
Abstract
There is increasing evidence that endogenous retroviruses (ERVs) play a significant role in central nervous system diseases, including amyotrophic lateral sclerosis (ALS). Studies of ALS have consistently identified retroviral enzyme reverse transcriptase activity in patients. Evidence indicates that ERVs are the cause of reverse transcriptase activity in ALS, but it is currently unclear whether this is due to a specific ERV locus or a family of ERVs. We employed a combination of bioinformatic methods to identify whether specific ERVs or ERV families are associated with ALS. Using the largest post-mortem RNA-sequence datasets available we selectively identified ERVs that closely resembled full-length proviruses. In the discovery dataset there was one ERV locus (HML6_3p21.31c) that showed significant increased expression in post-mortem motor cortex tissue after multiple-testing correction. Using six replication post-mortem datasets we found HML6_3p21.31c was consistently upregulated in ALS in motor cortex and cerebellum tissue. In addition, HML6_3p21.31c showed significant co-expression with cytokine binding and genes involved in EBV, HTLV-1 and HIV type-1 infections. There were no significant differences in ERV family expression between ALS and controls. Our results support the hypothesis that specific ERV loci are involved in ALS pathology.
Similar content being viewed by others


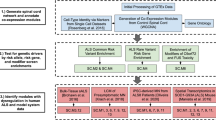
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease of cortical, bulbar and spinal motor neurons resulting in progressive weakness and ultimately death, typically within 3–5 years, as a result of neuromuscular respiratory failure. Genetic risk factors have been identified: up to 10% of people with apparently sporadic ALS have a pathologically expanded hexanucleotide repeat in the gene C9orf721,2; mutations in the genes TARDBP, FUS and SOD1 account for a further 4–5%. For most, the cause of the disease is unknown.
Our incomplete understanding of the causes of ALS is reflected in two major obstacles to progress. The first is that only a small proportion of the measured heritability of ALS is accounted for by observed genetic variation. While advances in modelling polygenicity and rare DNA variants may explain some of this missing heritability in the future, other genomic elements, not captured using genome-wide association (GWAS) and whole genome sequencing (WGS) studies, play a role in disease3,4. Among such genomic features are mobile genetic elements (including endogenous retroviruses)3,5, DNA structural variation4, and epigenetics6.
The second obstacle is that there is a significant non-genetic component that contributes to the risk of ALS7,8,9, and despite concerted efforts there is little consensus or replicated evidence as to what this is.
Research into several neurological diseases has shown that human endogenous retroviruses (ERVs) play a significant role in aetiology10,11. ERVs are retroviruses that have integrated into genomic DNA and become components of the mammalian genome over millions of years of evolution. It is estimated that 8–9% of the human genome consists of ERVs in comparison to the 1–2% comprising protein-coding genes. Most ERVs are unable to transcribe, translate or replicate, due to the accumulation of mutations in their DNA sequence. However, a significant proportion still retain full-length sequences that encode retroviral proteins. Furthermore, ERVs can regulate the expression of genes nearby through genetic components such as non-coding RNAs, and enhancer and promotor regions that include long terminal repeats (LTRs) that contain TATA-box promoters and AATAAA motifs for polyadenylation12,13.
Aberrant ERV transcription and protein expression have been implicated in several central nervous system diseases, including multiple sclerosis (MS)14,15,16, Alzheimer’s disease (AD)17,18, schizophrenia19,20,21, chronic inflammatory demyelinating polyneuropathy17 and ALS22,23,24,25,26. ERVs can be regulated by DNA polymorphisms, variation in methylation and chromatin state, and transactivation. Viral transactivation of ERV expression by exogenous viruses can have consequences on disease susceptibility and progression, such as changes in HERV-W expression due to herpes simplex and Epstein–Barr viruses in MS15,27,28,29,30,31,32 and changes in HERV-K expression in response to HIV Type 133,34,35. There has been renewed interest in the role exogenous viruses in neurodegeneration, with findings implicating herpes viruses in AD36,37,38.
Over the last 20 years, a number of studies have identified a signature of retrovirus expression in the form of increased reverse transcriptase activity in ALS serum samples22,23,24,39. There is elevated HERV-K RNA and protein expression in post-mortem ALS brain in comparison with the levels detected in healthy and disease-related controls25,40, although this is not confirmed in all studies41,42. A key finding is that HERV-K colocalises, coregulates and binds with TDP-4325,40, a protein that shows intracellular aggregation in up to 95% of people with ALS, post-mortem. In vivo and in vitro models indicate that upregulation of ERVs, as a consequence of TDP-43 overexpression, contribute to a more severe form43 which is likely to be cell-specific44. However, it remains unclear whether the association of ERVs with ALS is due to a single ERV locus, a specific ERV family or due to multiple ERV families.
