
Abstract
Cotton leaf curl disease (CLCuD), caused by begomoviruses in combination with betasatellite molecule, has adversely affected cotton industry of Indian subcontinent. To devise a CLCuD-control strategy, RNAi-mediated approach was followed in this study. Gossypium hirsutum cv. HS6 plants were transformed with intron-hairpin RNAi (ihpRNAi-C4) construct carrying silencing suppressor C4 gene of Cotton leaf curl Multan virus (CLCuMuV). Efficacy of the construct in imparting CLCuD resistance was evaluated in transgenic (T0, T1) cotton lines. Accumulation of CLCuMuV/betasatellite and attenuation of CLCuD symptoms in the transgenic lines were monitored at different times interval after virus inoculation. Northern hybridization revealed the expression of C4-gene derived siRNA. Expression of the ihpRNAi transcript was recorded higher in transgenic lines expressing siRNA which supposedly targeted the C4 gene. A significant delay in detection of virus as well as betasatellite was observed in the transgenic lines. At 30 days post inoculation (dpi), none of the lines tested positive. At 45 dpi, however, it could be detected in few lines having much lower titre as compared to non-transformed control plants. Notably, till 60 dpi, no significant progression of the virus/betasatellite DNA was observed and the plants did not exhibit any characteristic CLCuD symptoms. A tolerance phenomenon leading to escape of CLCuD symptoms in the transformed cotton was described.
Similar content being viewed by others

Introduction
Cotton (Gossypium hirsutum) is the most important source of natural fibre for textile, paper and vegetable oil industries worldwide. India is the second largest producer as well as exporter of cotton1. Most commercially grown cotton (~ 90%) in India belongs to different cultivars of G. hirsutum, whereas other species like G. arboreum, G. barbedense and G. harbaceum collectively contribute about 10% of total cultivation2. Cotton leaf curl disese (CLCuD), caused by begomoviruses in association with betasatellite, is endemic in north-western states (Punjab, Haryana, and Rajasthan). It inflicts enormous losses to the crop by reducing yield and compromising fibre quality. All cultivated varieties of G. hirsutum in the Indian subcontinent are susceptible to distinct begomoviruses associated with CLCuD (BAC) and Cotton leaf curl Multan betasatellite (CLCuMB).
Presently, there are five BAC species belonging to single-stranded (ss), circular DNA viruses (family Geminiviridae). The most widespread are Cotton leaf curl Multan virus (CLCuMuV) and Cotton leaf curl Kokhran virus (CLCuKoV), while Cotton leaf curl Bangalore virus, Cotton leaf curl Alabad virus, Cotton leaf curl Gezira virus and Papaya leaf curl virus are less frequently encountered in the Indian subcontinent3,4. They are transmitted exclusively by whitefly (Bemisia tabaci) in a circulative, persistent manner. The monopartite CLCuMuV and CLCuKoV genomes in association with Cotton leaf curl Multan betasatellite molecule have been demonstrated to cause CLCuD5,6. The initial symptoms of CLCuD are characterized by darkening and thickening of leaf veins, subsequently causing upward (sometimes downward) curling of leaf, formation of cup-shaped leaf-like structures (enations) on the underside of the leaf. The infected plant becomes stunted due to reduced inter-nodal distance. Furthermore, flowering, formation and maturation of bolls are affected leading to lower yield and poor fibre quality7. Over the last three decades, CLCuD remains the most disastrous disease of cotton and a major limiting factor for its production in the Indian subcontinent.
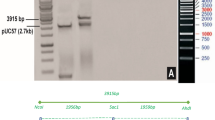
The CLCuMuV genome is monopartite, circular, ss DNA with approx. 2.7 kb in size (Fig. 1A). It is transcribed bidirectionally in virion sense and complimentary-sense orientations via intergenic region (IR) present between the first ORF of the virion sense and complimentary-sense strand. DNA elements required for replication and transcription are present in the IR. While V1 and V2 genes are transcribed from the virion sense, C1, C2, C3, C4 and C5 genes from the complimentary-sense strand8. The C1 encodes the replication associated protein (Rep) that participates in rolling circle replication and regulation of complimentary sense gene. Transcriptional activator protein, expressed by the C2 gene, regulates transcription of coat protein and movement protein encoding genes. Furthermore, it is involved in gene silencing. C3 encodes the replication enhancer protein that leads to accumulation of viral DNA inducing symptom development and assists to enhance Rep-mediated ATPase activity. The C4 gene encodes a multifunctional protein which is involved in symptom determination, systemic movement of virus and suppressing RNA silencing synergistically with transcriptional activator protein. In East African cassava mosaic virus and African cassava mosaic virus, AC4 protein has been shown to suppress gene silencing following binding of siRNA9. A comparative study on silencing suppressor genes of CLCuMuV demonstrated that the C4 protein may prevent siRNA from binding to RNA induced silencing complex (RISC) both prior to duplex binding as well as dicer mediated cleavage of long dsRNA10. Though C5 protein may participates in replication of DNA, it remains insignificant in viral infection. The CP (V1), which is multifunctional, is involved in genome packaging, insect-mediated transmission and spread of the virus. The V2 gene expresses pre-coat protein that interacts with silencing suppressor genes such as C2 and C4.

