-θ is reported to target Activator protein 1 (AP-1), Nuclear Factor Kappa B (NF-κB), and nuclear factor of activated T-cells (NFAT) which are the transcription factors of the IL-2 gene. The suppression in Filamin A synthesis is reported to result in the reduction of IL-2 production. This predicts an outcome when the cancer cell is successfully evading the immune cell and induced immune tolerance to cancer cell lineage. The demand for regulatory T-cells is increased because the need to suppress the immune response of cancer cells is increased. This required more IL-2 production and PKC-θ activation. Thus, need more Filamin A synthesis which results in overexpress of FLNA gene in PBMCs. This suggests that FLNA gene expression in the immune cell might be linked to the survival of cancer cells. However, multiple studies also report on the tumor-suppressive ability of the Filamin A42. It suppresses tumor growth and reduces tumor invasion by inhibiting transcription of the oncogenes when it has transmigrated into the nucleus.
CLU gene encodes Clusterin protein. This protein is involved in the clearance of cell debris and apoptosis pathway43. Its main function is to be a chaperone molecule that helps prevent cells to undergoes apoptosis by preventing the pro-apoptotic protein to bind to the mitochondria44. It has been reported that Clusterin is involve in many tumorigeneses’ activity. The protein is shown in vitro to involved in cell survival and aggregation45, promoting metastasis of HCC46,47, protecting the HCC cells from endoplasmic reticulum stress induced apoptosis48, regulating the NF-κB pathway which controls the innate immune response of the cell, and affecting resistance to drugs (such as Sorafenib)49,50. Clusterin is also reported to play a role in immunotolerance to the autoimmune associated antigen. After antigen presenting cells (APCs) clear the apoptotic cells by phagocytosis, it will present the antigen derive from the apoptotic cells. Any antigen that got present this way led to an increase in immune tolerance to it. The APCs also produce immunoregulatory cytokine (such as Transforming Growth factor-beta (TGF-β), interleukin-10 (IL-10)) to suppress local inflammatory response and prevent the development of an autoimmune disease51. Without a clearance of apoptotic cells, it will undergo necrosis and trigger an inflammatory response from APCs which led to the different pathway of presenting the antigen. When an antigen got present via inflammatory response, it will be recognized as a foreign element and the immune system will produce antibodies to combat it. If the autoimmune associated antigen got recognized then it could cause the autoimmune disease. Clusterin is reported to enhance the apoptotic way of presenting the antigen within T-cell, APCs and also delayed the transformation of apoptotic cells to necrosis cells. This suggests the association between the cancer cell and Clusterin protein level of the immune cell. The cancer cells could undergo apoptosis by themselves from over-mutation or by other causes and the apoptotic cancer cells got phagocytosis by APCs but get recognized as self-antigen. This led to the immune tolerance to the cancer cells and increase cancer cells’ survival. Then, the survival cancer cell multiplied, and more undergoes apoptosis. This causes more apoptotic cell clearance and the need for Clusterin protein. Thus, could lead to the up-regulation of the CLU gene in PBMCs.
CAP1 gene encodes Cyclase-Associated Protein 1(CAP1 protein). A study of the CAP1 gene reported its involvement in the metastasis of hepatocellular carcinoma due to its relationship to the actin filaments turnover cycle and also hypothesized that the cancer cell invasion will be accelerated when the gene is over-expressed52. It is also thought to be involved in the localization of cell polarity and mRNA53. Another study has reported that CAP1 gene expression increases in other cancers, such as ovarian cancer, and is involved in cell proliferation54. Moreover, CAP1 protein plays a part in stimulating monocyte and cause local inflammation in human55. CAP1 protein is a receptor to the resistin protein. Resistin is a cytokine involved in chronic inflammatory diseases such as atherosclerosis and insulin resistance. When resistin bind to CAP1 protein in monocyte it will activate cAMP-dependent signaling pathway, PKA, and NF-kB. These cause the increase in the production of pro-inflammatory cytokines such as IL-6, TNFα, and IL-1β. Up-regulate CAP1 in monocyte enhance the resistin-induced activity and cause low-level inflammation to develop into chronic inflammation. Cancer is associated with chronic inflammation56. By repeatedly undergoes inflammation, the cells will accumulate both DNA damages and mutations which could cause the transmutation from normal cells to cancer cells. If low-level inflammation accumulates in the cancer cells region, it will promote cancer cells’ development. This suggests the association between CAP1 gene expression in PBMCs and the development of cancer.