We therefore applied a combination of bioinformatic methods to multiple post-mortem ALS cohorts to test the hypothesis that specific ERVs or ERV families are associated with ALS. If confirmed, this would allow the development of treatments specifically targeting this group of retroviruses.
Results
ERV locus HML6_3p21.31c is differentially expressed in ALS
We used the curated ERVMap database and protocol to test for differentially expressed ERVs comparing ALS (n = 80) and controls (n = 28) using RNA-sequence data from King’s College London (primary motor cortex); see Table 1. 1654 of 3237 ERV transcripts had sufficient read-counts (> 10) across samples for testing. ERVs previously associated with ALS but not included in ERVMap were analysed independently (see Supplementary Table S1).
One ERV (HML6_3p21.31c) showed a significant increase in expression in ALS with respect to age-sex matched non-ALS controls after correcting for multiple testing (log2 fold-change = 0.691, standard error = 0.163, p value = 2.29 × 10−5, adjusted p value = 0.03). On human genome assembly 38 (hg38), HML6_3p21.31c is located at chr3:46426676–46433564 (see Fig. 1, Table 2, and Supplementary Table S2a).

Jitter boxplots comparing HML6_3p21.31c expression between ALS and controls across seven post-mortem RNA-sequence datasets. We found increased expression of HML6_3p21.31c in all motor cortex and cerebellum cohorts, but not in the frontal cortex tissue. X-axes: Disease status. Y-axes: Normalised expression of HML6_3p21.31c. Dots: HML6_3p21.31c expression for each sample. Boxplot: Boxplot of the distribution of HML6_3p21.31c. Boxplot colour: Disease status (ALS: Red; Control: Blue). Figure titles: Data source and tissue. Log2 FC: Log2 Fold-change, SE: Standard error, p value: Derived from differential expression analysis.
We then performed the same analysis using lateral motor cortex taken from the GSE137810 dataset (ALS n = 32, Controls n = 6). HML6_3p21.31c showed a significant increase in expression in ALS with a similar fold-change to the KCL cohort (log2 fold-change = 0.642, standard error = 0.220, p value = 0.003). See Fig. 1, Table 2, and Supplementary Table S2a.
To test if other ERV transcripts were significantly differentially expressed across both King’s College London and GSE137810 datasets we performed a Stouffer’s meta-analysis, with log2 fold-change as effect direction and sample size used as weights. Only HML6_3p21.31c was significant after correcting for multiple-testing, with Stouffer’s Z = 5.039, p value = 4.675 × 10−7 (Bonferroni threshold = 3.546 × 10−5). See Table 2 and Supplementary Table S2a.
To test if HML6_3p21.31c was significantly expressed in other brain areas we performed the same analysis in medial motor cortex, cerebellum and across frontal cortex regions using RNA-seq data from GSE137810 and GSE67196. We found a similar relationship between HML6_3p21.31c and ALS in the medial motor cortex (log2 fold-change = 0.645, standard error = 0.326, p value = 0.04). In addition, using Stouffer’s method, we found significant increased expression in cerebellum tissue using GSE137810 and GSE67196 datasets (Z = 2.721, p value = 0.006) but not in frontal regions (Z = 1.22, p value = 0.22). Please note that there are overlapping samples across cerebellum and prefrontal cortex datasets. See Fig. 1, Table 2, and Supplementary Table S2b,c.
The HML6 ERV family in general, modelled as a collapsed transcript element using TETranscripts, did not show significantly increased expression in ALS (see Supplementary Table S3). Previous research has identified specific ERV loci in the HML2 family as showing differential expression in ALS40. We did not find differential expression of these loci in motor cortex, cerebellum or prefrontal cortex. HML2 as a family of ERVs did not show differential expression between ALS and controls across tissue and studies (see Supplementary Table S3).
To assess whether differences in expression could be driven by differences in cell composition between ALS and control donors, we used the BRETIGEA45 cell marker database to estimate relative proportions of neurons across KCL samples. We did not find significant differences in estimated neuron proportions between ALS and controls, while controlling for covariates gender, age, post-mortem delay, RIN and surrogate variables (beta = 0.035, standard error = 0.023, p value = 0.150).
ERV locus HML6_3p21.31c co-expresses with genes involved in cytokine pathways and HIV
To assess the functional involvement of HML6_3p21.31c in post-mortem ALS we performed weighted co-expression network analysis integrating gene expression with ERV expression. Our network analyses included both ALS cases and controls. We performed network analyses using the KCL primary motor cortex cohort as well as the GSE137810 lateral motor cortex.
