
Abstract
The four adenosine receptors (ARs) A1AR, A2AAR, A2BAR, and A3AR are G protein-coupled receptors (GPCRs) for which an exceptional amount of experimental and structural data is available. Still, limited success has been achieved in getting new chemical modulators on the market. As such, there is a clear interest in the design of novel selective chemical entities for this family of receptors. In this work, we investigate the selective recognition of ISAM-140, a recently reported A2BAR reference antagonist. A combination of semipreparative chiral HPLC, circular dichroism and X-ray crystallography was used to separate and unequivocally assign the configuration of each enantiomer. Subsequently affinity evaluation for both A2A and A2B receptors demonstrate the stereospecific and selective recognition of (S)-ISAM140 to the A2BAR. The molecular modeling suggested that the structural determinants of this selectivity profile would be residue V2506.51 in A2BAR, which is a leucine in all other ARs including the closely related A2AAR. This was herein confirmed by radioligand binding assays and rigorous free energy perturbation (FEP) calculations performed on the L249V6.51 mutant A2AAR receptor. Taken together, this study provides further insights in the binding mode of these A2BAR antagonists, paving the way for future ligand optimization.
Similar content being viewed by others
Introduction
Adenosine receptors (ARs) are a family of G protein-coupled receptors (GPCR) for which an exceptional amount of structural and experimental data is available1,2. Still, the number of therapeutic agents on the market that specifically target this family of receptors remains relatively low3. On the other hand, selective targeting of any of the four adenosine receptor subtypes (A1, A2A. A2B and A3) provides an interesting avenue to address not only unmet therapeutic needs4 and limited off-target effects5, but also to help elucidating the (patho)physiological role of the different receptors within the family. One topic that is receiving increasing interest is the molecular mechanisms by which the two A2AR subtypes regulate the immune response to tumor growth and metastasis6.
Over the last years, different AR ligands have been developed with optimized selectivity profiles7,8,9. Within these AR ligand design programs, the generation of potent and selective antagonists has allowed the identification of powerful chemical tools to characterize each of the members of this receptor family. Examples include the A2AAR selective antagonist ZM241385, and the A2BAR selective antagonist ISAM-140, the latter originating from our in-house optimization program (Fig. 1)7,9,10,11. The development of ISAM-140 was done following careful structure-affinity relationship (SAR) modeling, based on a computational binding mode of this chemotype, which suggested an important role of the stereogenic center in the heterocyclic scaffold in its high binding affinity (Fig. 1)11,12. The prediction of the active stereoisomer for this chemotype was later confirmed indirectly by experimental characterization of the active stereoisomers for representative compounds of a series of cyanopyrimidines10, fluorinated tricyclic derivatives13 and aza-bioisosteres of the pentagonal heterocycle14. This binding model proposed that the stereospecific complementarity to the A2BAR cavity was due to the optimal accommodation of the thiophene/furan ring around the chiral center of the core scaffold (Fig. 1), with the A2BAR specific residue V2506.51 (Ballesteros Weinstein numbering in superscripts)15. Indeed, this valine is replaced by a leucine in all other AR subtypes, which could explain the highly selective profile of these series of non-planar antagonists towards the A2BAR.

2D representation of the chemical structures of the AR ligands used in this work, i.e. ZM241385, ( ±) ISAM-140, (R)-ISAM-140 and (S)-ISAM-140. The chiral center in ISAM-140 is indicated with an asterisk.
In this work, we report the chiral separation of ISAM140 and confirm its stereospecific binding mode to the A2BAR. An A2AAR construct was designed to include the corresponding A2BAR valine sidechain (L249V6.51 A2AAR mutant), which in line with the starting hypothesis partially recovered the affinity for ISAM-140. Interestingly, this effect was observed for both stereoisomers of the antagonist, and is herein explained on the basis of structure-energetic modeling via rigorous free energy perturbation (FEP) calculations. These results validate the proposed role of V2506.51 in the A2BAR subtype selectivity of these stereospecific chemotype, and paves the road for further design of selective antagonists as well as dual A2AR ligands.