It may be possible to think that cancer cells reshape the PBMC with various mechanism. An experiment was conducted in our laboratory in breast cancer cells which showed the actual “reshaped” by cancer cell was carried out, although the detailed mechanism is unknown. The same phenomenon seemed to be occurring also in the case of HCC. Future study to clarify the mechanism are needed.
The benefits of this study (qRT-PCR assay) are apparent. Currently, the diagnosis of HCC has been done with imaging tests (Ultrasound, CT, MRI), conventional markers like AFP, or biopsy. However, all of them have limitations: imaging tests are quite expensive and not very suitable for wide screening and some tests are operator dependent; conventional markers have unsatisfiable accuracy; the pathological diagnosis using biopsy is highly invasive. Our FLNA and CLU combination markers, on the contrary, can be attained by less invasive blood test, yield high performance, and could be done with lower cost. Additionally, our genes can differentiate between patient with HCC from the patient with predisposing condition such as hepatic fibrosis, and non-alcoholic fatty liver (Table 4) but the gene combination suffer a little reduction in the performances (Fig. 4). Our markers could be expected to contribute in both screening and diagnosis of HCC in future clinical application.
The limitations of this study include the absence of in vitro investigation of this experiment. This is because the researchers have discussed and theorized on the processes behind the alteration in PBMCs’ gene expression to be a multifactorial process and determine that a co-culture between HCC cell line and PBMCs is not necessarily required. This is because the co-culture could only reflect one of the three processes that led to the change in gene expression and such a test could only produce a short-term interaction between the cancer cell and white blood cell. Moreover, the HCC cell line and PBMCs will be coming from a different source which might lead to the different reactions from the immune cells and result in the different genes being up-regulated. Therefore, the researcher judged that this experiment did not require an in vitro investigation of co-culture between HCC cells and PBMCs. Even though, this study did compare samples of patients with predisposing factor such as HF and NAFL, the experiment still not include other malignant disease from the nearby organ (such as cholangiocarcinoma, pancreatic cancer, etc.) that may share similar genes expression. In which case, the accuracy may be affected in clinical application. Therefore, in the future, investigating whether elevated expression of the gene is specific to patients only with HCC is warranted.
Methods
Method statement
All research was performed in accordance with relevant guidelines and regulations.
Bioinformatics analysis
In this study, we recruited a bioinformatics approach to narrow the candidate genes for potential markers. In the Gene Expression Omnibus (GEO) repository of NCBI, microarray analysis results submitted by worldwide researchers are made available. From the NCBI database, data sets of gene expressions in PBMC were searched. Search terms were (Homo sapiens) AND (HCC OR (hepatocellular carcinoma)) AND (PBMC OR (peripheral blood mononuclear cell) OR (white blood cell) OR (WBC))”. Inclusion criteria were (1) PBMCs or any other white blood cells’ expression file (2) Including both healthy donor cases and HCC cases (3) Datasets of homo sapiens. As a result, two gene expression datasets that compared between PBMC samples from healthy individuals and HCC patients were selected, GSE49515 and GSE58208. We conducted t-tests in each gene expression of each dataset using “Connection Up and Down Regulation Expression Analysis of Microarrays (CU-DREAM) http://pioneer.netserv.chula.ac.th/~achatcha/CU-DREAM/)”,57 to evaluate the intersection genes and obtained 187 upregulated genes from both datasets. Then, three genes with highly significant p-values (p < 0.001) were selected and used to observed gene expressions in our samples.