In the KCL primary motor cortex, HML6_3p21.31c showed significant co-expression in a network enriched for cytokine signalling and binding, viral protein interaction, and responses to EBV and HIV Type 1 infection (see Table 3; Fig. 2). For the genes driving the enrichment for these categories see Supplementary Table S4 and for an extended list of enrichment categories see Supplementary Table S5. This network also showed significant correlation with disease status (Pearson’s r = 0.19, p = 0.05) indicating network-wide upregulation in ALS. HML6_3p21.31c was the most significant genomic element to associate with ALS in this network (see Table 3). For genes and ERVs that show differential expression in this network (with an uncorrected p value < 0.05) see Supplementary Table S6.

Gene function enrichment analyses of yellow network-module. This analysis identified significant enrichment66,67,68 of cytokine binding and signalling, as well as genes involved in HIV Type 1, in the yellow network-module of which HML6_3p21.31c is a member. Upper X-axes: − log(10) p value of the gene function enrichment analysis. Y-axes: Most significant gene function enrichment ontological categories. Bar-chart titles: Database used for gene function enrichment analyses.
For the GSE137810 lateral motor cortex cohort HML6_3p21.31c also showed significant co-expression in a network enriched for cytokine binding and receptor activity, and genes involved in HIV Type 1 infection (see Supplementary Fig. S2; Supplementary Table S7).
The HML6_3p21.31c locus is immediately adjacent to the chemokine receptor cluster on chromosome 3, which has been shown to influence retrovirus HIV viral load and is approximately 50 kb downstream of the HIV coreceptor gene CCR5, which is required for HIV entry into the cell. To explore the transcriptional relationship between HML6_3p21.31c and the genes proximal to it we performed Pearson’s correlation analyses using normalised expression estimates in the KCL primary motor cortex dataset. HML6_3p21.31c expression significantly correlated with expression of the HIV coreceptor gene CCR5 (r = 0.346, p value = 0.0002), as well as CCR1 (r = 0.248, p value = 0.009) and Lactoferrin (LTF) (r = 0.750, p value < 2.2 × 10−16), the latter of which have also shown association with HIV. Although HML6_3p21.31.c showed significant co-expression with CCR5, CCR1 and LTF, they do not show significant differential expression between ALS and control donors after multiple testing correction (see Supplementary Table S8).
Previous research has demonstrated that ERVs that show significant differential expression in ALS correlate with TARDBP expression. We performed expression correlation analyses between a list of 14 genes that associate with ALS and HML6_3p21.31c. A small but significant correlation was identified with TARDBP (Pearson’s r = 0.21, p value = 0.04) and optineurin (Pearson’s r = − 0.25, p value = 0.01). See Supplementary Fig. S3.
ERVs and genetic risk of ALS
To assess if ERVs that showed increased expression in post-mortem ALS increased genetic risk of the disease, we performed ERV-set analysis using MAGMA and three ALS GWAS datasets. For each post-mortem RNA-seq dataset analysed, ERVs were divided by their direction of fold-change and filtered by their disease status association with a p value ≤ 0.05. ERVs that showed significant increased expression in the KCL primary motor cortex dataset were marginally enriched for SNPs that increased ALS risk (beta = 0.315, S.D. = 0.032, p value = 0.039). No other tissue or tissue-source showed enrichment for ALS risk SNPs (see Supplementary Table S9). In addition, we tested whether ERVs belonging to family HML6 show significant enrichment of ALS risk SNPs (beta = 0.199, S.D. = 0.025, p value = 0.087).
Discussion
Using RNA-sequence data from post-mortem motor cortex, we found that an individual ERV locus of the HML6 family, HML6_3p21.31c, showed significantly increased expression in ALS. We tested six additional post-mortem ALS RNA-sequence datasets and found a significant increase in expression of HML6_3p21.31c in four of them. Increased expression of the ERV locus was found in motor cortex and cerebellum, but not in prefrontal cortex. To understand the potential functional involvement of HML6_3p21.31c in ALS we performed co-expression network analyses. HML6_3p21.31c significantly co-expressed with genes involved in cytokine binding and signalling, interleukin, inflammatory responses, EBV infection, viral protein interaction, HTLV-1 and HIV Type-1 infection.
Previous research has reported differential expression of specific HERV-K HML2 loci on chromosome bands 3q21.2, 7q34, and 10p14 in post-mortem ALS, as well as differential expression of HML2 as a family25,26. We did not find evidence that these specific loci or the HML2 family were differentially expressed in ALS motor cortex, cerebellum or prefrontal cortex. Our findings concur with recent studies using qPCR in post-mortem ALS that also found no differential expression of these loci or the HML2 ERV family41,42. The qPCR primers used across all previous studies examining HML2 in ALS would not have amplified HERV-K HML6 loci and therefore could not show association with ALS. Two key differences exist between our study and the studies that originally found differential expression of HML2 loci. We have used methods that are specifically designed to quantify ERV loci-specific transcripts, while accounting for sequence alignment ambiguity and repetitive genomic elements. In addition, the sample-size of our study is larger and has greater power to account for variation across ALS and control populations.