Genome organization of Cotton leaf curl Multan virus (CLCuMuV) (A) and associated Cotton leaf curl Multan betasatellite (CLCuMuB) (B). (A) Genome representing complementary-sense genes C1, C2, C3, C4 (silencing suppressor) and virion sense V1, V2 genes. (B) Betasatellite βC1 gene encoding C1 protein. Abbreviations: A rich-Adenine-rich region; IR-Intergenic region; SCR-satellite conserved region. (C) Development of Cotton leaf curl Multan virus-C4 intron-hairpin RNAi construct. The linear T-DNA map of intron-hairpin (ihp) RNAi construct in binary expression vector pFGC1008, in sense and antisense orientation separated by GUS intron. The complete RNAi cassette was under regulatory control of CaMV 35S promoter and OCS terminator, and having hpt as a plant selectable marker. Abbreviations: CaMV 35S—Cauliflower mosaic virus 35S promoter, GUS intron—β-glucuronidase intron, OCS—Octopine synthase terminator, MAS I promoter—Mannopine synthetase I promoter, and hpt—hygromycin phosphotransferase.
The begomoviruses are associated with three satellite molecules namely alphasatellite, betasatellite and newly-characterized deltasatellite4,11. These satellites depend on their helper virus genome for encapsidation and systemic infection. Alphasatellite encodes a single Rep protein which is non-pathogenic and enhances capability of autonomous replication in host plant12. While several distinct alphasatellites have been identified in CLCuD—affected cotton, they have not been shown essential either for development of the disease or virus/betasatellite activity. The betasatellites, ssDNA molecules, do not reveal any sequence homology with their helper viruses except stem loop structure having nonanucleotide (TAATATTAC) sequence13,14. The stem loop structure structure is present within highly conserve sequence called as satellite conserved region. Furthermore, adenine rich sequence and a single coding sequence (βC1) is located in the complimentary sense of the betsatellites (Fig. 1B). Betasatellite encodes a single βC1 protein which is a pathogenicity determinant, suppressor of host defence and involved in upregulating the level of viral DNA in planta10,15,16. Deltasatellites, small (~ 675 nts) non-coding satellites, are rarely encountered and not associated with CLCuD. Similar to C4 protein, βC1 acts as a suppressor of RNA silencing following binding to host’s DICER-like proteins required for biogenesis of small RNA molecules namely small interfering RNA (siRNA) and microRNA (miRNA)17.
Efforts are being made to develop cotton variety resistant to CLCuD. Control of CLCuD through resistance breeding has not been promising due to availability of limited resistance source and frequent emergence of recombinant begomoviruses18. RNAi based approaches have shown remarkable success against begomovirus infection following homology-based degradation of virus gene and its transcript RNA through transcriptional gene silencing (TGS) and post-transcriptional gene silencing (PTGS) mechanism in host plants19,20,21. RNAi is a highly conserved mechanism known to control gene expression in sequence specific manner by short non-coding RNAs (siRNA, miRNA) molecules22,23,24. These non-coding RNAs are closely related in their biogenesis and mode of action17. siRNA is originated from long double-stranded RNA (dsRNA) which is further processed into short (21–25 nts) single-stranded mature siRNA. Apart from regulating gene expression, siRNA is implicated in a variety of processes, such as defence against viruses, establishment of hetero-chromatin formation, DNA methylation, silencing of transgenes and post-transcriptional regulation of mRNA25,26. Interestingly, siRNA target the genes from which they are derived and repress any target mRNA showing substantial sequence complementarity.
In begomoviruses, both TGS and PTGS, involve processing of dsRNA by Dicer-like protein into siRNA which are subsequently incorporated into an enzyme complex termed as RISC. The RISC guides its argonaute protein to degrade target mRNA homologous to the siRNA in sequence specific manner. RNAi based antiviral defense mechanism in plants is designed following transgene expression of a virus specific dsRNA cognate to viral genome. A potential RNAi triggers molecules which can be successfully transformed into plants via expression of intron hairpin (ihp) construct carrying viral genome sequence into siRNA. In this study, G. hirsutum cv. HS6 was genetically transformed with the ihpRNAi construct harbouring the silencing suppressor gene (C4) of CLCuMuV to impart CLCuD resistance. The transgenic cotton lines, thus produced, were evaluated for resistance to CLCuMuV/CLCuMuB infection.
Results
Transformation of G. hirsutum cv. HS6
A total of approx. 1000 embryo apices co-cultivated with Agrobacterium tumefaciens GV3101 led to the production of nine transgenic (T0) lines (namely T0.3, T0.4, T0.6, T0.8, T0.10, T0.25, T0.26, T0.27 and T0.28 ) (Suppl Fig. SF1).
Production of T1 progeny
Seeds (10–12 per line) of G. hirsutum cv. HS6 (T0) lines were allowed to germinate on MS selection media supplemented with hygromycin. The germinated (considered as CLCuMuV-resistant) and non-germinated (supposedly CLCuMuV-sensitive) seeds, maintained a germination ratio of 3:1 against hygromycin resistance on the selection medium. The presence of transgene in the T1 plants was confirmed in a total of 43 plants reflecting ~ 72% positive. This ratio was consistently observed in most of the lines (Supplementary Table ST2), and may be correlated with the Mendelian segregation (3:1) of a single dominant gene27.
Molecular characterization of transgenic lines
Detection of hpt and transgene (C4) insert
The integration of transgenes (CLCuMuV-C4, hpt) in the nine putative transformed (T0) plants was checked by PCR. Among them, six plants (T0.3, T0.4, T0.6, T0.8, T0.10 and T0.26) were found PCR positive carrying copies of C4 and hpt genes (Fig. 2A, B).