Results
Generating A2AAR-ligand models
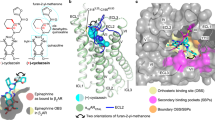
The binding mode of (S)-ISAM-140 was obtained by superposition of the previously published complex of this molecule with our A2BAR homology-based model11 onto a modeled L249V6.51 A2AAR mutant, i.e. introducing the A2BAR sidechain in this position. Such a construct was built and equilibrated on the basis of the high-resolution crystal structure of the ZM241385 — A2AAR complex (see “Methods”)16. The binding mode obtained included the two key interactions typical of ARs antagonists: (i) hydrogen bond(s) with N2536.55 and (ii) π–π stacking with F168EL2, both residues completely conserved among ARs1. The high-affinity A2AAR antagonist ZM241385 showed an optimal shape complementarity with the A2AAR WT residue L2496.51 (Fig. 2A), whereas the corresponding L249V6.51 mutant is expected to minimally disrupt this shape complementarity due to a reduced volume (Fig. 2B). On the other hand, the obtained binding modes for (S)-ISAM-140 on the WT A2AAR (also obtained assuming the same binding mode as in the A2BAR homology-based model11) showed a non-optimal fit, in accordance with the lack of affinity exhibited for the A2AAR receptor by this derivative and other compounds within the series8,9,10,11. In particular, the presence of the native L2496.51 in the A2AAR appeared to introduce a steric clash with either the 2-furyl or 3-thienyl substituents of the ligands, which we hypothesized would reduce binding affinities (Fig. 2C). Conversely, introducing the A2BAR sidechain on the modeled L249V6.51 A2AAR mutant provided a better shape complementarity (Fig. 2D), allowing us to hypothesize that the binding affinity of these antagonists might be recovered to some extent.

Binding mode of two ligands, ZM241385 (in blue, panels A and B) and (S)-ISAM-140 (orange, panels C and D), to the WT (panels A and C) and the L249V6.51 mutant (panels B and D) A2AAR. Volumetric occupancies are shown as surface. Figure created with Pymol v2.0.
Chiral separation of ISAM-140
The racemic mixture of ISAM-140, obtained as previously described11, was resolved into its enantiopure forms. A combination of chiral HPLC, circular dichroism (CD) spectroscopy and X-ray crystallography was employed to separate and unequivocally assign the configuration of the heterocyclic stereocenter in each stereoisomer. Semipreparative HPLC separation of ( ±) ISAM-140 on a chiral stationary phase (see “Experimental information”) provided the expected enantiomers (Fig. 3) with excellent stereochemical purity (> 97%), the analytical and spectroscopy data provided in the Supplementary Information. As described previously for 3,4-dihydropyrimidin-2-ones17,18,19, the characteristic CD activity of the enamide chromophore (300–350 nm) allowed the unambiguous assignment of the absolute configuration of each enantiomer (Fig. 3) by comparison with the reported CD data for enantiopure 3,4-dihydropyrimidin-2(1H)-ones of known configuration. In the structures shown in Fig. 3, enantiomers that show a negative Cotton effect (blue line) contain the furan ring pointing backwards, which corresponds to (S)-ISAM-140. In contrast, the stereoisomers giving a positive Cotton effect (red line) contain the pentagonal heterocycle pointing forward, which corresponds to (R)-ISAM-140. Single crystals suitable for X-ray analysis were grown by slow evaporation of each enantiomer in ethanol. The structures were solved and the data extracted from X-ray crystallography of both monocrystals presented in the Supporting Information (Supplementary Table S1)13. The crystal structures of (S)-ISAM-140 and (R)-ISAM-140 (monoclinic, Fig. 3) confirmed the configuration assignment established by circular dichroism. The benzimidazole moiety is essentially planar in both enantiomers, while the dihydropyrimidine core adopts a pseudo envelope conformation, with the C4 atom being lightly displaced by 0.26 Å.

Chiral HPLC separation, circular dichroism spectra and crystal X-ray structure of compounds (R)-ISAM-140 and (S)-ISAM-140.
Determination of ligand binding affinities
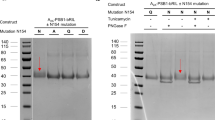
To further confirm the role of position 6.51 as a receptor selectivity hotspot, we attempted to express L249V/A6.51 A2AAR and V250L/A6.51 A2BAR mutant receptors. Whilst both A2AAR mutant receptors were successfully expressed (Supplementary Fig. S1), none of the A2BAR mutants could be expressed using standard (non-viral) transfection methods, and consequently the A2BAR mutants designed had to be excluded from further experimentation. Thereafter, we determined the binding affinity of ISAM-140, both as a racemate and pure enantiomers, together with the prototypical antagonist ZM241385 at both WT and mutant A2AARs, as well as at the WT A2BAR (Fig. 4 and Table 1). The affinities determined for ZM241385 and racemic ISAM-140 on WT A2BAR (pKi of 6.78 and 7.86, respectively, see Table 1) were in line with previous reports10. As expected from the modeling, the corresponding data for the enantiopure forms of ISAM-140 showed that the affinity of the racemic mixture was due to (S)-ISAM-140, with even a gain in binding affinity as compared to the racemic mixture (∆pKi = 0.19), which was dramatically reduced for the low-affinity (R)-ISAM-140 (∆pKi = 1.31 between both enantiomers, Fig. 4A and Table 1).

Displacement of (A) specific [3H]PSB-603 binding from A2BAR and (B) specific [3H]ZM241385 binding from the WT and the L249V6.51 mutant A2AAR at 25 °C by ZM241385 (blue), ( ±) ISAM-140 (yellow), (R)-ISAM-140 (black) and (S)-ISAM-140 (red). Combined graphs are from three individual experiments performed in duplicate.
