
Abstract
We assembled new plastomes of 19 species of Mikania and of Ageratina fastigiata, Litothamnus nitidus, and Stevia collina, all belonging to tribe Eupatorieae (Asteraceae). We analyzed the structure and content of the assembled plastomes and used the newly generated sequences to infer phylogenetic relationships and study the effects of different data partitions and inference methods on the topologies. Most phylogenetic studies with plastomes ignore that processes like recombination and biparental inheritance can occur in this organelle, using the whole genome as a single locus. Our study sought to compare this approach with multispecies coalescent methods that assume that different parts of the genome evolve at different rates. We found that the overall gene content, structure, and orientation are very conserved in all plastomes of the studied species. As observed in other Asteraceae, the 22 plastomes assembled here contain two nested inversions in the LSC region. The plastomes show similar length and the same gene content. The two most variable regions within Mikania are rpl32-ndhF and rpl16-rps3, while the three genes with the highest percentage of variable sites are ycf1, rpoA, and psbT. We generated six phylogenetic trees using concatenated maximum likelihood and multispecies coalescent methods and three data partitions: coding and non-coding sequences and both combined. All trees strongly support that the sampled Mikania species form a monophyletic group, which is further subdivided into three clades. The internal relationships within each clade are sensitive to the data partitioning and inference methods employed. The trees resulting from concatenated analysis are more similar among each other than to the correspondent tree generated with the same data partition but a different method. The multispecies coalescent analysis indicate a high level of incongruence between species and gene trees. The lack of resolution and congruence among trees can be explained by the sparse sampling (~ 0.45% of the currently accepted species) and by the low number of informative characters present in the sequences. Our study sheds light into the impact of data partitioning and methods over phylogenetic resolution and brings relevant information for the study of Mikania diversity and evolution, as well as for the Asteraceae family as a whole.
Similar content being viewed by others


Introduction
Chloroplasts are organelles linked to photosynthesis, which have many essential functions in plants, such as carbon fixation and biosynthesis of starch, fatty acids, amino acids, and pigments1,2. The chloroplast genome (plastome) in angiosperms usually has a circular shape, with 120 to 180 kb in size, divided in four main regions: two Inverted Repeat (IR) regions, one Large Single Copy (LSC), and one Small Single Copy (SSC) region3. Plastome gene composition and order are generally conserved among land plants4,5, but recent studies have documented that variation at many levels can occur6,7. It has been widely accepted that plastomes are uniparentally inherited and do not present recombination, with the whole genome frequently being interpreted as a single locus in phylogenetic analysis, implying in all genes evolving concertedly8. However, in the last decade, evidence has accumulated that this organelle can be biparentally inherited, copies with different sequences can occur9 and that different portions of the genome can evolve at different paces10. Recent studies recommend analyzing plastome genes individually in phylogenetic inferences, through methods like the multispecies coalescent, which also accounts for possible incongruence between gene trees and species trees10,11.
Mikania Willd. is the most diverse genus within the tribe Eupatorieae and the largest genus of climbing plants in Asteraceae, with around 450 species12,13. It has a pantropical distribution, mainly neotropical, with most of the diversity concentrated in South America12,13. The large number of species makes carrying out taxonomic revisions and molecular studies for the genus difficult14. Nevertheless, Mikania is easily morphologically recognized by its four-flowered heads surrounded by four involucral bracts; its circumscription has been indisputable since its description in 174215. A reevaluation of the current infrageneric classification of the genus is needed16, but the lack of broadly sampled phylogenies prevents the elaboration of a classification based on evolutionary relationships and monophyletic groups15. Several regional taxonomic studies of Mikania (e.g.17,18,19) are available, but large-scale taxonomic treatments are still a challenge. Some taxa, such as Mikania glomerata Spreng. and M. laevigata Sch.Bip. ex Baker, have known pharmacological uses, especially in the treatment of respiratory diseases20. Consequently, the genus is well represented in phytochemical studies, which have been conducted with approximately 12% of all Mikania species20. The main chemical compounds linked to pharmacological activities, which are found in different parts of Mikania plants, are coumarins and derivatives, sesquiterpenes, sesquiterpenes lactones, diterpenes, phytosterols/terpenoids, and flavonoids20. Some species are considered invasive, including some widespread weeds, such as Mikania micrantha Kunth16.