HML6_3p21.31c belongs to the HERV-K(HML6) family of ERVs. HML-6 expression has been found throughout the genome in healthy and diseased tissue46. The most recent clinical association was found comparing HIV-1 infected cell cultures with control T cells47. In a similar bioinformatic approach to that used in our paper47, Grandi et al. found upregulation of HML6_ 19q13.43 in HIV-positive cells, which was the highest expressed ERV of the 3250 tested. HML-6 has also shown association with breast cancer48 and melanoma49.
HML6_3p21.31c is proximal to and shows significant co-expression with HIV associated genes CCR5, CCR2 and LTF. Outside of the HLA locus, CCR5 is the only gene that associates with HIV susceptibility50 and viral load51 through genetic inheritance. CCR5 encodes for a HIV co-receptor52, where genetic mutation (CCR5-delta 32) reduces receptor functionality and inhibits HIV’s capacity to infect cells53. Lactoferrin also exhibits anti-HIV effects54, with preliminary evidence of LTF polymorphisms influencing maternal transmission of HIV-1 to offspring. Using network analyses, we found genes that co-expressed with HML6_3p21.31c were enriched for genes that associate with HIV Type-1 infection. These included IL4R, CCL3L1, CXCR1 and CCL2, in addition to genes proximal to the ERV locus (CCR5, CCR2, CCR1 and LTF).
Comorbidity exists between ALS and HIV and was first identified in 198555. Estimates indicate that 3.5 in 1000 HIV patients develop ALS-like symptoms56, and this is now referred to as HIV-associated ALS. Clinical studies have found that HIV-associated ALS symptoms may be reversible through antiretroviral therapies56, and there are clinical trials57 in ALS patients without HIV infection, currently ongoing. Our results present the first locus-specific link between ALS, ERVs and HIV associated genes. Given that none of our post-mortem samples had known HIV-associated ALS, our results support evidence that ERV and HIV-related pathways are important in the wider ALS clinical population.
One limitation of our study is the uncertainty about what is causing upregulation of HML6_3p21.31c in ALS. It may be driven by expression of HIV-associated genes nearby, however HML6_3p21.31c alone showed significant upregulation in this locus, whereas HIV-associated genes did not. HML6_3p21.31c co-expressed with cytokine and HIV-associated genes as part of a wider network. Within this network, HML6_3p21.31c was the most significant genomic element to associate with ALS. Given that ERVs are known to regulate gene expression, our results highlight the potential that HML6_3p21.31c may be key genomic elements in modifying HIV associated and cytokine pathways in ALS.
Enrichment analyses of genes that co-expressed with HML6_3p21.31c in a network included genes associated with infection by several exogenous viruses, including HIV-1, HTLV-1, and EBV, as well as other viral-associated immune response genes. A limitation to our study is that we cannot be certain that this enrichment is not driven by immunological pathways in response to ALS, that parallel immune responses to viruses.
We found HML6.3p21.31c co-expressed with a network that correlated with both neuronal and non-neuronal cell-types. While it cannot be certain that differential expression of HML6.3p21.31c is not influenced by differences in cell composition between ALS and control donors, we integrated surrogate variable analyses into our model which controls for cell heterogeneity across samples58. In addition, we found no significant differences in neuron estimates between ALS and controls donors using cell composition analyses.
Anti-retroviral drugs are currently being trialled in ALS57. These trials are based upon decades of research showing differential expression of reverse transcriptase activity and ERV transcripts in ALS. While ERV families, and reverse transcriptase activity have shown association with the disease, findings regarding specific ERV transcripts have been less consistent. We found significant upregulation of a specific ERV locus (HML6_3p21.31c) in four post-mortem ALS RNA-sequence datasets across three studies. To date, this is the largest analysis of its kind in ALS. HML6_3p21.31c is located at the chemokine receptor gene cluster on chromosome 3 that modifies HIV susceptibility, viral entry into the cell and viral load. Network analyses of HML6_3p21.31c reveal significant correlation with genes involved in cytokine and interleukin activity, and infection with HIV and other exogenous viruses. In summary, our results (1) support the hypothesis that ERVs are involved in ALS pathology, (2) identify a specific ERV locus that shows consistent upregulation in ALS, and (3) identify major pathological pathways in the disease in which ERVs may play a significant role.