For the A2AAR, we first established whether the L249V/A6.51 mutants still sufficiently bound ZM241385, to validate the viability of using it as a radioligand in the homologous displacement assays. Of note, the resulting KD values could then be used to obtain Ki values from the IC50 values (see Methods), which enabled us to compare affinity values for WT and mutant A2AARs. Moreover, the resulting Bmax values showed that the A2AAR L249V6.51 mutant had a lower expression level than the WT A2AAR. A slight reduction in affinity of both [3H]ZM241385 and ZM241385 was observed on this mutant (Table 1), which was in line with our hypothesis that the shape complementarity between ZM241385 and L249 is mostly preserved with a smaller Val. However, a substantial hydrophobic side chain was important for the binding of this antagonist to the A2ARs, since its affinity to the A2AAR L249A6.51 mutant was completely lost (Supplementary Fig. S2), in line with previous reports20. The results of the displacement assays for ISAM-140 (racemate and both stereoisomers) are illustrated in Fig. 4B and Table 1. Although one data point for ( ±) ISAM-140 at the concentration of 10−5 M was excluded from the curve of WT A2AAR, due to low water solubility, in all cases the binding affinity for the WT A2AAR was very low (within micromolar range). Notably, it followed the same trend as observed on WT A2BAR, i.e. the highest affinity for (S)-ISAM-140 and the lowest for (R)-ISAM-140. The selectivity ratio between A2B and A2A ARs was substantial for ( ±) ISAM-140, (∆pKi = 1.33), in line with the previous reports for this ligand11. This difference was maintained for the eutomer (S)-ISAM-140 (∆pKi = 1.29) and, to a lower extent, even for (R)-ISAM-140 (∆pKi = 0.79), which is expected due to its already low affinity for A2BAR. Notably, the affinity values were significantly recovered at the A2AAR L249V6.51 mutant, i.e. when the receptor was more “A2BAR-like”, thus supporting the initial modeling hypothesis. The moderate affinity gains observed for the A2AAR L249V6.51 mutant as compared to the A2AAR WT [0.39, 0.41 and 0.51 log unit for ( ±) ISAM-140, (S)-ISAM-140, and (R)-ISAM-140, respectively, see Table 1] did not restore the affinity values as in the WT A2BAR.
Computational characterization of binding free energies
Finally, we investigated the observed shifts in binding affinities for (S)-ISAM-140, (R)-ISAM-140 and ZM241385 in the context of the structural binding model of these molecules to the A2AAR. The approach was to compare the WT and L2496.51V mutant (A2B equivalent) versions of A2AAR using the Q-FEP protocols21,22. This strategy consists on the simulation of the mutation (Leu to Val) both in the presence and absence of each of the docked ligands. While the structure of the ZM241385 — A2AAR complex is experimentally known16, the binding mode of each enantiomer of ISAM140 was inferred from our previous work on this chemotype7. Figure 5 summarizes the calculated shift in the free energy of binding due to the L249V6.51 mutation for each enantiomer of ISAM-140 and for ZM241385. It can be observed a very good agreement between the calculations and the experimental affinity data here reported in Fig. 4B, with a very low mean average error (MAE = 0.25 kcal/mol, numerical data provided in Supplementary Table S2). Thus, the simulation of this mutation resulted in a predicted increase in affinity (negative ∆∆Gbind (mut − WT) values in Fig. 5) for both enantiomers of ISAM-140, with values proportional to those extracted from the experimental data. Conversely, the experimental affinity of ZM241385 is decreased for the L249V6.51 mutant A2AAR, which is also captured by our modeling as a mild positive value for the calculated ∆∆Gbind (mut − WT).

Experimental (grey) and calculated (orange) relative changes in binding free energies to the L249V6.51 mutant A2AAR for the two enantiomers of ISAM-140 and ZM241385.
Discussion
In this work, we investigated the role of position 6.51 in determining the specificity for A2BAR binding of a series of chiral antagonists recently developed for this receptor. The modeling hypothesis behind the design of the potent antagonist ISAM-140 placed the S-stereoisomer in perfect shape complementarity with Val2506.51 in the A2BAR, while analogous docking in the high resolution A2AAR bearing a bulkier Leu in the same position showed initial steric clashes. This allowed us to propose this sidechain as a landmark for A2BAR selectivity for this ligand class, and the (S)-ISAM-140 as the active stereoisomer. To experimentally validate this hypothesis, the ISAM-140 enantiomers were separated and their absolute configuration unequivocally assigned. Besides this goal, the enantiomeric separation and pharmacological characterization of this reference A2BAR antagonist allowed to confirm the expected higher affinity of the S enantiomer, in line with the original modeling hypothesis11 and recent similar results obtained with derivatives of this scaffold10,13,14.