The first attempt to investigate phylogenetic relationships within Mikania with molecular data was based on AFLP markers, the intronic region of the plastid gene rps16, and ribosomal ITS and ETS, but included only representatives of generic sections proposed for Brazilian species of Mikania21. More molecular studies are needed to evaluate infrageneric limits within this genus, as well as to explore its morphological and chemical evolution, biogeographic history, and diversification. The only genomic resources reported in the literature for the genus are the complete plastome (NC031833.122) and the chromosome-scale genome23 of Mikania micrantha, an invasive plant well known for causing significant damage to natural ecosystems and crops in several parts of the world. For the tribe Eupatorieae as a whole, which has ~ 180 genera and 2200 species24, only three other plastomes were published to date, besides M. micrantha: i.e., Ageratina adenophora (Spreng.) R.M.King & H.Rob. (NC01562125), Praxelis clematidea R.M.King & H.Rob. (NC02383326), and Ageratum conyzoides L. (MK90523827).
The family Asteraceae is one of the most species-rich families of flowering plants, including an impressive morphological and ecological diversity28. While the backbone relationships within the family have recently become clearer, internal relationships at tribal and generic levels still need a lot of attention28,29. Although many Asteraceae plastomes were recently published, most studies characterize the plastome of a single taxon and focus in comparative genomic analyses at higher taxonomic levels (e.g.22,25,26,27,30,31). These studies are important to improve the understanding of plastome variation in the family as a whole27,31 and to provide more information on their phylogenetic utility. Yet, larger infrageneric samplings are essential to explore evolution and phylogenetic relationships in Asteraceae at lower taxonomic levels (e.g.32,33).
In this study, we sequenced new complete plastomes of 19 species of Mikania representative of the morphological diversity of the genus. The sampled taxa are: M. additicia B.L.Rob., M. brevifaucia W.C.Holmes & McDaniel, M. burchelii Baker, M. decora Poepp., M. decumbens Malme, M. fasciculata C.T.Oliveira & Pirani, M. glomerata Spreng., M. haenkeana DC., M. lehmanii Hieron., M. neurocaula DC., M. oblongifolia DC., M. obtusata DC., M. parvifolia Baker, M. purpurascens (Baker) R.M.King & H.Rob., M. salviifolia Gardner, M. smaragdina Dusén ex Malme, M. sylvatica Klatt., M. ternata (Vell.) B.L.Rob., and M. triangularis Baker (Table 1). The plastomes of three species from other genera of Eupatorieae, namely Ageratina fastigiata (Kunth) R.M.King & H.Rob., Litothamnus nitidus (DC.) W.C.Holmes, and Stevia collina Gardner, were also sequenced and used as outgroups. The previously published plastome of Mikania micrantha (NC031833.122) was included in the analyses as well. This study aims to characterize and compare the plastomes within Mikania and among closely related genera within tribe Eupatorieae to improve our understanding about the evolution of this genome and investigate different methods of phylogenetic reconstruction with this dataset. More specifically, we: (i) sequenced, assembled, and characterized the overall plastome structure; (ii) performed comparative genomic analyses within Mikania, and among Mikania and other Eupatorieae genera; (iii) identified putative repeated regions; and (iv) investigated phylogenetic relationships using both concatenation and multispecies coalescent methods with different data partitions.