PCR amplification of: (A) silencing suppressor Cotton leaf curl Multan virus CLCuMuV-C4 gene, (B) hygromycin phosphotransferase (hpt) gene in transformed T0 G. hirsutum cv. HS6 plants. T0.3, T0.4, T0.6, T0.8, T0.10 and T0.26 denote PCR positive lines, while T0.25, T0.27 T0.28 were found PCR negative. M is 100 bp DNA marker, PC is positive control, NT is non-transformed control and NC is negative controls. (C) CLCuMuV-C4 gene in T1 G. hirsutum cv. HS6- T1.3, T1.4, T1.6, T1.8, T1.10 and T1.26 lines employing C4 gene specific primers. M is100 bp DNA marker, In PC, construct plasmid DNA served as the template; In NC/NT, genomic DNA from non-transformed control was employed as the template in PCR.
Similarly, PCR was performed to confirm the presence of the transgene (C4) in the transgenic T1 lines. All the six lines were found PCR positive for the C4 gene (Fig. 2C).
Southern hybridization
Southern blot hybridization using a DNA probe specific to the C4 gene was performed. The transgene integrated in T0 lines was detected at a single locus in the lines T0.6, and T0.26, while double loci integration event was observed in the T0.3, T0.4, T0.8 and T0.10 lines (Suppl Fig. SF2A).
Analysis of the T1 lines also showed low copies (2–3) of transgene insertions. The T1 lines, namely, T1.3.1, T1.6.1, T1.8.1 and T1.26.1 showed two copies of the transgene integration events, while T1.4.1and T1.10.1 carried three integration events (Suppl Fig. SF2B).
Semi-quantitative RT-PCR analysis
Different level of dsRNA expression was observed in all the six transgenic (T0) lines. While, transcript level appeared to be higher in T0.8, relatively a lower expression was exhibited in T0.10 and T0.26 transgenic lines.
Likewise, in the T1 lines, intensity of the transcript level appeared to be higher in the line T1.8.1. In contrast, lines T1.10.1 and T1.26.1 exhibited lower intensity bands among all the T1 lines. An intensity of relatively intermediate level was observed in T1.3.1, T1.4.1 and T1.6.1 lines. The expression of the transgene was found to be comparable with that of internal control GAPDH gene, yielding more or less of equal intensity amplicons.
qRT-PCR
The Quantitative Real-time data (Ct values) revealed that ihpRNA-C4 was transcribed into dsRNA in all the transgenic (T0 and T1) lines. Taking GAPDH gene as an internal control, the RQ value of the dsRNA was found to be higher in T0 as compared to control plant. T0.8 being the highest, the lowest transcription was observed in T0.26 (Suppl Fig. SF3A).
The RQ value of the dsRNA transcripts was also exhibited high in T1 transgenic lines as compared to NT (non transformed) control plants (Suppl Fig. 3B). Transgenic lines T1.8.1, T1.6.1,T1.10.1, T1.4.1, and T1.3.1 exhibited 12, 12, 12, 11 and 11-fold, increase respectively, while T1.26.1 showed lowest expression (nine-fold) as compared to the NT control plants (Suppl Fig. SF3B).
Northern hybridization
It was performed to check the expression of siRNA levels in the transgenic lines. The C4 gene-based DIG-11-dUTP-labeled probe yielded positive signals corresponding to the 20–25 nt region of ultra-low range marker, indicating the processing of ihpRNAi-C4 construct subsequently producing siRNAs in the transgenic lines. The intensity of siRNA bands appeared to be slightly higher in T0.3, T0.4, T0.8, T0.10 and T0.26 lines as compared to the line T0.6 (Fig. 3A, B).

(A) Detection of small RNAs population of transgenic (T0) G. hirsutum cv. HS6- T0.3, T0.4, T0.6, T0.8, T0.10 and T0.26 and non-transformed (NT) control resolved through denaturing polyacrylamide gel electrophoresis. (B) Northern blot analysis of siRNAs generated in transgenic T0 lines transferred onto HybondN membrane and hybridized with DIG-11-dUTP-labelled C4-derived probe.
Intensity of siRNA signals seemed to be of equal intensity in all the T1 lines, except T1.3 which was slightly higher (Suppl Fig. SF4A, B).
Whitefly-mediated virus inoculation of CLCuMuV and assessment of CLCuD symptoms
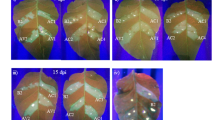
Following CLCuMuV/CLCuMB inoculation of transgenic lines with viruliferous whiteflies, symptom expression were periodically evaluated over a period of two months under strict greenhouse conditions (Fig. 4A–C). There were no visible CLCuD symptoms in the transgenic lines as compared to control showing severe symptoms (Fig. 4A, B). The initial symptoms of CLCuD appeared in the non-transformed control plant at 15 dpi. While the transgenic plants were apparently seen healthy without visible symptoms at 30 dpi when the characteristic symptoms were fully expressed in the control plants. The symptoms consisted of vein thickening, leaf curling, leafy enations and stunting of plants with no or few ball settings as observed in the control plants (Fig. 4A, C). Contrary, all the transgenic lines (T0 and T1) remained apparently symptom free maintaining their natural vegetative and reproductive growth (Suppl Fig. SF8).