Site-directed mutagenesis of position 6.51 was performed on the A2AAR to replace the WT Leu by the Val specific of A2BAR, as the reverse mutation of the A2BAR appeared unfeasible in our hands, somehow in contrast to previous report of Müller and co-workers who managed to express the corresponding Ala mutant (V250A6.51) in the A2BAR23. It is worth noting that, while there had been reports of the Alanine scan of position 6.51 in both A2A20 and A2BARs23, this is the first time that the introduction of the A2BAR characteristic Val sidechain on the A2AAR is evaluated.
The L249V6.51 A2AAR mutant partially recovered the affinity of ISAM-140 lost for this receptor, supporting the initial modeling hypothesis. This partial recovery in affinity, consistently observed for all three forms of this molecule (i.e., racemic mixture and both eutomers) is in line with recent reports on ‘selectivity hotspots’ between A1AR and A2AAR, where a single-point mutation clearly affecting the experimental binding mode could only partially explain the observed selectivity profile of the A1AR selective xanthines under investigation24. On the other hand, the opposed effect was observed for ZM241385 (i.e. decrease in affinity for the L249V6.51 A2AAR mutant) in line with the well-described preference of this ligand for the A2AAR.
To further assess the amino acid conservation between the A2A and A2BARs binding sites, a pseudo-sequence alignment is presented in Fig. 6. One can observe that, in addition to position 6.51 here studied, only two sidechains vary within the 5 Å cut-off distance with the ligand: Ala2536.54 in A2BAR, situated one helix turn below position 6.51, is an Ile in A2AAR. This residue, however, is not in contact with the ligand and instead involved in the TM packing as shown in the Fig. 6. In the EL3 region, His2647.31 in A2AAR is making a salt bridge interaction with Glu1695.30 in EL2, a role that in our A2BAR model is undertaken by Lys2677.31 (Fig. 6). While this residue has been shown to be involved in ligand binding kinetics1, we should not rule out an additional role of the more variable EL regions in the selectivity profile of this antagonist. This analysis also allows to explore potential indirect effects of the V6.51L mutation on neighbouring residues conserved in the ARs, like His6.52 that has been shown to be involved in both agonist and antagonist binding1. As it can be seen in Fig. 6., this residue is not predicted to change conformation between A2A and A2B ARs, which is supported by the water-mediated interaction with Asn5.42 previously characterized by MD simulations of this pair of receptors25.

Pseudo-sequence alignment of the residues within 5 Å of any atom of (S)-ISAM140, as predicted by docking on the A2BAR, between this receptor and the A2AAR. The location of each sidechain is shown in the 3D superposition of the (S)-ISAM140-A2BAR (gray sidechains and cartoon, ligand in orange sticks) with the A2AAR crystal structure (cyan sidechains). Position 6.51 is highlighted on a yellow box. Figure created with Pymol v2.0.
In the lack of a crystal structure of the A2BAR, the observed effects were rationalized back in the modeled structures, by means of first-principle FEP simulations of this mutation. The QresFEP protocol has been broadly applied to investigate the A2AAR mutational landscape26,27,28, showing exceptional sensitivity to capture the correct affinity shifts for different chemotypes. The binding model of (S)-ISAM-140 to the WT and L249V6.51 mutant versions of A2AAR was here assumed to be the same as our docking model of this compound to the WT A2BAR13. That model suggested that the high A2BAR affinity of ( ±) ISAM-140 was due to the stereoselective optimal fitting of the (S) isomer to the A2BAR binding site, facilitated by the Val sidechain in position 6.51 of this receptor13. The calculated recovery of the binding affinity of (S)-ISAM-140 upon the L249V6.51 mutation in the A2AAR, which is in line with the experimental design of this A2B-like mutation on the A2AAR, further confirms the validity of the binding model for this chemotype on the A2BAR.
Overall, both experimental and computational results of this study clearly support the binding mode used to design this study, providing useful structural insights in the selective recognition of these A2BAR antagonists that should aid in future structure-based optimization.
Methods
HPLC separation and characterization of ISAM-140 enantiomers
The chiral resolution of the razemic ligand ISAM-140 was performed following procedures recently described13. Briefly, chiral resolution was performed using a Water Breeze 2 (binary pump 1525, detector UV/Visible 2489, 7725i Manual Injector Kit 1500 Series). Compound ISAM-140 enantiomers were separated using a 250 mm × 20 mm Chiralpak 5 µm IE-3 (DAICEL). All the separations were performed at 25 ºC with hexane/isopropanol 7:3 as mobile phase. The enantiomers [(R)-ISAM-140 (3 mg, tR = 17.90 min), (S)-ISAM-140 (3.1 mg, tR = 20.31 min)] were isolated, their stereochemical purity analyzed by chiral HPLC (ee: 97–99% for each enantiomer) and then characterized by NMR (see Supplementary Material).