Material and methods
Samples and cohorts
Samples with post-mortem RNA-sequence data were used from three sources: (1) King’s College London (KCL) and the MRC London Neurodegenerative Diseases Brain Bank, (2) Target ALS Human Post-mortem Tissue Core and the New York Genome Centre (NYGC) published by Conlon et al.59 and Tam et al.60 (NCBI GEO ID: GSE137810), and (3) the Florida Mayo Clinic published by Prudencio et al.61 (NCBI GEO ID: GSE67196). Hereafter, datasets will be referred to by their respective GEO (GSE) accession number. ALS samples and controls were matched by age and sex where possible for each individual dataset. For a summary of sample frequencies, stratified by project and anatomical location, please see Table 1.
For KCL and the MRC London Neurodegenerative Diseases Brain Bank, frozen human post-mortem tissue was taken from primary motor cortex. For GSE137810, frozen human post-mortem tissue was taken from motor cortex (medial and lateral), frontal cortex (various locations) and cerebellum. For GSE67196, frozen human post-mortem tissue was taken from the lateral hemisphere of the cerebellum, the prefrontal cortex (Brodmann area 9/44), and the primary motor cortex (Brodmann area 4). For KCL and GSE6719661, all controls had Braak stage ≤ 2 with the exception of one control in GSE6719661.
RNA-sequencing of post-mortem samples
Library preparation and sequencing of RNA are described in https://metronome.nygenome.org/NYGC-RNA-Seq-QC-README_29Jun2017.pdf for GSE137810 data and Prudencio et al.61 for the GSE67196 dataset.
For the MRC London Neurodegenerative Diseases Brain Bank please see supplementary methods.
Experimental design and statistical analyses
Differential expression analysis of individual ERVs
All differential expression analyses were performed using DESeq2 controlling disease status, gender, age, post-mortem delay, RIN and surrogate variables, where available. All log2 fold-change values reflect changes in expression in comparison to non-ALS controls. All p values reported are raw p values unless reported otherwise. For multiple testing correction we used independent hypothesis weighting62 and Bonferroni corrections, which are reported where applicable.
Co-expression and cell-type analyses
To identify which genes significantly co-expressed with ERVs (defined by ERVMap transcript IDs), we performed variance stabilizing transformations (VST) for all required datasets using DESeq263. We corrected normalised expression estimates by covariates sex, age, post-mortem delay, RIN and surrogate variables, using jaffelab function cleaningY64 to regress them out. Next we used BRETIGEA to estimate cell composition per individual sample using BRETIGEA’s cell marker dataset (50 markers)45.
We used weighted correlation network analysis (WGCNA)65 alongside VST normalised expression data for the KCL primary motor cortex and GSE137810 lateral motor cortex datasets. WGCNA was performed using a deepsplit of 0 and a minimum module size of 30 genes\ERVs. Next, we analysed if WGCNA modules correlated with disease by performing Pearson’s r correlation between disease status and module eigengene values. We performed the same analysis for cell-type estimates by sample.
To perform co-expression analysis of individual ERVs and genes we used Pearson’s r analyses using PerformanceAnalytics in R (https://github.com/R-Finance/PerformanceAnalytics).
Gene function enrichment analyses
Enrichment analyses of gene function was performed using g:profiler and Enrichr, which include KEGG pathways66,67,68. Ontological categories with a term size (number of genes) greater than 1000 are not reported.
ERV-set enrichment analysis
To asses if ERVs were enriched for SNPs that modify ALS disease risk we used MAGMA69 and three ALS GWAS summary statistics from Nicolas et al.70 and van Rheenen and Shatunov et al.9. Note that these GWAS have overlapping samples but use alternative statistical models (please see references for more details).
We performed annotation of the 1000 genomes reference build GRCh37/hg19 Phase 3 using a 50 kb flanking window. A 50 kb flanking region was selected as the median length of a haplotype for chromosome 171 and ERV loci regions are small in comparison to protein-coding genes, which diminishes the capacity to identify an enrichment of SNPs that associate with disease. Gene-level analyses were performed on an ad-hoc ERV loci file using ERVMap mapping coordinates lifted over to reference genome GRCh37/hg19 Phase 3. ERV-set analyses were then performed using standard MAGMA protocols.
Family-level ERV expression analyses
To test if ERV families were differentially expressed in post-mortem ALS we used TETranscripts72. TETranscripts uses an expectation maximization algorithm to quantify multi-mapped reads, which is designed to estimate abundance of transcribed transposable elements (like ERVs) that have high sequence fidelity (like the sequence overlap that occurs between ERVs of the same family).
To perform TETranscripts we used RNA-seq bam files derived from all seven datasets tested. ERV families with total read-count less than 10 were discarded. All differential expression analyses of ERV families were performed using DESeq2 controlling disease status, gender, age, post-mortem delay, RIN, surrogate variables, where available.