(A) Assessment of cotton leaf curl disease (CLCuD) symptoms in transgenic G. hirsutum cv. HS6 (T0.8) and non-transformed control plants after inoculation with viruliferous whiteflies (Bemisia tabaci) under greenhouse conditions. (B) No characteristic CLCuD symptoms developed on the leaves of transgenic lines 60 days post inoculation and transgenic plant showed normal phenotype. (C) CLCuD symptoms were developed on non-transformed control G. hirsutum var. HS6 plant such as vein thickening, leaf curling and enations were observed.
Remarkably, 8-weeks post-inoculation none of the transgenic lines exhibited any CLCuD symptoms. Apparently, no phenotypic differences were observed among the transgenic and NT control plants, except appearance of severe symptoms in the control plants.
Detection of CLCuMuV and betasatellite DNA
PCR failed to detect CLCuMuV and CLCuMuB in T0 lines eight weeks post-inoculation. While, the control plants yielded DNA fragments corresponding to the CP gene (~ 780 bp) and betasatellite (~ 1.3 kb) (Suppl Fig. SF5A, B).
Similarly, PCR did not lead to amplification of CLCuMV and betasatellite in (T1) lines at eight weeks post-inoculation. However, non-transformed control plants yielded DNA fragments of CLCuMuV and CLCuMuB (Suppl Fig. SF6A, B).
Rolling circle amplification
Total genomic DNA isolated from the transgenic lines and NT control plants was subjected to Rolling Circle Amplification (RCA). The RCA products served as templates for PCR amplification of the virus and betasatellite using primers specific to the CP gene and betasatellite as described in above section. RCA products, derived from the T0 lines at 15 dpi, did not detect the virus and betasatellite, though it tested positive in control plants (Suppl Fig. SF7). Thus, clearly demonstrating that there was no detection of virus/betasatellite DNA in any of transgenic (T0) lines.
However, at 30 dpi, very faint DNA fragments of CLCuMuV and betasatellite were observed in lines T0.10 and T0.26 (Suppl Fig. SF7A, B).RCA analysis at 60 dpi, revealed higher intensity DNA fragments CLCuMuV/betasatellite in control plants, as compared to all other T0 transgenic plants (Suppl Fig. SF7A, B).
The progression of CLCuMuV/CLCuMuB in the T1 lines was also assayed by RCA. No virus/betasatellite DNA fragments were identified at 30 dpi, while they could be detected in the NT control plants. In lines (T1.4.1, T1.10.1 and T1.26.1), CLCuMuV/betasatellite could be detected at 45 dpi (Fig. 5A, B).

RCA-PCR based detection of: (A) CLCuMuV-coat protein (CP) gene, and (B) CLCuMuB. Total genomic DNA of T1 progeny G. hirsutum cv. HS6- T1.3, T1.4, T1.6, T1.8, T1.10 and T1.26 plants and non-transformed control plants were isolated at 30, 45 and 60 dpi with viruliferous whitefly, and RCA was performed. The RCA product served as a template in PCR employing primers specific to CLCuMuV-CP and betasatellite. Amplification of the CP gene (~ 780 bp) and the CLCuMuB (~ 1.3 kb) was detected in non-transformed control plants only at 30 dpi. A low degree amplification of CP gene and betasatellite was recorded in T1.4, T1.10 and T1.26 lines at 45 dpi and in transgenic HS6- T1.3, T1.4, T1.6, T1.10 and T1.26 lines at 60 dpi. M is 1 kb DNA marker, NT is non-transformed control and Mock is uninoculated control.
However, a low accumulation of the virus and betasatellite DNA was detected in all the lines except in T1.8.1 at 60 dpi (Fig. 5A, B).
Discussion
In this study, ihpRNAi construct carrying the C4 gene of CLCuMuV was evaluated for its efficacy in combating the CLCuD infection following transformation in G. hirsutum cv. HS6 plants. The expression of ihp dsRNA-C4 -derived siRNA, assessment of CLCuD resistance and progression of the CLCuMV/CLCuMB levels in transgenic cotton were studied. The expression of siRNA was high in the transgenic lines exhibiting no visible CLCuD symptoms. They showed resistance against CLCuD following viruliferous whitefly-mediated inoculation. All the transgenic lines exhibited significantly lower level of virus as well as associated betasatellite in comparison to the control plants. A low number of transgene integration was observed in transgenic lines. In contrast to T0 lines, a slight variation in the number of transgene integration was evident in the T1 lines which could be attributed to minor variations in the genetic background or differences in the plant microenvironments27. Insertion of multiple copy transgene in the plant genome favours resistance leading to higher production of transgene derived siRNA generation.
qRT-PCR analysis showed that the level of ihp dsRNA was unexpectedly high in the transgenic lines, displaying high level of siRNA expression. A high level of dsRNA could be due to its incomplete processing28.
A significant lower level of the virus/betasatellite DNA as compared to the control plants was attributed to the silencing of CLCuMuV-C4 gene by hpRNAi-derived siRNA. It may be mentioned that the C4 gene is fully contained inside the C1 gene in a different ORF. The C1 gene encodes Rep protein which is absolutely required for virus replication. Thus, additional targeting of C1 transcript by the C4-derived siRNA may not be overlooked. That may explain the detection of a low virus titer.
The C4 and βC1 genes appear to be important targets for the control of the begomovirus infections as they are strong RNA silencing suppressors and play a vital role in the survival of viruses. AC4/C4 gene encodes a multifunctional protein which acts as pathogenicity determinants in monopartite and bipartite geminiviruses. In a previous study, role of individual genes encoded by four begomoviruses (mono/bipartite) on infection cycle and level of miRNAs involved in plant development, was investigated. A disruption of the C4 gene (monopartite begomovirus) resulted in attenuation of symptoms and low infectivity. The C4 protein was responsible to change the levels of miRNAs and was probably involved in modulating gene expression to create an environment favourable for virus proliferation29. In CLCuKoV, it is involved in virus spread and in modulating symptoms, but dispensible for maintenance of the betasatellite suggesting a lack of suppressor activity. If present, does not overcome the silencing based resistance to virus movement30.