Circular dichroism
Circular dichroism spectra were recorded on a Jasco-815 system equipped with a Peltier-type thermostatic accessory (CDF-426S, Jasco). Measurements were carried out at 20 °C using a 1 mm quartz cell in a volume of 600 µL. Compounds (0.5 mg) were dissolved in MeOH (1.0 mL) and then diluted 10-fld in MeOH. The instrument settings were bandwidth, 1.0 nm; data pitch, 1.0 nm; speed, 500 nm/min; accumulation, 10; wavelengths, 400–190 nm.
X-ray crystallography of ISAM-140 enantiomers
Crystals of (S)-ISAM-140 and (R)-ISAM-140 were grown by slow evaporation from ethanol solutions. For the crystal structure determination13, the data were collected by applying the omega and phi scans method on a Bruker D8 VENTURE PHOTON III-14 diffractometer using Incoatec multilayer mirror monochromated with Cu-Kα radiation (λ = 1.54178 Å) from a microfocus sealed tube source at 100 K with detector resolution of 7.3910 pixels mm-1. Computing data and reduction were made with the APEX3 v2018.7–2 (BRUKER AXS, 2005). The structure was solved using SHELXT2018/22 and finally refined by full-matrix least-squares based on F2 by SHELXL2018/3.3 An empirical absorption correction was applied using the SADABS2016/2 program. Software used for molecular graphics: ORTEP for Windows. Software used to prepare material for publication: WinGX2018.3 publication routines4 and Mercury.
The obtained structures were refined following recently described methods as follows13: All non-hydrogen atoms were refined anisotropically and the hydrogen atom positions were included in the model on the basis of Fourier difference electron density maps. All aromatic CH hydrogen (C-H = 0.95 Å), methine hydrogen (C-H = 1.0 Å) and methylene hydrogen (C-H = 0.99 Å) atoms were refined using a riding model with Uiso(H) = 1.2 Ueq(C). The methyl hydrogen (C–H = 0.98 Å) atoms were refined as a rigid group with torsional freedom [Uiso(H) = 1.5 Ueq(C)] and the hydrogens atom of NH groups (HiN) as a free atom with Uiso(H) = 1.2 Ueq(C).
Site-directed mutagenesis
Site-directed mutants of the A2AAR were generated by polymerase chain reaction (PCR) mutagenesis as described previously29. pcDNA3.1(+)-hA2AAR with N-terminal HA and FLAG tags and a C-terminal His tag was used as the template. Primers for mutants L249V6.51 and L249A6.51 were designed by the QuikChange Primer Design Program of Agilent Technologies (Santa Clara, CA, USA) and primers were obtained from Eurogentec (Maastricht, The Netherlands). All DNA sequences were verified by Sanger sequencing at LGTC (Leiden, The Netherlands).
Cell culture and transient transfection
CHO cells stably expressing the human A2BAR (CHO-spap-hA2BAR) were cultured in Dulbecco’s modified Eagle’s medium: Nutrient Mixture F-12 (DMEM/F12) supplemented with 10% newborn calf serum, 50 µg/mL streptomycin, and 50 IU/mL penicillin at 37 °C and 5% CO2 atmosphere. Cells were subcultured twice a week at a confluency of 80–90%. For transient transfections, human embryonic kidney (HEK) 293 cells were cultured as monolayers in DMEM supplemented with stable glutamine, 10% newborn calf serum, 50 µg/mL streptomycin, and 50 IU/mL penicillin at 37 °C and 7% CO2 atmosphere as reported previously29,30. The cells were seeded on 10 cm ø plates and transfected with 10 μg plasmid DNA of wild-type (WT) or mutant hA2AAR using the calcium phosphate precipitation method31, followed by a 48 h incubation.
Membrane preparation
HEK293 cells transiently expressing WT or mutant human A2AAR (HEK293-hA2AAR) were detached from the plates 48 h post-transfection by scraping into phosphate-buffered saline (PBS) and collected by centrifugation at 1,000 × g for 5 min. The pellets from 10 plates were pooled and resuspended in ice-cold Tris–HCl buffer (50 mM, pH 7.4) and then homogenized with an UltraTurrax homogenizer (Heidolph Instruments, Schwabach, Germany). The cell membrane suspensions were centrifuged at 100,000×g at 4 °C for 20 min in a Beckman Optima LE-80 K ultracentrifuge. The pellet was resuspended in ice-cold Tris–HCl buffer, and the homogenization and centrifugation steps were repeated one more time. After this, Tris–HCl buffer was used to resuspend the pellet of HEK293 cell membranes. Membrane preparation for CHO-spap-hA2BAR cells followed a similar procedure after they were grown to 90% confluence in 15 cm plates, and membranes pellets were finally resuspended in Tris–HCL buffer containing 10% (w/v) CHAPS. In both cases, 0.8 IU/ml adenosine deaminase was added to break down endogenous adenosine and membranes were aliquoted into 250 μL and stored at -80 °C until further use. Membrane protein concentrations were determined using the BCA method32.