Ethics declaration
Post-mortem tissue samples from King’s College London were collected under the ethical approval of the MRC London Neurodegenerative Diseases Brain Bank and under the regulations of the Human Tissue Act UK 2014. All post-mortem tissue was donated to the MRC London Neurodegenerative Diseases Brain Bank under standard ethical and Human Tissue Act procedures, with informed consent provided by the next of kin. Data generated from this material were anonymized and analysed on a high-performance computing cloud (https://www.maudsleybrc.nihr.ac.uk/facilities/rosalind/) with data protection protocols in accordance with Department of Health Policy (UK) and the security standards set by the National Data Guardian. Ethical approval to process and analyse post-mortem samples stored at King’s College London was provided by a local ethics committee at the Institute of Psychiatry, Psychology & Neuroscience, King’s College London, and the MRC London Neurodegenerative Diseases Brain Bank.
Data availability
Dataset are available, on reasonable request. The post-mortem genetic and RNA-sequence datasets are available from the corresponding author, the GWAS survival datasets are available through Dr Isabella Fogh, and the Braineac eQTL datasets are available through the UK Brain Expression Consortium.
Change history
19 August 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41598-022-18488-y
References
Majounie, E. et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330 (2012).
Renton, A. E., Chiò, A. & Traynor, B. J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23 (2014).
Payer, L. M. & Burns, K. H. Transposable elements in human genetic disease. Nat. Rev. Genet. 20, 760–772 (2019).
Ho, S. S., Urban, A. E. & Mills, R. E. Structural variation in the sequencing era. Nat. Rev. Genet. 21, 171–189 (2020).
Savage, A. L. et al. Retrotransposons in the development and progression of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 90, 284 (2018).
Cortijo, S. et al. Mapping the epigenetic basis of complex traits. Science (80-.) 343, 1145–1148 (2014).
Al-Chalabi, A. et al. An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry 81, 1324–1326 (2010).
Fogh, I. et al. A genome-wide association meta-analysis identifies a novel locus at 17q11.2 associated with sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet 23, 2220–2231 (2014).
van Rheenen, W. et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 48, 1043–1048 (2016).
Küry, P. et al. Human endogenous retroviruses in neurological diseases. Trends Mol. Med. 24, 379–394 (2018).
Tam, O. H., Ostrow, L. W. & Gale Hammell, M. Diseases of the nERVous system: retrotransposon activity in neurodegenerative disease. Mob. DNA 10, 1–4 (2019).
Benachenhou, F., Blikstad, V. & Blomberg, J. The phylogeny of orthoretroviral long terminal repeats (LTRs). Gene 448, 134–138 (2009).
Benachenhou, F. et al. Conserved structure and inferred evolutionary history of long terminal repeats (LTRs). Mob DNA 4, 5 (2013).
Perron, H. et al. Human endogenous retrovirus (HERV)-W ENV and GAG proteins: physiological expression in human brain and pathophysiological modulation in multiple sclerosis lesions. J. Neurovirol. 11, 23–33 (2005).
Perron, H. et al. Human endogenous retrovirus type W envelope expression in blood and brain cells provides new insights into multiple sclerosis disease. Mult. Scler. J. 18, 1721–1736 (2012).
Van Horssen, J., Van Der Pol, S., Nijland, P., Amor, S. & Perron, H. Human endogenous retrovirus W in brain lesions: rationale for targeted therapy in multiple sclerosis. Mult. Scler. Relat. Disord. 8, 11–18 (2016).
Guo, C. et al. Tau activates transposable elements in Alzheimer’s disease. Cell Rep. 23, 2874–2880 (2018).
Protasova, M. S. et al. Quantitative analysis of L1-retrotransposons in Alzheimer’s disease and aging. Biochemistry 82, 962–971 (2017).
Karlsson, H. et al. Retroviral RNA identified in the cerebrospinal fluids and brains of individuals with schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 98, 4634–4639 (2001).
Yao, Y. et al. Elevated levels of human endogenous retrovirus-W transcripts in blood cells from patients with first episode schizophrenia. Genes Brain Behav. 7, 103–112 (2008).
Duarte, R. R. R. et al. Schizophrenia risk from locus-specific human endogenous retroviruses. bioRxiv https://doi.org/10.1101/798017 (2019).
Steele, A. J. et al. Detection of serum reverse transcriptase activity in patients with ALS and unaffected blood relatives. Neurology 64, 454–458 (2005).
MacGowan, D. J. L. et al. A controlled study of reverse transcriptase in serum and CSF of HIV-negative patients with ALS. Neurology 68, 1944–1946 (2007).
McCormick, A. L., Brown, R. H., Cudkowicz, M. E., Al-Chalabi, A. & Garson, J. A. Quantification of reverse transcriptase in ALS and elimination of a novel retroviral candidate. Neurology 70, 278–283 (2008).