In a first report of a begomovirus requiring four suppressor genes to establish infections under field conditions, efficacy and relative strength of CLCuMuV and CLCuMB-encoded suppressors to suppress RNAi silencing were compared. While C4 protein exhibited silencing suppressor activity, V2 being the strongest. It could bind both to long dsRNA as well as siRNA31. In another report, CLCuKoV C4 protein was demonstrated to prevent the spread of systemic silencing. It could bind to long dsRNA and both long ds/ssDNA, but not to sRNA/DNA32.
In the present study, transgenic G. hirsutum lines did not display any symptoms at two months post-inoculation of CLCuMuV/CLCuMB. While conventional PCR failed to detect virus DNA and betasatellite in any transgenic line at 60 dpi, RCA-PCR was positive in all except one. However, level of the virus accumulation was much lower in comparison to non-transformed plants, and the plants displayed no characteristic CLCuD symptoms.
G. hirsutum plants following transformation led to generation of siRNA responsible for suppressing CLCuD symptoms, though CLCuMuV/betasatellite could be detected at a later stage. This type of observed resistance may be termed as tolerance, where a balance between the host defence response and virus counter defence seems to exist that allows the virus to survive in the host without causing apparent symptoms. These results are consistent with earlier studies where RNAi has imparted tolerance33. In a previous study, intergenic region (CLCuMuV) carrying ihpRNAi construct was transformed in Indian cotton cv. ‘Narsimha’. Transformed (T0) cotton plants did not develop symptoms at 90 dpi. However, second (T1) generation was not assessed. A tolerance phenomenon was observed as some of the transformed plants revealed the presence of virus34. In a preliminary study, tobacco plants transformed with βC1 exhibited no or delayed CLCuD symptoms following CLCuMuV/betasatellite inoculation. Titre of the virus or presence of βC1-derived siRNA could not be tested in the transgenic lines35.
In the absence of an immune cultivar, availability of virus/betasatellite tolerant variety may be an alternative to minimise CLCuD losses. In this study, cotton lines showing RNAi-mediated tolerance resistance against CLCuD infection have not been tested in fields. Presence of virus in the tolerant plants, and the variation in virus complex causing CLCuD, however, may pose a risk development of recombinant virus mutants that may produce novel viruses capable of overcoming resistance and spread to other crop plants36,37. Reference may be made of cassava mosaic disease in which interaction between two distinct begomoviruses (African cassava mosaic virus-CM and East African cassava mosaic virus) are responsible for developing severe disease symptoms. The synergism is due to the action of silencing suppressor AC2 and AC4 genes that target different steps in RNA silencing and overcome host defense38.
Conclusion
In this study, transgenic G. hirsutum cv. HS6 lines significantly reduced the virus DNA accumulation and attenuated CLCuD symptoms following CLCuMuV-C4 derived RNAi strategy. Further, siRNAs were triggered at a high level and symptoms remained escaped. However at the later stage, the transgenic plants were mildly infected, supporting CLCuMuV/CLCuMB replication. Role of C4 gene-derived siRNA in developing virus resistance was investigated for the first time in transgenic G. hirsutum cultivar against CLCuD infection.
Materials and methods
ihpRNAi construct preparation
ihpRNAi construct harbouring C4 gene sequence in sense-antisense orientation was produced in binary vector pFGC1008. Full-length clone of Cotton leaf curl Multan virus-Raj (CLCuMuV-Raj, Accession No. GQ220850) was used as a template for PCR-based amplification of C4 gene followed by its cloning in binary vector pFGC1008 in sense orientation at AscI and SwaI, and in antisense orientation at BamHI and SpeI restriction sites (Sambrook and Russell 2012). The orientations of the gene were separated by a GUS-intron under the transcriptional control of constitutive CaMV 35S promoter containing hpt gene as a plant selection marker (Fig. 1C). The developed ihpRNAi cassette was mobilized into Agrobacterium tumefaciens strain GV3101 using freeze thaw method. The integrity of the construct was tested by sequencing (Xcelris genomics, India).
Transformation of Gossypium hirsutum cv. HS6
The young embryo apex explants excised from germinating seeds of G. hirsutum cv. HS6 were, were transformed with A. tumefaciens strain GV3101 harbouring ihpRNAi-C4 construct. Cotton transformation was done as described earlier39,40. A total of 1000 number of explants (in different batches) were transferred on the solid MS basal medium supplemented with cefotaxime (250 mg/L) (Suppl Fig. SF1A). The explants were placed on selection medium supplemented with hygromycin (50 mg/L) and cefotaxime (250 mg/L) (B), sub-cultured on shooting medium for 3 weeks (C). The regenerated shoots were subsequently shifted on root induction medium (D, E). For 2–3 weeks, plantlets with well-developed roots were placed in Hoagland medium (F). The hardened plants were transferred to pots in greenhouse under controlled conditions (G, H).