Radioligand binding assays
Radioligand binding experiments on CHO-spap-hA2BAR membranes were adjusted from previously reported data33. Membrane aliquots containing 30 µg of protein were incubated in a total volume of 100 µL of assay buffer. Nonspecific binding was determined with 10 µM ZM241385. Then 25 µL cell membrane suspension, 25 µL of 1.5 nM radioligand [3H]PSB-603, 25 µL of assay buffer [50 mM Tris–HCl, 0.1% (w/v) CHAPS, pH 7.4 at 25 °C] and 25 µL of the indicated compounds in increasing concentrations in the same assay buffer were added to each well and followed by a 120 min incubation at 25 °C. Radioligand displacement experiments with transient HEK293-hA2AAR cell membranes were performed as described previously34. Briefly, membrane aliquots containing 5–7.5 µg of protein were incubated in a total volume of 100 µL of assay buffer to adjust the assay window to approximately 2000 DPM. Nonspecific binding was determined in presence of 100 µM NECA and represented less than 10% of the total binding. Then 25 µL membrane suspension (5–7.5 µg of protein), 25 µL of 5.0 nM radioligand [3H]ZM241385, 25 µL of assay buffer [50 mM Tris–HCl, pH 7.4] and 25 µL of the indicated compounds at different concentrations in the same assay buffer were added to each well, with final assay concentration of radioligand of 5 nM. For homologous displacement experiments, radioligand displacement experiments were performed in presence of three concentrations of [3H]ZM241385 (1.7 nM, 5.0 nM and 9.5 nM) and increasing concentrations of unlabeled ZM241385. After 120 min at 25 °C, incubations were terminated by rapid vacuum filtration through GF/B filter plates (PerkinElmer, Groningen, Netherlands) using a Perkin Elmer Filtermate-harvester. Filterplates were subsequently washed ten times with ice-cold assay buffer. Filter-bound radioactivity was determined by scintillation spectrometry using a Microbeta2 2450 microplate counter (PerkinElmer).
Data analysis
Data analyses were performed using GraphPad Prism 7.0 software (GraphPad Software Inc., San Diego, CA). pKD values and Bmax were obtained by non-linear regression analysis using “one-site homologous” model. pIC50 values were determined by fitting the data using non-linear regression to a sigmoidal concentration–response curve equation. pKi values were calculated from pIC50 values using the Cheng-Prusoff equations35.
Computational modeling
The high resolution crystal structure of A2AAR (PDB code 4EIY16) was used as a starting point for the calculations. The protein was prepared for MD simulations as follows: (i) removing co-factors and fused proteins employed for crystallization, (ii) reverting the crystal construct to the wild-type (WT) A2AAR receptor, (iii) the assignment of protonation states of ionizable residues, (iv) mutation of the WT Leu2496.51 to Val as in the corresponding A2BAR and (v) membrane insertion using PyMemDyn25. The latter stage involves embedding of the protein in a pre-equilibrated POPC membrane, soaking of the system with bulk water and a short (5 ns) equilibration period with GROMACS 4.636 using the OPLS-AA force field37 and Berger parameters for the lipids38. Thereafter, ligands were manually docked to the equilibrated receptor using as a reference the putative binding mode of SYAF0147 to the A2BAR previously described. In the case of ZM241385, the coordinates of the crystal structure ligand were retained during the equilibration process. Subsequently, each equilibrated L249V6.51 A2AAR-ligand complex was transferred to the MD software Q39 for free energy perturbation (FEP) calculations under spherical boundary conditions using QligFEP21. A 25 Å radius sphere centered on the center of geometry of the ligand was constructed for these MD simulations. Solvent atoms were subject to polarization and radial restraints using the surface-constrained all-atom solvent (SCAAS)40 model to mimic the properties of bulk water at the sphere surface. Atoms lying outside the simulation sphere were tightly constrained (200 kcal/mol/Å2 force constant) and excluded from the calculation of non-bonded interactions. Long range electrostatic interactions beyond a 10 Å cut off were treated with the local reaction field method41, except for the atoms undergoing the FEP transformation, where no cutoff was applied. Solvent bond and angles were constrained using the SHAKE algorithm42. All titratable residues outside the sphere were neutralized as reported elsewhere21. Residue parameters were translated from the OPLS-AA/M force field43 and the parameters for the lipids were inherited from the previous MD stage, while ligand parameters were generated using the ffld_server as implemented in the Schrödinger suite. The simulation sphere was warmed up from 0.1 to 298 K, during a first equilibration period of 0.61 ns, where an initial restraint of 25 kcal/mol/Å2 imposed on all heavy atoms was slowly released for all complexes. Thereafter the system was subject to 10 parallel replica MD simulations, in which the FEP protocol was applied for each residue transformation. Each of these MD replicates started with a 0.25 ns unbiased equilibration period, with different initial velocities. The FEP protocol for the L → V mutation was generated by combing the QresFEP22 protocol for residue mutations with a dual topology approach inspired from QligFEP21, where the effective topology along the transformation is a linear combination of the two original sidechain topologies. Each FEP transformation consisted of 51 evenly distributed λ-windows with 10 ps MD sampling each. In order to fulfill a thermodynamic cycle and calculate relative binding free energies, parallel FEP transformations were run for the apo-structure, i.e. the protein structure without ligand. In these simulations the same parameters were applied (i.e., sphere size, simulation time, etc.), and a total of 10 replicates × 2 (apo/holo) states × 2 (WT and mut) annihilations × 51 λ-windows × 10 ps = 20.4 ns sampling was performed for each mutation simulation. The relative binding free energy shift between WT and mutant receptors for each ligand was estimated by solving the thermodynamic cycle utilizing the Bennett acceptance ratio (BAR)44. All 3D images were produced in PyMOL45.