Results
Plastome assembly and characterization
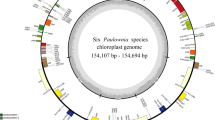
Approximately 1–2 GB of data and 10,488,036–20,546,020 paired-end raw reads for each plastome were obtained. The 19 Mikania plastomes range in length from 151,773 (M. glomerata) to 152,229 bp (M. salviifolia) (Table 1, Supplementary Fig. S1). All assembled plastomes show the general structure found in most angiosperms, divided in four main regions, which in Mikania consists of a LSC (83,527–83,878 bp), a SSC (18,163–18,347 bp), and a pair of IR (24,877–25,014 bp) regions (Table 1, Supplementary Fig. S1). As observed in other Asteraceae taxa, the 22 plastomes assembled here contain two inversions in the LSC region34: a large inversion (22,211–22,423 bp, M. salviifolia and Ageratina fastigiata, respectively) including 16 genes from trnSGCU-trnCGCA to trnGUCC-trnTGGU and a small inversion (3223–3304 bp, Litothamnus nitidus and A. fastigiata, respectively) nested within the former, which includes six genes located between trnSGCUtrnCGCA and trnEUUC (Table 1, Supplementary Fig. S1). All plastomes sequenced in this study encode 113 unique genes, including 79 protein-coding genes (CDS), 17 of which contain introns, 30 tRNA genes, and four rRNA genes (Table 2). The plastomes of Mikania and the other three Eupatorieae have identical structure and order. The boundaries between the four main plastome regions are very conserved within Mikania species and among the three Eupatorieae genera sampled here: the LSC/IRb border is within rps19, the IRb/SSC is within ycf1, the SSC/IRa is between ndhF and a partial ycf1 (ψycf1), and the IRa/LSC is between a truncated rps19 (†rps19) and trnHGUG (Supplementary Figs. S1, S2).
Variable regions across plastomes
Pairwise comparison of divergent regions performed in mVISTA within three selected Mikania plastomes and between the Eupatorieae genera sequenced here reveals low intra-generic sequence divergence within Mikania and higher sequence variation among genera and in noncoding regions, except for the ycf1 gene (Supplementary Fig. S3). The nucleotide variability (π) values within 800 bp across the plastomes range from 0 to 0.013, with a mean value of 0.0036. We identified only two regions with π > 0.01 (rpl32-ndhF and rpl16-rps3) and six regions with π values around 0.009 (rbcL, ycf1, petN-psbM, rps16-trnQUUG, trnHGUG-psbA, atpI-atpH) (Fig. 1A).

(A) Sliding window analysis of the chloroplast genomes of 20 Mikania plastomes (i.e., 19 sequenced here plus M. micrantha, NC031833.1) (window length: 800 bp, step size: 200 bp). X-axis, position of the midpoint of a window; Y-axis, nucleotide diversity (π) in each window. (B,C) Most variable protein-coding genes within the 20 Mikania plastomes. (B) Genes with up to 1% of variable sites. (C) Genes with up to five variable sites.
In alignments of the 19 complete Mikania plastomes assembled here, the noncoding regions are more variable (i.e., 3.15% of the intergenic regions and 2.24% of the introns) than the coding regions (i.e., 1.34% of the protein-coding genes; Table 3). Among the 79 protein-coding genes, the ten genes with the highest percentage of variable sites are: ycf1 (3.83%), rpoA (3.43%), psbT (2.94%), ndhF (2.43%), rpl32 (2.42%), ccsA (2.37%), rbcL (2.33%), matK (2.24%), rps3 (2.13%), and rpl20 (2.1%) (Fig. 1B,C; Supplementary Tables S1, S2).