Collection and germination of seeds of transgenic T0 lines
The transformed (T0) G. hirsutum cv. HS6 plants were self-pollinated under strict greenhouse conditions for the production of seeds. Second generation transgenic (T1) plants were raised following collection and subsequent germination of transgenic (T0) plants seeds. The 10–12 seeds of each (T0) transformed line were germinated on MS selection medium containing hygromycin (50 mg/L), cefotaxime (250 mg/L) and BAP (2.0 mg/L).
Confirmation of transformed G. hirsutum cv. HS6 plants
Total genomic DNA from leaves of putative transformed (T0) G. hirsutum cv. HS6 plants was isolated using DNeasy plant mini kit according to manufacturer’s instructions (Qiagen, Germany). The integration of transgenes in putative T0 transformed plants was confirmed by PCR and Southern hybridization.
PCR
PCR was performed employing C4 gene specific (C4 For/Rev primers, Supplementary Table ST1) using genomic DNAs (T0 plants) as templates. PCR mixture (200 ng of template DNA, 10X Taq buffer A, 2.5 mM each dNTP, 50 pmol of each primer, 3 U/μL Taq DNA polymerase) with parameters: initial denaturation at 95 °C for 7 min followed by 30 cycles of final denaturation at 95 °C for 50 s, annealing at 50 °C for 50 s, initial extension at 72 °C for 30 s and final extension of 72 °C for 5 min.
Southern hybridization
For Southern hybridization, 10 µg of total genomic DNA isolated from the PCR positive transgenic (T0, T1 plants) and non-transformed G. hirsutum cv. HS6 control plants was subjected to restriction digestion with 10U BamHI (Fermentas, USA). The digested genomic DNA was electrophoresed and transferred on Hybond-N nylon membrane (GE healthcare, UK)41. DIG labelled DNA probe representing C4 gene was prepared using labelling kit (Roche Diagnostics, Germany). The membrane was hybridized with the DIG labelled probe and subjected to the development of colour following incubation in in a freshly prepared Colour-substrate solution Nitro blue tetrazolium/5-Bromo-4-chloro-3-indolyl phosphate (NBT/BCIP).
Expression of transgene transcript
Total RNA was isolated from the transgenic and non-transformed plants using RNeasy Plant Mini kit (Qiagen, Germany). cDNA was synthesized with M-MuLV RT-PCR Kit (Merck, Germany). Expression of the transgene transcript was analysed through Reverse transcriptase (RT)-PCR and Real Time (qRT)-PCR.
RT-PCR
It was performed to compare relative expression of the transgene transcript level with that of GAPDH gene in transgenic (T0 and T1) lines using Qiagen Long Range 2Step RT-PCR kit, Germany with oligo primers specific to the C4 (RT-C4 For/Rev) and GAPDH (RT-GA For/Rev) genes (Supplementary Table ST1). PCR mixture was subjected to PCR amplification with parameters: initial denaturation at 94 °C for 5 min, followed by 35 cycles of final denaturation at 94 °C for 45 s, annealing at 55 °C for 45 s and initial extension at 72 °C for 30 s and final extension at 72 °C for 3 min.
Quantitative real-time PCR
It was conducted to quantify the level of expression of ihpRNA dsRNA transcript, in comparison to that of GAPDH gene in the transgenic lines and non-transformed control plants. The housekeeping gene GAPDH served as an internal control. The mixture contained template cDNA (100 ng), 20 pmol of each (RT-C4 For/Rev and RT-GA For/Rev (Supplementary Table ST1) and Real Time master mix including SYBR green dye (2X KAPA SYBR® FAST Universal and 50X ROX high) (KAPA Biosystems, USA). The qRT-PCR programming conditions were as follow: initial denaturation at 95 °C for 5 min, followed by 40 cycles of final denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s and extension at 72 °C for 30 s. All the target samples, internal control and negative control were taken in triplicates and the experiment was conducted twice.
The qRT-PCR generated data was statistically analysed to calculate the relative quantification (RQ) of ihpRNA transcript level in the transgenic and NT control plants. The StepOnePlus Real-Time PCR System, having StepOne™ Software v2.3 was used for the calculation of threshold cycle (Ct) values of all the target samples (Applied Biosystems, USA). The RQ of the target samples was calculated by Livak’s 2−ΔΔCt method using mean Ct values of the target and the internal control gene from triplicate samples42. Standard deviation (SD) and standard error (SE) were determined from the triplicate samples.
Detection of siRNAs in transgenic lines
Northern hybridization
Total small RNA population was isolated from the leaves of transgenic and NT control plants43. It was performed to assess the expression level of siRNA in transgenic (T0, T1) plants with respect to corresponding non-transformed cotton plants. The low molecular weight RNA were separated on denaturing polyacrylamide gel (PAGE 15%) and transferred onto a Hybond-N nylon membrane (GE Healthcare, UK). Hybridization was performed with CLCuMuV-C4 gene-based DIG-11-dUTP labelled probe at 42 °C in DIG Easy Hyb solution using DIG DNA labelling and detection kit (Roche Diagnostics, Germany). The membrane was equilibrated in Detection buffer containing NBT/BCIP-T under Gel Doc™ EZ System (BioRad, USA) using the Image Lab™ software Version 4.1. siRNA fragments (20–25 nt long) were visualized, which were compared with the original PAGE exhibiting ultra-low range RNA marker (Thermo Scientific, Germany).
Screening and evaluation of transgenic lines for resistance to CLCuD
Transgenic (T0, T1) plants were inoculated (at 4–5 leaf stage) with CLCuMuV/CLCuMB through whitefly (Bemisia tabaci)44. Approximately, 100 viruliferous whiteflies were placed on each transgenic and non-transformed control plants for 72 h inside insect proof chamber under controlled conditions. The plants were periodically assessed for 60 dpi for the development of symptoms.