Study population
All samples were recruited from King Chulalongkorn Memorial Hospital, Bangkok, Thailand and included 2 cohorts as the following:
Cohort 1: Samples were collected from June 2018 to January 2019 and included 83 HCC cases, 52 healthy donors, 10 with hepatic fibrosis, 10 with non-alcoholic fatty liver.
Cohort 2: Samples were collected from January 2020 to July 2020 and included 70 HCC cases, 24 healthy donors, 10 with hepatic fibrosis, 10 with non-alcoholic fatty liver.
A total of 153 HCC cases, 76 healthy donors, 20 hepatic fibrosis, and 20 non-alcoholic fatty liver participated in this study. Patients with hepatitis viral infection were excluded from this study. HCC staging was recorded according to current BCLC guidelines. All subjects in this study were of Asian descent, further bioinformation is provided in Table 2.
We then used the preliminary results from both GSE 49515 and GSE 58208 to find the appropriate sample size with the following formula:
n = sample size.
d = Different of value in each group.
\(\overline{X }\) d = Different of mean in each group.
σ2d = Different of variance in each group.
Zα/2 = Standard normal variate for level of significance.
Zβ = Standard normal variate for power.
We calculated and found that the sample size for our study was 24.44 samples, confirming that our study has recruited enough samples for the experimentation.
Blood sampling and PBMC extraction
Two ml of EDTA blood was extracted from all patients. Lymphocyte isolation medium was added to a 15 ml tube and centrifuged at 1600 rpm at 16 °C for 12 min and the plasma was separated. Whole blood (diluted 1:1) with PBS was carefully layered on a tube of lymphocyte separation medium and centrifuged at 2800 rpm for 15 min at 16 °C. The cell interface layer was carefully separated into 1.5 ml tubes and cells were washed with 1 ml PBS for 15 min at 1700 rpm 16 °C and 500 ml PBS for 5 min at 4 °C. The research methodology employed in this project was approved by The Institutional Review Board of the Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand (IRB No. 108/60 and 438/60). All study subjects provided written informed consent.
RNA extraction
PBMCs were mixed with 1 ml of TRIzol reagent (ThermoFisher Scientific, MA, USA) and incubated at room temperature for 5 min, then 200 μl of chloroform was added and incubated at room temperature for 3 min. Thereafter, it was separated into three phases by centrifugation at 8760 rpm at 4 °C for 15 min. The colorless upper aqueous phase was transferred to a new RNA tube, supplemented with 4 μL of glycogen (20 mg / mL) and 500 μl of 100% isopropanol, incubated for 10 min at room temperature, then centrifuged at 8760 rpm at 4 °C for 15 min. The supernatants of the centrifuged tubes were discarded, and the RNA pellets were washed with 1 ml of 75% ethanol, mixed by vortexing, and centrifuged at 6930 rpm at 4 °C for 5 min. Thereafter, supernatant was discarded again, and RNA pellet was dried by vacuum for 8 min and resuspended with 30 μL of DEPC water. RNA concentration and integrity were confirmed by Nanodrop and bioanalyzer.
Complementary DNA (cDNA) synthesis
After, the total RNA was extracted from PBMCs using TRIzol reagent (Thermo Scientific) according to the manufacturer’s protocol. Then, cDNA was synthesized using RevertAid First Strand cDNA Synthesis (Thermo Scientific). The process of cDNA synthesis is as follows: thaw, mix and centrifuge the components of the kit then add the template RNA 0.1 ng—5 µg, primer 1 µL, nuclease-free water up to 12 µL, 5X reaction buffer 4 µL, Ribolock RNAse inhibitor 1 µL, 10 mM dNTP mix 2 µL, RevertAid M-MuLV RT 1 µL. After mixing and brief centrifuging, the samples were incubated for 5 min at 25 °C followed by 60 min at 42 °C. Finally, terminate the reaction by heating at 70 °C for 5 min. The product of the first strand cDNA synthesis can be used directly in PCR or qPCR.