Analyses of SSR and tandem repeats
In the 19 Mikania plastomes, the total number of SSRs range from 34 to 44 SSRs, while 51, 49, and 38 SSRs are recovered in A. fastigiata, L. nitidus, and S. collina, respectively (Supplementary Fig. S4A–C). The most abundant SSRs are A or T mononucleotide repeats, which account for 54.3–69% of the total SSRs in Mikania, 70.6% in A. fastigiata, 53.1% in L. nitidus, and 52.6% S. collina; G or C repeats, on the other hand, are rare (Supplementary Table S3). Among the total number of SSR motifs in Mikania, 20–29 (57.5–69%) are mono-repeats, 4–6 (9.5–14.3%) are di-repeats, 2–5 (5.4–12.5%) are tri-repeats, 5–7 (12.2–17.6%) are tetra-repeats, 0–1 (0–2.3%) is penta-repeat, and 0–1 (0–2.9%) is hexa-repeat (Supplementary Fig. S4B, Supplementary Table S3). Furthermore, most of the SSRs in the Mikania species are located in the LSC region (70.3–83.8%), while the IR regions include between 0 and 12.2% of the SSRs, and the SSC region includes between 9.1 and 29.7%. Yet, the relative density of SSRs in the LSC is somewhat similar to that found in the SSC when considering the size of each region (Supplementary Fig. S4A–C, Supplementary Table S3). In the three other Eupatorieae genera sequenced here, 73.5–74.5% of the SSRs are located in the LSC, 2–11.8% in the IRs, and 13.7–24.5% in the SSC region (Supplementary Fig. S4A–C, Supplementary Table S3).
We also used REPuter to identify tandem repeat sequences longer than 30 bp in the plastomes sequenced here. In all 22 plastomes, repeats with 30–33 bp are the most common. Most repeats are found in the LSC, a few in the IRs, and none in the SSC (Supplementary Fig. S4D,E, Supplementary Table S4).The total number of repeats in Mikania range between 17 and 45, with maximum sizes of 48 bp in all Mikania species (Supplementary Fig. S4D,E, Supplementary Table S4). The Mikania plastomes contain 8–23 forward repeats, 6–14 palindrome repeats, 0–13 reverse repeats, with complement repeats being rare, 0–2 (Supplementary Table S4). The total number of repeats in A. fastigiata is 31 bp, while in L. nitidus and S. collina is 17 bp. The maximum repeat size in A. fastigiata is 46 bp, in L. nitidus is 58 bp, while in S. collina is 48 bp (Supplementary Fig. S4D,E, Supplementary Table S4).
Phylogenetic relationships of twenty Mikania species
Phylogenetic analyses using two different methods, concatenated maximum likelihood and multispecies coalescent, and three datasets (only coding regions, only non-coding regions and both combined) generated six different topologies with different degrees of support (Fig. 2). Ageratina fastigiata, Litothamnus nitidus, Stevia collina, and Helianthus annuus (NC007977; Heliantheae) were used as outgroups and the trees were rooted using H. annuus (Fig. 3). Mikania emerges as monophyletic and all trees present three generally well-supported main clades (bootstrap support (BS) ≥ 90%, local posterior probabilities (LPP) ≥ 0.95) containing the same species: Clade I (Mikania sylvatica and M. lehmanii), Clade II (M. brevifaucia, M. salviifolia, M. fasciculata, M. purpurascens, M. ternata, M. micrantha, M. decumbens) and Clade III (M. parvifolia, M. smaragdina, M. triangularis, M. additicia, M. obtusata, M. neurocaula, M. burchelii, M. oblongifolia, M. glomerata, M. haenkeana, M. decora). Within each clade, the relationships between some species pairs are stable, but the position of some taxa (e.g., M. smaragdina, M. ternata) consistently change, especially in Clade III (Fig. 2). The adjusted Robinson-Foulds distances fitted in a multidimensional scaling model show that topologies are considerably different among themselves, especially the three coalescent trees (Fig. 4, Supplementary Table S5). Although not directly comparable, support values are generally higher in the concatenated analyses (BS) than in the multispecies coalescent analyses (LPP), and in the total dataset in comparison with the coding-only or non-coding-only datasets (Fig. 2). Gene discordance analyses ran with the coalescent trees show a high level of incongruence between the species tree and the gene trees, especially in the dataset containing only coding regions (Fig. 5, Supplementary Fig. S5A).