Detection of CLCuMuV/CLCuMB DNA in the transgenic lines
The presence of virus DNA and betasatellite molecules in transgenic plants were assayed by PCR and RCA.
PCR
Total genomic DNA was isolated from the transgenic and non-transformed plants. It was subjected to PCR for the specific detection of the CP gene of CLCuMuV and betasatellite molecules employing primer pairs For/Rev and Beta For/Rev specific for the CP gene of CLCuMuV and betasatellite molecules, respectively (Supplementary Table ST1), as described in previous sections.
Rolling circle amplification based PCR
Total genomic DNA was subjected to RCA using TempliPhi DNA amplification kit (GE Healthcare, UK). The RCA products served as the templates for amplification of the virus DNA and betasatellite employing primers specific for the CLCuMuV-CP gene and betasatellite (Supplementary Table T1). The PCR parameters remained same as described above.
References
Anonymous. Ministry of Textiles, Government of India, http://texmin.nic.in/textile-data (2020).
Rajagopalan, P. A. et al. Dominance of resistance-breaking cotton leaf curl Burewala virus (CLCuBuV) in northwestern India. Adv. Virol. 157, 855–868 (2012).
Briddon, R. W. & Markham, P. G. Cotton leaf curl virus disease. Virus Res. 71, 151–159 (2000).
Qadir, R., Khan, Z. A., Monga, D. & Khan, J. A. Diversity and recombination analysis of Cotton leaf curl Multan virus, a highly emerging begomovirus in northern India. BMC Genomics 20, 274 (2019).
Mansoor, S. et al. Identification of a novel circular single-stranded DNA associated with cotton leaf curl disease in Pakistan. Virology 259, 190–199 (1999).
Briddon, R. W. et al. Identification of DNA components required for induction of cotton leaf curl disease. Virology 285, 234–243 (2001).
Sattar, M. N., Kvarnheden, A., Saeed, M. & Briddon, R. W. Cotton leaf curl disease–an emerging threat to cotton production worldwide. J. Gen. Virol. 94, 695–710 (2013).
Hanley-Bowdoin, L., Settlage, S. B., Orozco, B. M., Nagar, S. & Robertson, D. Geminviruses: Models for plant DNA replication, transcription, and cell cycle regulation. Crit. Rev. Plant Sci. 18, 71–106 (1999).
Chellappan, P., Vanitharani, R., Pita, J. & Fauquet, C. M. Short interfering RNA accumulation correlates with host recovery in DNA virus-infected hosts, and gene silencing targets specific viral sequences. J. Virol. 78, 7465–7777 (2004).
Amin, I. et al. Suppressors of RNA silencing encoded by the components of the cotton leaf curl begomovirus-betasatellite complex. Mol. Plant-Microb. Interact. 24, 973–983 (2011).
Fiallo-Olive, E., Tovar, R. & Navas-Castillo, J. Deciphering the biology of deltasatellites from the New World: Maintenance by new world begomoviruses and whitefly transmission. New Phytol. 212, 680–692 (2016).
Idris, A. M. et al. An unusual alphasatellitebassociated with monopartite begomoviruses attenuates symptoms and reduces betasatellite accumulation. J. Gen. Virol. 92, 706–717 (2011).
Briddon, R. W. et al. Diversity of DNA beta, a satellite molecule associated with some monopartite begomoviruses. Virology 312, 106–121 (2003).
Saunders, K., Briddon, R. W. & Stanley, J. Replication promiscuity of DNA-β satellites associated with monopartite begomoviruses; deletion mutagenesis of the Ageratum yellow vein virus DNA-β satellite localizes sequences involved in replication. J. Gen. Virol. 89, 3165–3172 (2008).
Briddon, R. W. & Stanley, J. Sub-viral agents associated with plant infecting single stranded DA viruses. Virology 344, 198–210 (2006).
Stanley, J. Subviral DNAs associated with geminivirus disease complexes. Vet. Microbiol. 98, 121–129 (2004).
Bartel, D. P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116, 281–297 (2004).
Mansoor, S. et al. Breakdown of resistance in cotton to cotton leaf curl disease in Pakistan. Plant Pathol. 52(6), 784 (2003).
Pooggin, M., Shivaprasad, P. V., Veluthambi, K. & Hohn, T. RNAi targeting of DNA virus in plants. Nat. Biotechnol. 21, 131–132 (2003).
Bonfim, K., Faria, J. C., Nogueira, E. O., Mendes, E. A. & Aragão, F. J. RNAi-mediated resistance to Bean golden mosaic virus in genetically engineered common bean (Phaseolus vulgaris). Mol. Plant Microb. Interact. 20, 717–726 (2007).
Ahmad, A. et al. Engineered disease resistance in cotton using RNA-interference to knock down Cotton leaf curl Kokhran virus-Burewala and Cotton leaf curl Multan betasatellite expression. Viruses 9(9), 257. https://doi.org/10.3390/v9090257 (2017).
Hamilton, A. J. & Baulcombe, D. C. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 286, 950–952 (1999).
Meister, G. & Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 431, 343–349 (2004).
Eamens, A., Wang, M. B., Smith, N. A. & Waterhouse, P. M. RNA silencing in plants: yesterday, today, and tomorrow. Plant Physiol. 147, 456–468 (2008).