References
Jespers, W. et al. Structural mapping of adenosine receptor mutations: Ligand binding and signaling mechanisms. Trends Pharmacol. Sci. 39, 75–89 (2018).
Jazayeri, A., Andrews, S. P. & Marshall, F. H. Structurally enabled discovery of adenosine A 2A receptor antagonists. Chem. Rev. 117, 21–37 (2017).
Gessi, S., Merighi, S. & Varani, K. Adenosine receptors: The status of the art. Receptors 34, 1–11 (2018).
Jacobson, K. A. & Gao, Z.-G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 5, 247–264 (2006).
Chen, J. F., Eltzschig, H. K. & Fredholm, B. B. Adenosine receptors as drug targets-what are the challenges?. Nat. Rev. Drug Discov. 12, 265–286 (2013).
Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012).
Crespo, A. et al. Discovery of 3,4-dihydropyrimidin-2(1 H )-ones as a novel class of potent and selective A2B adenosine receptor antagonists. ACS Med. Chem. Lett. 4, 1031–1036 (2013).
Yaziji, V. et al. Pyrimidine derivatives as potent and selective A3 adenosine receptor antagonists. J. Med. Chem. 54, 457–471 (2011).
Jespers, W. et al. Structure-based design of potent and selective ligands at the four adenosine receptors. Molecules 22, 1–17 (2017).
Carbajales, C. et al. Enantiospecific Recognition at the A2B adenosine receptor by alkyl 2-cyanoimino-4-substituted-6-methyl-1,2,3,4-tetrahydropyrimidine-5-carboxylates. J. Med. Chem. 60, 3372–3382 (2017).
El Maatougui, A. et al. Discovery of potent and highly selective A 2B adenosine receptor antagonist chemotypes. J. Med. Chem. 59, 1967–1983 (2016).
Crespo, A. et al. Discovery of 3,4-dihydropyrimidin-2(1H)-ones as a novel class of potent and selective A2B adenosine receptor antagonists. ACS Med. Chem. Lett. 4, 1031–1036 (2013).
Mallo-Abreu, A. et al. Trifluorinated pyrimidine-based A2B antagonists: Optimization and evidence of stereospecific recognition. J. Med. Chem. 62, 9315–9330 (2019).
Mallo-Abreu, A. et al. Nitrogen-walk approach to explore bioisosteric replacements in a series of potent A2BAdenosine receptor antagonists. J. Med. Chem. 63, 7721–7739 (2020).
Ballesteros, J. A. & Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428 (1995).
Liu, W. et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236 (2012).
Lacotte, P., Buisson, D. A. & Ambroise, Y. Synthesis, evaluation and absolute configuration assignment of novel dihydropyrimidin-2-ones as picomolar sodium iodide symporter inhibitors. Eur. J. Med. Chem. 62, 722–727 (2013).
Krenn, W., Verdino, P., Uray, G., Faber, K. & Kappe, C. O. Determination of absolute configuration in 4-aryl-3, 4-dihydro-2(1H)-pyrimidones by high performance liquid chromatography and CD spectroscopy. Chirality 11, 659–662 (1999).
Uray, G., Verdino, P., Belaj, F., Kappe, C. O. & Fabian, W. M. F. Absolute configuration in 4-alkyl- and 4-aryl-3,4-dihydro-2(1H)-pyrimidones: A combined theoretical and experimental investigation. J. Org. Chem. 66, 6685–6694 (2001).
Jaakola, V.-P. et al. Ligand binding and subtype selectivity of the human A(2A) adenosine receptor: Identification and characterization of essential amino acid residues. J. Biol. Chem. 285, 13032–13044 (2010).
Jespers, W., Esguerra, M., Åqvist, J. & Gutiérrez-de-Terán, H. QligFEP: An automated workflow for small molecule free energy calculations in Q. J. Cheminform. 11, 1–16 (2019).
Jespers, W. et al. QresFEP: An automated protocol for free energy calculations of protein mutations in Q. J. Chem. Theory Comput. 15, 5461–5473 (2019).
Thimm, D. et al. Ligand-specific binding and activation of the human adenosine A2B receptor. Biochemistry 52, 726–740 (2013).