Phylogenetic analyses of Mikania using different data partitions and inference methods. RAxML refers to concatenate maximum likelihood analyses and Astral refers to multispecies coalescent inference. Support values are color-coded and nodes without symbols correspond to unsupported branches (BS < 50%, LPP < 0.8). (a) Analyses carried out with the whole plastome sequence with one of the IRs removed. (b) Analyses conducted only with the coding regions (CDS) of the plastome. (c) Analyses carried out only with non-coding regions of the plastome (intergenic regions and introns).

Representative topology of phylogenetic relationships in Mikania, showing three main clades. This tree represents the concatenated maximum likelihood analysis conducted with the whole plastome sequence with one of the IRs removed. Bootstrap support values shown in each node.

Multidimensional scaling of adjusted Robinson-Foulds values calculated from the pairwise comparison of all rooted trees. Coding: analyses conducted only with the coding regions (CDS). Non-coding: analyses conducted with non-coding regions (intergenic regions and introns). Total: whole plastome sequence with one IR removed.

Gene tree discordance analysis conducted with the total dataset and multispecies coalescent inference (Astral total). The number above the branch indicates the number of concordant gene trees and the one below the number of conflicting gene trees. Pie charts indicate the proportion of gene trees supporting that clade (blue), the proportion that supports the main alternative topology for that clade (yellow), and the proportion that supports all other topologies (orange).
Discussion
In this study, we assembled 19 complete plastomes of Mikania species and of three other species from tribe Eupatorieae (i.e., Ageratina fastigiata, Litothamnus nitidus, and Stevia collina), and conducted phylogenetic analyses with different datasets and inference methods. The organization of Mikania plastomes is similar across the studied species and to other Asteraceae plastomes. The overall genomic structure among Mikania plastomes is very conserved, including similar lengths, boundaries between the SC/IR regions, and number of duplicated genes in the IRs (Tables 1, 2, Supplementary Figs. S1–S3). All plastomes assembled here show the two inversions in the LSC present in most Asteraceae taxa, except for the early diverging tribe Barnadesieae30,35,36. These inversions present a very conserved structure, including the same genes and similar sizes, in all of the 22 plastomes reported here (Table 1), as well as when comparing with more distantly related Asteraceae genomes, such as Helianthus annuus (NC00797730) and Lactuca sativa (NC00757830). On the other hand, in Ageratina adenophora25 and Praxelis clematidea26, these inversions do not start between the trnSGCU and trnCGCA genes, as in other Asteraceae taxa, but between the trnCGCA and petN genes25,26. We also noticed an inversion within the ycf1 gene in the SSC region in the Ageratina adenophora25 and Praxelis clematidea26 plastomes, which was not observed in the plastomes assembled here, nor in M. micrantha (NC031833.122) or Ageratum conyzoides (MK90523827) (Supplementary Fig. S6).
The gene content found in the 22 plastomes assembled here resembles that found in other Asteraceae genomes25,31,33. They encode 113 unique genes, including 79 protein-coding genes (CDS), 30 tRNA genes, and four rRNA genes. All plastomes include 17 intron-containing genes (14 contain one, while three contain two introns; Table 3). Within Eupatorieae, a duplication of the trnFGAA gene was detected in Ageratina adenophora25 and Praxelis clematidea26, which was previously reported for other Asteraceae subfamilies (Carduoideae, Cichorioideae, Asteroideae, and Heliantheae alliance)37. The rpoC1 gene in Ageratina adenophora25, Ageratum conyzoides27, and Praxelis clematidea26 contains two introns, while in all plastomes assembled here it contains only one intron, similarly to other Asteraceae plastomes sequenced to date25,33. Previous plastome comparative studies within Heliantheae detected a ~ 450 bp deletion in the ycf2 gene for some taxa30,33, which was not observed in the previously published Eupatorieae plastomes or those newly sequenced here.