Baulcombe, D. C. RNA silencing in plants. Nature 431, 356–363 (2004).
Mello, C. C. & Conte, D. Jr. Revealing the world of RNA interference. Nature 431, 338–342 (2004).
Budar, F., Thia-Toong, L., Von Montagu, M. & Hernalsteens, J. P. Agrobacterium-mediated gene transfer results mainly in transgenic plants transmitting T-DNA as a single Mendelian factor. Genetics 114, 303–313 (1986).
Dutta, R. U., Banakar, T. K. & Rao, U. The status of RNAi-based transgenic research in plant nematology. Front. Microbiol. 5, 760 (2015).
Amin, I., Patil, B. L., Briddon, R. W., Mansoor, S. & Fauquet, C. M. Comparison of phenotypes produced in response to transient expression of genes encoded by four distinct begomoviruses in Nicotiana benthamiana and their correlation with the levels of developmental miRNAs. Virol. J. 8, 238 (2011).
Iqbal, Z., Sattar, M. N., Kvarnheden, A., Mansoor, S. & Briddon, R. W. Effects of the mutation of selected genes of Cotton leaf curl Kokhran virus on infectivity, symptoms and the maintenance of Cotton leaf curl Multan betasatellite. Virus Res. 169, 107–116 (2012).
Amin, I. et al. Suppressors of RNA silencing encoded by the components of the cotton leaf curl begomovirus-betasatellite complex. Mol. Plant Microb. Interact. 24, 973–983 (2011).
Saeed, M., Briddon, R., Dalakouras, A., Krczal, G. & Wassenegger, M. Functional analysis of Cotton leaf curl Kokhran virus/Cotton leaf curl Multan betasatellite RNA silencing suppressors. Biology 4, 697 (2015).
Ammara, U. et al. RNA interference-based resistance in transgenic tomato plants against Tomato yellow leaf curl virus-Oman (TYLCV-OM) and its associated betasatellite. Virol. J. 12, 38 (2015).
Khatoon, S., Kumar, A., Sarin, N. B. & Khan, J. A. RNAi-mediated resistance against Cotton leaf curl disease in elite Indian cotton (Gossypium hirsutum) cultivar Narasimha. Virus Genes 52(4), 530–537 (2016).
Akhtar, S., Akmal, M. & Khan, J. A. Resistance to cotton leaf curl disease in transgenic tobacco expressing βC1 gene derived intron-hairpin RNA. Indian J. Biotechnol. 16, 56–62 (2017).
Takahashi, H., Fukuhara, T., Kitazawa, H. & Kormelink, R. Virus latency and the impact on plants. Front. Microbiol. 10, 2764 (2019).
Pooggin, M. M. RNAi-mediated resistance to viruses: A critical assessment of methodologies. Curr. Opin. Virol. 26, 28–35 (2017).
Vanitharani, R., Chellappan, P., Pita, J. S. & Fauquet, C. M. Differential roles of AC2 and AC4 of cassava geminiviruses in mediating synergism and suppression of posttranscriptional gene silencing. J. Virol. 78, 9487–9498 (2004).
Wamiq, G., Akhtar, S., Khan, Z. A., Alam, P. & Khan, J. A. Development of an efficient method for regeneration and Agrobacterium-mediated transformation of cotton (Gossypium hirsutum L.) cv HS6. Indian J. Biotechnol. 15, 39–47 (2016).
Wamiq, G. & Khan, J. A. Overexpression of ghr-miR166b generates resistance against Bemisia tabaci infestation in Gossypium hirsutum plants. Planta 247, 1175–1189 (2018).
Green, M. R. & Sambrook, J. Molecular Cloning: A Laboratory Manual 241–242 (Cold Spring Harbor Laboratory Press, 2012).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Peng, J. et al. Rapid and efficient isolation of high-quality small RNAs from recalcitrant plant species rich in polyphenols and polysaccharides. PLoS ONE 9, e95687 (2014).
Khan, J. A. & Ahmad, J. Diagnosis, monitoring and transmission characteristics of Cotton leaf curl virus. Curr. Sci. 88, 1803–1809 (2005).
Acknowledgements
University Grants Commission (Govt. of India) is acknowledged for PhD fellowships to MSB and SA. The authors thanks Dr. D.K. Monga for seeds of G. hirsutum cv. HS6 seeds.
Author information
Authors and Affiliations
Contributions
M.S.B. performed all experiments (except designing, cloning, production and validation of ihpRNAi construct) and wrote the MS. S.A. cloned, developed and validated ihpRNAi construct in tobacco. J.A.K. supervised the whole study, designed experiments, partly wrote the MS, revised and edited manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baig, M.S., Akhtar, S. & Khan, J.A. Engineering tolerance to CLCuD in transgenic Gossypium hirsutum cv. HS6 expressing Cotton leaf curl Multan virus-C4 intron hairpin. Sci Rep 11, 14172 (2021). https://doi.org/10.1038/s41598-021-93502-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-93502-3
This article is cited by
-
Development, Design, and Application of Efficient siRNAs Against Cotton Leaf Curl Virus-Betasatellite Complex to Mediate Resistance Against Cotton Leaf Curl Disease
Indian Journal of Microbiology (2024)
-
Whole genome characterization and diagnostics of prunus necrotic ringspot virus (PNRSV) infecting apricot in India
Scientific Reports (2023)
-
A novel tasi RNA-based micro RNA-induced gene silencing strategy to tackle multiple pests and pathogens in cotton (Gossypium hirsutum L.)
Planta (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