Douville, R., Liu, J., Rothstein, J. & Nath, A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann. Neurol. 69, 141–151 (2011).
Li, W. et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 7, 307ra153 (2015).
Perron, H. et al. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 94, 7583–7588 (1997).
Sotgiu, S. et al. Multiple sclerosis-associated retrovirus and progressive disability of multiple sclerosis. Mult. Scler. 16, 1248–1251 (2010).
Garson, J. A., Tuke, P. W., Giraud, P., Paranhos-Baccala, G. & Perron, H. Detection of virion-associated MSRV-RNA in serum of patients with multiple sclerosis [1]. Lancet 351, 33 (1998).
Sutkowski, N., Conrad, B., Thorley-Lawson, D. A. & Huber, B. T. Epstein-Barr virus transactivates the human endogenous retrovirus HERV-K18 that encodes a superantigen. Immunity 15, 579–589 (2001).
Ruprecht, K. et al. Regulation of human endogenous retrovirus W protein expression by herpes simplex virus type 1: implications for multiple sclerosis. J. Neurovirol. 12, 65–71 (2006).
Mameli, G. et al. Expression and activation by epstein barr virus of human endogenous retroviruses-W in blood cells and astrocytes: inference for multiple sclerosis. PLoS ONE 7, e44991 (2012).
Garrison, K. E. et al. T cell responses to human endogenous retroviruses in HIV-1 infection. PLoS Pathog. 3, 1617–1627 (2007).
Tandon, R. et al. Identification of human endogenous retrovirus-specific T cell responses in vertically HIV-1-infected subjects. J. Virol. 85, 11526–11531 (2011).
SenGupta, D. et al. Strong human endogenous retrovirus-specific T cell responses are associated with control of HIV-1 in chronic infection. J. Virol. 85, 6977–6985 (2011).
Jamieson, G. A., Maitland, N. J., Wilcock, G. K., Craske, J. & Itzhaki, R. F. Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J. Med. Virol. 33, 224–227 (1991).
Itzhaki, R. F. et al. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 349, 241–244 (1997).
Readhead, B. et al. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 99, 64-82.e7 (2018).
Andrews, W. D. et al. Detection of reverse transcriptase activity in the serum of patients with motor neurone disease. J. Med. Virol. 61, 527–532 (2000).
Li, W. et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl. Med. 7, 307ra153 (2015).
Mayer, J. et al. Transcriptional profiling of HERV-K(HML-2) in amyotrophic lateral sclerosis and potential implications for expression of HML-2 proteins. Mol. Neurodegener. 13, 1–25 (2018).
Garson, J. A. et al. Quantitative analysis of human endogenous retrovirus-K transcripts in postmortem premotor cortex fails to confirm elevated expression of HERV-K RNA in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 7, 45 (2019).
Krug, L. et al. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet. 13, e1006635 (2017).
Manghera, M., Ferguson-Parry, J. & Douville, R. N. TDP-43 regulates endogenous retrovirus-K viral protein accumulation. Neurobiol. Dis. 94, 226–236 (2016).
McKenzie, A. T. et al. Brain cell type specific gene expression and co-expression network architectures. Sci. Rep. 8, 1–19 (2018).
Seifarth, W. et al. Comprehensive analysis of human endogenous retrovirus transcriptional activity in human tissues with a retrovirus-specific microarray. J. Virol. 79, 341–352 (2005).
Grandi, N., Pisano, M. P., Scognamiglio, S., Pessiu, E. & Tramontano, E. Comprehensive analysis of HERV transcriptome in HIV+ cells: absence of HML2 activation and general downregulation of individual HERV loci. Viruses 12, 481 (2020).
Frank, O. et al. Variable transcriptional activity of endogenous retroviruses in human breast cancer. J. Virol. 82, 1808–1818 (2008).
Thonnard, S., Joëlle, F., Didier, C., Thierry, B. & Coulie, P. G. A human endogenous retroviral sequence encoding an antigen recognized on melanoma by cytolytic T lymphocytes. Cancer Res. 1, 5510–5516 (2002).
McLaren, P. J. et al. Association study of common genetic variants and HIV-1 acquisition in 6,300 infected cases and 7,200 controls. PLoS Pathog. 9, e1003515 (2013).
McLaren, P. J. et al. Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc. Natl. Acad. Sci. U.S.A. 112, 14658–14663 (2015).
Liu, R. et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86, 367–377 (1996).
Dean, M. et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science (80-.) 273, 1856–1862 (1996).
Berkhout, B. et al. Characterization of the anti-HIV effects of native lactoferrin and other milk proteins and protein-derived peptides. Antiviral Res. 55, 341–355 (2002).