The nucleotide variability is relatively low within Mikania plastome sequences (mean π value of 0.0036). Yet, another comparative plastome study in Asteraceae, with 36 species of Aldama (Heliantheae), found an even lower mean π value, 0.0011833. The rpl32-ndhF and rpl16-rps3 intergenic regions are the most variable loci found within Mikania plastomes, making them candidate markers for phylogenetic studies at the species level within the genus. Other regions with higher nucleotide variability within Mikania are: rbcL, ycf1, petN-psbM, rps16-trnQUUG, trnHGUG-psbA, and atpI-atpH (Fig. 4A). The noncoding regions are more variable that the coding regions, as expected38 (Supplementary Fig. S3, Supplementary Table S2). Some of the noncoding regions that are variable within Mikania have been reported to be likely useful for molecular studies at lower taxonomic levels39,40. Considering only the coding regions and the percentage of variable sites, the ycf1 gene is the most variable (3.83%) followed by rpoA (3.43%) (Fig. 4B,C, Supplementary Table S1). The ycf1 gene is well known as a variable coding region at lower taxonomic levels, including within Asteraceae, and has been used in phylogenetic studies within distinct plant groups33,41. In addition, the rpo genes (rpoA, rpoB, rpoC1, and rpoC2) have been previously reported to be relatively rapidly evolving42,43 and divergent within Asteraceae25,33,41.
The number of Single Sequence Repeats (SSRs), 34 to 44, identified within Mikania plastomes is similar to that reported for other Asteraceae, such as within Heliantheae, where 38–57 SSRs were found in a study with 15 species33. In all plastomes assembled here, most SSRs found are mononucleotide repeats (59–61% within Mikania and 22–37% in the other genera), followed by tetranucleotide repeats (5–7% within Mikania and 57–72% in the other genera). The A or T motifs are the most common SSR repeat, in agreement with other studies31,44 (Supplementary Fig. S4A–C, Supplementary Table S3). Dispersed repeats are considered to have important influence in genome structure, size, recombination, and rearrangements25. The number of repeats ≥ 30 bp found in the plastomes sequenced in this study range from 17 to 45. The maximum repeat size found within all Mikania species was 48 bp (Supplementary Fig. S4D,E, Supplementary Table S4). In Myripnois dioica (Pertyeae) 58 repeats ≥ 20 bp were found and the maximum repeat length was the same found for Mikania (48 bp)31. In Lactuca sativa and Helianthus annuus, 15 and 33 repeats ≥ 23 bp were found, respectively, of which most were smaller than 40 bp, with only two larger than 90 bp30. In Ageratina denophora, 59 repeats ≥ 15 bp were found, most ranging between 15 and 50 bp, but repeats > 100 bp were also present25.
The phylogenetic analyses performed here sampled only 20 out of ~ 450 Mikania species (Fig. 3). Yet, the relationships within this genus were never investigated using complete plastomes and represent an advance in our knowledge of infrageneric evolutionary relationships. The only phylogenetic study of the genus to date21 was focused on species delimitation of a few highly variable taxa, such as M. micrantha and M. cordifolia, based on AFLPs and two nuclear ribosomal markers. The divergent sampling, with only four species overlapping between both studies, precludes a proper comparison between the topologies from the previous study and the ones found here. The differences in the genomic compartment used by both studies further hinders a proper comparison, given the frequent occurrence of discrepancies between nuclear and plastidial phylogenies. The comparison among trees obtained with different reconstruction methods and datasets show a scenario of incongruence among topologies, especially in the higher nested clades (Figs. 2, 4). The backbones of most trees show Clade I as sister to a clade formed by Clade II and Clade III, except for the Astral coding tree, which shows Clade II as sister to Clade I and Clade III (Fig. 2B). Clade II presents the same relationships in all three concatenated analyses, while in the coalescent trees the relative positions of some taxa, such as Mikania ternata and M. purpurascens, change in all trees, but especially when comparing the coding tree with the non-coding and total trees (Fig. 2). The relationships within Clade III are responsible for most of the incongruence among all six trees, as they change in each topology. Some species emerge as sisters in most topologies, such as M. decora + M. haenkeana, M. oblongifolia + M. burchelii, M. obtusata + M. neurocaula and M. additicia + M. triangularis. Similarly, M. parvifolia emerges as sister to all other species in Clade III in all analyses, but the relative positions of M. smaragdina and M. glomerata are variable across all topologies, usually with M. smaragdina being close to M. additicia and M. triangularis, and M. glomerata close to M. decora and M. haenkeana (Fig. 2).