Primer preparation
Primers were designed using Primer3plus58 (for FLNA) and Primer Blast59 (for CAP1 and CLU). Primers were synthesized by BIONEER. Each primer sequence, melting temperature, and product length are shown in Table 5. Prior to quantitative PCR, conventional PCR and electrophoresis for finding optimal temperature for each primer was conducted.
Quantitative Real-time PCR (qRT-PCR) analysis
The quantitative PCR contained 10 µl SensiFast (Bioline), 0.8 µl of forward and reverse primers, cDNA(1 µl for FLNA, 0.5 µl for CLU and CAP1), and 7.4 µl distilled water in a total volume of 20 µl. The reactions were carried out on QuantStudio 6 (Thermo Fisher Scientific) according to the manufacturer’s protocol. PCR conditions were as follows: denaturation at 95 °C for 2 min with 45 cycles, annealing at 59 °C, 55 °C, 59 °C for CLU, FLNA, CAP1, respectively for 30 s. Fluorescence signals from the amplified product were detected at the end of the annealing step. Duplications were done on available and unamplified samples. The Ct value was set to 45 if the sample did not show any amplification twice. In this study, the housekeeping gene or the reference gene, that was used is glyceraldehyde 3-phosphate dehydrogenase (GAPDH). We used this gene to test and analyze alongside our interested gene (CLU, FLNA, and CAP1).
The calculation is as follows:
The final result is represented in the folds of change (thus, the equation is in the power of 2 or 2−ΔΔCt) of the interested gene expression in the sample against the reference sample60.
Statistical analysis
Box plot, summary of the dataset (including t-test results of Ct mean of each gene), benchmarks (Accuracy, Sensitivity, and Specificity) heatmaps (confusion matrices) and the Receiver Operating Characteristic (ROC) curves were drawn with python 3.9 program with packages (scipy61, pandas62 and matplotlib63). Ct values of each gene were included into the dataset. For the evaluation of performance, the entire dataset was used for the test. The p-value cut-off for each test was at < 0.05 for results to be statistically significant.
Ethical statement
The research methodology employed in this project was approved by The Institutional Review Board of the Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand (IRB No. 108/60 and 438/60). All study subjects provided written informed consent.
Abbreviations
- AFP:
-
Alpha-Fetoprotein
- APCs:
-
Antigen Presenting Cells
- AP-1:
-
Activator protein 1
- cAMP:
-
Cyclic adenosine monophosphate
- CAP1:
-
Cyclase Associated Actin Cytoskeleton Regulatory Protein 1
- CLU:
-
Clusterin
- Ct:
-
Cycle threshold
- CTLA-4:
-
Cytotoxic T-Lymphocytes-Associated protein 4
- FLNA:
-
Filamin A
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GEO:
-
Gene Expression Omnibus
- HCC:
-
Hepatocellular Carcinoma
- HF:
-
Hepatic Fibrosis
- IL-1β:
-
Interleukin 1 beta
- IL-2:
-
Interleukin-2
- IL-6:
-
Interleukin-6
- IL-10:
-
Interleukin-10
- MHC:
-
Major Histocompatibility Complex
- NAFL:
-
Non-Alcoholic Fatty Liver
- NFAT:
-
Nuclear factor of activated T-cells
- NF-κB:
-
Nuclear Factor Kappa B
- PBMCs:
-
Peripheral Blood Mononuclear Cells
- PD-L1:
-
Program death ligand 1
- PD-L2:
-
Program death ligand 2
- PKA:
-
Protein kinase A
- PKC-θ:
-
Protein kinase C-θ
- qRT-PCR:
-
Quantitative Real Time – Polymerase Chain Reaction
- ROC:
-
Receiver operating characteristic
- TCR:
-
T-cell receptor
- TGF-β:
-
Transforming Growth factor beta
- TNFα:
-
Tumor necrosis factor alpha
- WBCs:
-
White Blood Cells
References
Yang, J. D. et al. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 16, 589–604. https://doi.org/10.1038/s41575-019-0186-y (2019).