Hoffman, P. M. et al. Isolation of LAV/HTLV-III from a patient with amyotrophic lateral sclerosis. N. Engl. J. Med. 313, 324–325 (1985).
Moulignier, A., Moulonguet, A., Pialoux, G. & Rozenbaum, W. Reversible ALS-like disorder in HIV infection. Neurology 57, 995–1001 (2001).
Gold, J. et al. Safety and tolerability of triumeq in amyotrophic lateral sclerosis: the lighthouse trial. Amyotroph. Lateral Scler. Front. Degener. 20, 595–604 (2019).
Brägelmann, J. & Lorenzo Bermejo, J. A comparative analysis of cell-type adjustment methods for epigenome-wide association studies based on simulated and real data sets. Brief. Bioinform. 20, 2055–2065 (2019).
Conlon, E. G. et al. Unexpected similarities between C9ORF72 and sporadic forms of ALS/FTD suggest a common disease mechanism. Elife 7, e37754 (2018).
Tam, O. H. et al. Postmortem cortex samples identify distinct molecular subtypes of ALS: retrotransposon activation, oxidative stress, and activated glia. Cell Rep. 29, 1164–1177 (2019).
Prudencio, M. et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 18, 1175–1182 (2015).
Ignatiadis, N., Klaus, B., Zaugg, J. B. & Huber, W. Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods 13, 577–580 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Commonly used functions by the Jaffe lab jaffelab. http://research.libd.org/jaffelab/.
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucl. Acids Res. 49, D545–D551 (2021).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 28, 27–30 (2000).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLOS Comput. Biol. 11, e1004219 (2015).
Nicolas, A. et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268-1283.e6 (2018).
Belmont, J. W. et al. A haplotype map of the human genome. Nature 437, 1299–1320 (2005).
Ying, J., Oliver, T. H., Eric, P. & Molly, H. TEtranscripts: a package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 31, 3593–3599 (2015).
Acknowledgements
We would like to thank and acknowledge the MRC London Neurodegenerative Diseases Brain Bank at the Institute of Psychiatry, Psychology and Neuroscience, King's College London. With special thanks to the people who have donated tissue to the advancement of clinical sciences.
Funding
This work was funded by the Motor Neurone Disease Association (Grant: Jones/Oct15/958-799), the ALS Association (Grant: 18-LGCA- 394 "HERV-K molecular studies in ALS") and MND Scotland (Grant: "Multi-omics analysis of the role of human endogenous retroviruses in ALS"). AAC is an NIHR Senior Investigator. This is an EU Joint Programme - Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organisations under the aegis of JPND - https://www.jpnd.eu (United Kingdom, Medical Research Council (MR/L501529/1; MR/R024804/1) and Economic and Social Research Council (ES/L008238/1)) and through the Motor Neurone Disease Association. This study represents independent research part funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London.
Author information
Authors and Affiliations
Contributions
Contribution to concept and design: A.R.J., A.I., B.N.A., C.T., J.A.G., A.L.M., A.A.C.; Contribution of data: A.R.J., A.S., C.T.; Contribution of intellectual input: A.R.J., A.I., B.N.A., H.B., A.S., C.T., J.A.G., A.L.M., A.A.C.; Contribution to write-up: A.R.J., J.A.G., A.L.M., A.A.C.
Corresponding author
Ethics declarations
Competing interests
Ammar Al-Chalabi reports grants from Motor Neurone Disease Association, grants from Betty Messenger Foundation, grants from Anonymous Family Trust, grants from Joint Programme on Neurodegeneration via MRC, grants from NIHR during the conduct of the study, other from Mitsubishi Tanabe Pharma, other from OrionPharma, other from Cytokinetics, other from Chronos Therapeutics, outside the submitted work. Ashley R Jones, Alfredo Iacoangeli, Brett N Adey, Harry Bowles, Aleksey Shatunov, Claire Troakes, Jeremy A Garson, and Adele L McCormick report no conflict of interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained an error in the Funding section. Full information regarding the corrections made can be found in the correction for this Article.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jones, A.R., Iacoangeli, A., Adey, B.N. et al. A HML6 endogenous retrovirus on chromosome 3 is upregulated in amyotrophic lateral sclerosis motor cortex. Sci Rep 11, 14283 (2021). https://doi.org/10.1038/s41598-021-93742-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-93742-3
This article is cited by
-
Locus specific reduction of L1 expression in the cortices of individuals with amyotrophic lateral sclerosis
Molecular Brain (2022)
-
Retroviral Elements in Pathophysiology and as Therapeutic Targets for Amyotrophic Lateral Sclerosis
Neurotherapeutics (2022)
-
Neuron-specific chromosomal megadomain organization is adaptive to recent retrotransposon expansions
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