Cheng, R. K. Y. et al. Structures of human A1 and A2A adenosine receptors with xanthines reveal determinants of selectivity. Structure 25, 1275-1285.e4 (2017).
Rodríguez, D., Piñeiro, Á. & Gutiérrez-de-Terán, H. Molecular dynamics simulations reveal insights into key structural elements of adenosine receptors. Biochemistry 50, 4194–4208 (2011).
Jespers, W. et al. X-Ray Crystallography And Free Energy calculations reveal the binding mechanism of A2A adenosine receptor antagonists. Angew. Chem. Int. Ed. 59, 16536–16543 (2020).
Keränen, H., Gutiérrez-de-Terán, H. & Åqvist, J. Structural and energetic effects of A2A adenosine receptor mutations on agonist and antagonist binding. PLoS ONE 9, e108492 (2014).
Keränen, H., Åqvist, J. & Gutiérrez-de-Terán, H. Free energy calculations of A 2A adenosine receptor mutation effects on agonist binding. Chem. Commun. 51, 3522–3525 (2015).
Guo, D. et al. Molecular basis of ligand dissociation from the adenosine A2A receptor. Mol. Pharmacol. 89, 485–491 (2016).
Guo, D., Mulder-Krieger, T., IJzerman, A. P. & Heitman, L. H. Functional efficacy of adenosine A 2A receptor agonists is positively correlated to their receptor residence time. Br. J. Pharmacol. 166, 1846–1859 (2012).
Dasgupta, S. et al. Adenosine A(2A) receptors modulate the binding characteristics of dopamine D2 receptors in stably cotransfected fibroblast cells. Eur. J. Pharmacol. 316, 325–331 (1996).
Smith, P. K. et al. Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85 (1985).
Schiedel, A. C. et al. The four cysteine residues in the second extracellular loop of the human adenosine A2B receptor: Role in ligand binding and receptor function. Biochem. Pharmacol. 82, 389–399 (2011).
Yang, X. et al. A covalent antagonist for the human adenosine A2A receptor. Purinergic Signal 13, 191–201 (2017).
Cheng, Y. & Prusoff, W. H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 (1973).
Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008).
Kaminski, G. A., Friesner, R. A., Tirado-Rives, J. & Jorgensen, W. L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 105, 6474–6487 (2001).
Berger, O., Edholm, O. & Jähnig, F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 72, 2002–2013 (1997).
Marelius, J., Kolmodin, K., Feierberg, I., Åqvist, J. & Aqvist, J. Q: A molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J. Mol. Graph. Model. 16, 213–225 (1998).
King, G. & Warshel, A. A surface constrained all-atom solvent model for effective simulations of polar solutions. J. Chem. Phys. 91, 3647 (1989).
Lee, F. S. & Warshel, A. A local reaction field method for fast evaluation of long-range electrostatic interactions in molecular simulations. J. Chem. Phys. 97, 3100 (1992).
Ryckaert, J.-P.J., Ciccotti, G. & Berendsen, H. J. H. Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Robertson, M. J., Tirado-Rives, J. & Jorgensen, W. L. Improved peptide and protein torsional energetics with the OPLS-AA force field. J. Chem. Theory Comput. 11, 3499–3509 (2015).
Bennett, C. H. Efficient estimation of free energy differences from Monte Carlo data. J. Comput. Phys. 22, 245–268 (1976).
The PyMOL Molecular Graphics System, Version 2.0. Schrödinger, LLC.
Acknowledgements
This work was financially supported by the Consellería de Cultura, Educacioń e Ordenacioń Universitaria of the Galician Government: (grant: ED431B2017/70), Centro singular de Investigacioń de Galicia accreditation 2016−2019 (ED431G/ 09), Xunta de Galicia (ED431C 2018/21) and the European Regional Development Fund (ERDF). Xuesong Wang thanks the China Scholarship Council (CSC) for her PhD scholarship. The authors are part of the EU COST action ERNEST (CA 18133) and the Swedish strategic research program eSSENCE (H.G.dT., W.J.), The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC).
Funding
Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Contributions
X.W., W.J., R.P.-D., E.S., L.H.H. and H.G.d.T. participated in the conceptualization of the research. X.W., W.J. and R.P.-D. performed the experiments.; X.W., W.J. and R.P.-D., H.G.dT, L.H. and E.S. analyzed the data. All authors contributed to writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Jespers, W., Prieto-Díaz, . et al. Identification of V6.51L as a selectivity hotspot in stereoselective A2B adenosine receptor antagonist recognition. Sci Rep 11, 14171 (2021). https://doi.org/10.1038/s41598-021-93419-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-93419-x
This article is cited by
-
Structural insight into the dual-antagonistic mechanism of AB928 on adenosine A2 receptors
Science China Life Sciences (2024)
-
Structural basis of selective cannabinoid CB2 receptor activation
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