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68(6), 394–424. https://doi.org/10.3322/caac.21492 (2018).
Somboon, K., Siramolpiwat, S. & Vilaichone, R. K. Epidemiology and survival of hepatocellular carcinoma in the central region of Thailand, Asian. Pac. J. Cancer Prev. 15(8), 3567–3570. https://doi.org/10.7314/apjcp.2014.15.8.3567 (2014).
Clark, T., Maximin, S., Meier, J., Pokharel, S. & Bhargava, P. Hepatocellular carcinoma: Review of epidemiology, screening, imaging diagnosis, response assessment, and treatment. Curr. Probl. Diagn. Radiol. 44(6), 479–486. https://doi.org/10.1067/j.cpradiol.2015.04.004 (2015).
Zhang, J. et al. The threshold of alpha-fetoprotein (AFP) for the diagnosis of hepatocellular carcinoma: A systematic review and meta-analysis. PLoS ONE 15(2), e0228857. https://doi.org/10.1371/journal.pone.0228857 (2020).
Sanai, F. M. et al. Assessment of alpha-fetoprotein in the diagnosis of hepatocellular carcinoma in Middle Eastern patients. Dig. Dis. Sci. 55(12), 3568–3575. https://doi.org/10.1007/s10620-010-1201-x (2010).
Chan, S. L. et al. Performance of serum α-fetoprotein levels in the diagnosis of hepatocellular carcinoma in patients with a hepatic mass. HPB (Oxford) 16(4), 366–372. https://doi.org/10.1111/hpb.12146 (2014).
Amit, S. & Jorge, A. M. Screening for hepatocellular carcinoma. Gastroenterol. Hepatol. (N.Y.) 4(3), 201–208 (2008).
Puttipanyalears, C., Kitkumthorn, N., Buranapraditkun, S., Keelawat, S. & Mutirangura, A. Breast cancer upregulating genes in stromal cells by LINE-1 hypermethylation and micrometastatic detection. Epigenomics 8(4), 475–486. https://doi.org/10.2217/epi-2015-0007 (2016).
Sturgeon, S. R. et al. White blood cell DNA methylation and risk of breast cancer in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial (PLCO). Breast Cancer Res. 19, 94. https://doi.org/10.1186/s13058-017-0886-6 (2017).
Kunadirek, P. et al. Identification of BHLHE40 expression in peripheral blood mononuclear cells as a novel biomarker for diagnosis and prognosis of hepatocellular carcinoma. Sci Rep. 11, 11201. https://doi.org/10.1038/s41598-021-90515-w (2021).
Chen, S. et al. Identification of human peripheral blood monocyte gene markers for early screening of solid tumors. PLoS ONE 15(3), e0230905. https://doi.org/10.1371/journal.pone.0230905 (2020).
Lei, C. J. et al. Change of the peripheral blood immune pattern and its correlation with prognosis in patients with liver cancer treated by sorafenib. Asian Pac. J. Trop. Med. 9(6), 592–596. https://doi.org/10.1016/j.apjtm.2016.04.019 (2016).
Shi, M. et al. A blood-based three-gene signature for the non-invasive detection of early human hepatocellular carcinoma. Eur. J. Cancer 50(5), 928–936. https://doi.org/10.1016/j.ejca.2013.11.026 (2014).
Boonsongserm, P. et al. Tumor-induced DNA methylation in the white blood cells of patients with colorectal cancer. Oncol. Lett. 18(3), 3039–3048. https://doi.org/10.3892/ol.2019.10638 (2019).
Jiang, J. X. et al. Insights into significant pathways and gene interaction networks in peripheral blood mononuclear cells for early diagnosis of hepatocellular carcinoma. J. Cancer Res. Ther. 12(2), 981–989. https://doi.org/10.4103/0973-1482.154081 (2016).
Puttipanyalears, C. et al. Quantitative STAU2 measurement in lymphocytes for breast cancer risk assessment. Sci. Rep. 11, 915. https://doi.org/10.1038/s41598-020-79622-2 (2021).
Hui, K. M. Expression profiling of PBMC from patients with hepatocellular carcinoma. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE49515 (2013).
Hui, K. M. Gene expression profiling of PBMC from normal individuals, chronic hepatitis B carriers and hepatocellular carcinoma patients. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE58208 (2014).
Chow, M. T. & Luster, A. D. Chemokines in cancer. Cancer Immunol. Res. 2(12), 1125–1131. https://doi.org/10.1158/2326-6066.CIR-14-0160 (2014).
Raman, D., Baugher, P. J., Thu, Y. M. & Richmond, A. Role of chemokines in tumor growth. Cancer Lett. 256(2), 137–165. https://doi.org/10.1016/j.canlet.2007.05.013 (2007).
Bhat, A. A. et al. Cytokine-chemokine network driven metastasis in esophageal cancer; promising avenue for targeted therapy. Mol. Cancer 20(1), 2. https://doi.org/10.1186/s12943-020-01294-3 (2021).
Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 4(1), 11–22. https://doi.org/10.1038/nrc1252 (2004).
Griffiths, J. I. et al. Circulating immune cell phenotype dynamics reflect the strength of tumor-immune cell interactions in patients during immunotherapy. Proc. Natl. Acad. Sci. U.S.A. 117(27), 16072–16082. https://doi.org/10.1073/pnas.1918937117 (2020).
Chappell, D. B. & Restifo, N. P. T cell-tumor cell: A fatal interaction?. Cancer Immunol. Immunother.: CII 47(2), 65–71. https://doi.org/10.1007/s002620050505 (1998).
Karwacz, K. et al. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol. Med. 3(10), 581–592. https://doi.org/10.1002/emmm.201100165 (2011).
Sansom, D. M. CD28, CTLA-4 and their ligands: Who does what and to whom?. Immunology 101(2), 169–177. https://doi.org/10.1046/j.1365-2567.2000.00121.x (2000).
de Lima, V. A. B. et al. Immune cell profiling of peripheral blood as signature for response during checkpoint inhibition across cancer types. Front. Oncol. 11, 558248. https://doi.org/10.3389/fonc.2021.558248 (2021).
Zheng, Y., Fang, Y. C. & Li, J. PD-L1 expression levels on tumor cells affect their immunosuppressive activity. Oncol. Lett. 18(5), 5399–5407. https://doi.org/10.3892/ol.2019.10903 (2019).
Connolly, P. H. et al. Effects of exercise on gene expression in human peripheral blood mononuclear cells. J. Appl. Physiol. 97(4), 1461–1469. https://doi.org/10.1152/japplphysiol.00316.2004 (2004).
Inoue, C., Takeshita, T., Kondo, H. & Morimoto, K. Cigarette smoking is associated with the reduction of lymphokine-activated killer cell and natural killer cell activities. Environ. Health Prev. Med. 1(1), 14–19. https://doi.org/10.1007/BF02931167 (1996).
Wieczfinska, J., Kowalczyk, T., Sitarek, P., Skała, E. & Pawliczak, R. Analysis of short-term smoking effects in PBMC of healthy subjects-preliminary study. Int. J. Environ. Res. Public Health 15(5), 1021. https://doi.org/10.3390/ijerph15051021 (2018).
Tseng, Y. M. et al. Effects of alcohol-induced human peripheral blood mononuclear cell (PBMC) pretreated whey protein concentrate (WPC) on oxidative damage. J. Agric. Food Chem. 56(17), 8141–8147. https://doi.org/10.1021/jf801034k (2008).
Sureshchandra, S. et al. Dose-dependent effects of chronic alcohol drinking on peripheral immune responses. Sci. Rep. 9(1), 7847. https://doi.org/10.1038/s41598-019-44302-3 (2019).
Roy, B., Shelton, R. C. & Dwivedi, Y. DNA methylation and expression of stress related genes in PBMC of MDD patients with and without serious suicidal ideation. J. Psychiatry Res. 89, 115–124. https://doi.org/10.1016/j.jpsychires.2017.02.005 (2017).
Nakamura, F., Stossel, T. P. & Hartwig, J. H. The filamins: Organizers of cell structure and function. Cell Adhes. Migr. 5(2), 160–169. https://doi.org/10.4161/cam.5.2.14401 (2011).
Su, W., Mruk, D. D. & Cheng, C. Y. Filamin A: A regulator of blood-testis barrier assembly during post-natal development. Spermatogenesis 2(2), 73–78 (2012).
Yue, J., Huhn, S. & Shen, Z. Complex roles of filamin-A mediated cytoskeleton network in cancer progression. Cell Biosci. 3, 7. https://doi.org/10.1186/2045-3701-3-7 (2013).
Donadon, M. et al. Filamin A expression predicts early recurrence of hepatocellular carcinoma after hepatectomy. Liver Int. 38(2), 303–311. https://doi.org/10.1111/liv.13522 (2018).
Ai, J. et al. FLNA and PGK1 are two potential markers for progression in hepatocellular carcinoma. Cell Physiol. Biochem. 27(3–4), 207–216. https://doi.org/10.1159/000327946 (2011).
Hayashi, K. & Altman, A. Filamin A is required for T cell activation mediated by protein kinase C-theta. J. Immunol. 177(3), 1721–1728. https://doi.org/10.4049/jimmunol.177.3.1721 (2006).
Savoy, R. M. & Ghosh, P. M. The dual role of filamin A in cancer: Can’t live with (too much of) it, can’t live without it. Endocr. Relat. Cancer. 20(6), R341–R356. https://doi.org/10.1530/ERC-13-0364 (2013).
Cunin, P. et al. Clusterin facilitates apoptotic cell clearance and prevents apoptotic cell-induced autoimmune responses. Cell Death Dis. 7(5), e2215. https://doi.org/10.1038/cddis.2016.113 (2016).
Zhang, H. et al. Clusterin inhibits apoptosis by interacting with activated Bax. Nat. Cell Biol. 7(9), 909–915. https://doi.org/10.1038/ncb1291 (2005).
Blaschuk, O., Burdzy, K. & Fritz, I. B. Purification and characterization of a cell-aggregating factor (clusterin), the major glycoprotein in ram rete testis fluid. J. Biol. Chem. 258(12), 7714–7720 (1983).
Lau, S. et al. Clusterin plays an important role in hepatocellular carcinoma metastasis. Oncogene 25, 1242–1250. https://doi.org/10.1038/sj.onc.1209141 (2006).
Wang, C. et al. Clusterin facilitates metastasis by EIF3I/Akt/MMP13 signaling in hepatocellular carcinoma. Oncotarget 6(5), 2903–2916. https://doi.org/10.18632/oncotarget.3093 (2015).
Zhong, J. et al. Therapeutic role of meloxicam targeting secretory clusterin-mediated invasion in hepatocellular carcinoma cells. Oncol. Lett. 15(5), 7191–7199. https://doi.org/10.3892/ol.2018.8186 (2018).
Zhong, J. et al. Downregulation of secreted clusterin potentiates the lethality of sorafenib in hepatocellular carcinoma in association with the inhibition of ERK1/2 signals. Int. J. Mol. Med. 41(5), 2893–2900. https://doi.org/10.3892/ijmm.2018.3463 (2018).
Zheng, W. et al. Silencing clusterin gene transcription on effects of multidrug resistance reversing of human hepatoma HepG2/ADM cells. Tumour Biol. 36(5), 3995–4003. https://doi.org/10.1007/s13277-015-3043-9 (2015).
Taylor, A., Verhagen, J., Blaser, K., Akdis, M. & Akdis, C. A. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology 117(4), 433–442. https://doi.org/10.1111/j.1365-2567.2006.02321.x (2006).
Ono, S. The role of cyclase-associated protein in regulating actin filament dynamics - more than a monomer-sequestration factor. J. Cell Sci. 126(Pt 15), 3249–3258. https://doi.org/10.1242/jcs.128231 (2013).
Liu, Y. et al. Upregulated expression of CAP1 is associated with tumor migration and metastasis in hepatocellular carcinoma. Pathol. Res. Pract. 210(3), 169–175. https://doi.org/10.1016/j.prp.2013.11.011 (2014).
Hua, M. et al. CAP1 is overexpressed in human epithelial ovarian cancer and promotes cell proliferation. Int. J. Mol.. Med. 35(4), 941–949. https://doi.org/10.3892/ijmm.2015.2089 (2015).
Lee, S. et al. Adenylyl cyclase-associated protein 1 is a receptor for human resistin and mediates inflammatory actions of human monocytes. Cell Metab. 19(3), 484–497. https://doi.org/10.1016/j.cmet.2014.01.013 (2014).
Singh, N. et al. Inflammation and cancer. Ann. Afr. Med. 18(3), 121–126. https://doi.org/10.4103/aam.aam_56_18 (2019).
Aporntewan, C. & Mutirangura, A. Connection up- and down-regulation expression analysis of microarrays (CU-DREAM): A physiogenomic discovery tool. Asian Biomed. 5(2), 257–262. https://doi.org/10.5372/1905-7415.0502.034 (2011).
Untergasser, A. et al. Primer3—New capabilities and interfaces. Nucleic Acids Res. 40(15), e115. https://doi.org/10.1093/nar/gks596 (2012).
Ye, J. et al. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 13, 134. https://doi.org/10.1186/1471-2105-13-134 (2012).
Rao, X., Huang, X., Zhou, Z. & Lin, X. An improvement of the 2ˆ(−delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinform. Biomath. 3(3), 71–85 (2013).
Virtanen, P. et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272. https://doi.org/10.1038/s41592-019-0686-2 (2020).
McKinney, W. Data Structures for Statistical Computing in Python. Preprint at https://conference.scipy.org/proceedings/scipy2010/mckinney.html (2010).
Hunter, J. D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 9(3), 90–95. https://doi.org/10.1109/MCSE.2007.55 (2007).
Acknowledgements
We would like to thank Department of Anatomy, Chulalongkorn University for equipment and Ms. Papitchaya Watcharanurak for technical laboratory assistance. We also thank Center of Excellence in Hepatitis and Liver Cancer, Faculty of Medicine, Chulalongkorn University for specimen collection.
Funding
This project was financially supported by the National Research Council of Thailand (NRCT); National Science and Technology Development Agency (Grant number FDA-CO-2561-8477-TH) and Ratchadapiseksomphot Fund for Postdoctoral Fellowship and Development of New Faculty Staff, Chulalongkorn University and the Thailand Research Fund (RTA6280004). The role of the funding bodies in this study is to provide financial support only.
Author information
Authors and Affiliations
Contributions
R.P.—acquisition of data, analysis and interpretation of data, drafting of the manuscript, statistical analysis; S.R.—acquisition of data, analysis and interpretation of data, statistical analysis; P.K.—acquisition of data, material support; N.C.—acquisition of data, material support; P.T.—acquisition of data, material support; A.M.—study concept and design, obtained funding, administrative support; C.P.—study concept and design, acquisition of data, drafting of the manuscript, critical revision of the manuscript for important intellectual content, technical support, study supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Patarat, R., Riku, S., Kunadirek, P. et al. The expression of FLNA and CLU in PBMCs as a novel screening marker for hepatocellular carcinoma. Sci Rep 11, 14838 (2021). https://doi.org/10.1038/s41598-021-94330-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-94330-1
This article is cited by
-
The high FKBP1A expression in WBCs as a potential screening biomarker for pancreatic cancer
Scientific Reports (2024)
-
Identification of a novel bile marker clusterin and a public online prediction platform based on deep learning for cholangiocarcinoma
BMC Medicine (2023)
-
Differential expression of immune-regulatory proteins C5AR1, CLEC4A and NLRP3 on peripheral blood mononuclear cells in early-stage non-small cell lung cancer patients
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