The gene tree discordance analyses (Fig. 5, Supplemental Fig. S5) show strong discordance across the three datasets (total, coding, non-coding), with few gene trees agreeing with the relationships shown in the species tree. The multispecies coalescent has been extensively used in the context of multi-locus phylogenies obtained from target capture data45, but few studies have applied it to plastid data, due to the widespread misconceptions about the lack of biparental inheritance and recombination in this organelle11. Among recent studies that used the multispecies coalescent in plastid data, three of them refer to higher-level phylogenies, i.e., among Angiosperms46, among Rosids10 and among tribes of Asteraceae47, while one deals with a single genus48. Most of these studies found incongruences between concatenated and coalescent analyses, but only two of them presented information about gene tree/species tree discordance46,47, both showing wide discordance between the inferred species tree and the gene trees.
Walker et al.46 proposes that uninformative genes are one of the reasons for discordance, and this is likely one of the issues in our trees. In the coding dataset, the number of variable sites in each gene across species of Mikania varies from 0 to 3.83%, while the non-coding dataset presents a little more variation, from 0.5% to 8.8% (Supplementary Tables S1, S2). The large variation in gene tree topologies, summarized by the gene tree discordance analysis (Fig. 5, Supplementary Fig. S5), leads to weakly resolved gene trees and consequently to poorly supported species trees, as the calculation of local posterior probabilities (LPP) depends on the concordance among the three possible topologies for a determined quartet of branches49. The length of each individual locus alignment also influences on the degree of conflict, as shorter loci tend to have less informative sites, contributing to the lack of resolution in gene trees46. In our dataset, these two factors seem to be correlated, with most of the shorter alignments presenting very few variable sites (Supplementary Fig. S7, Supplementary Table S1, S2).
Recent studies have shown that in concatenated analysis, a few outlier genes can drive topology inference45,50. Our concatenated analyses present topologies more similar to each other than the coalescent topologies, regardless of the dataset (Fig. 4). The concatenated coding tree is more similar to the concatenated total topology (Figs. 2, 4), possibly indicating that one or more specific genes are responsible for defining most of the topology, whereas the three coalescent topologies are all different from each other, due to lack of resolution in individual gene trees. Incomplete lineage sorting (ILS) is usually considered a source of conflict in the multispecies coalescent45, and one metric that can be used to assess its occurrence is the normalized quartet score of a coalescent tree, which measures the percentage of quartet trees found in the species tree from all calculated quartet trees51. The normalized quartet scores calculated for the three Astral topologies are considered very high (total: ~ 52%, coding: ~ 49%, non-coding: ~ 57%), but considering the sparse sampling in our study, which included ca. 0.45% of Mikania species, it is difficult to delimit the occurrence of ILS in relation to the lack of actual sampling. Further studies with more complete sampling could help untangle cases of ILS and lack of resolution due to uninformative genes, by also increasing the likelihood of sequence variation.
Although applying the multispecies coalescent methods to chloroplast sequences makes sense biologically, due to the possibility of evolutionary process that could lead to different parts of the genome evolving in different rates, in practice the results tend to be confounding, as seen here and in previous studies10,46. The causes of plastome conflict are still poorly understood46, and in lower-level phylogenies, as the case presented here, it might be hard to untangle sources of conflict inherent to plastome biology from lack of sequence variability due to rapid radiations over short evolutionary times. In Mikania, where the phylogenetic relationships are poorly known, especially in relation to the nuclear genome, it is difficult to map out other potential root causes for conflict, such as hybridization, plastome capture, or incomplete lineage sorting. An expanded sampling, both in terms of species and genome compartment (e.g., adding nuclear markers), could bring a clearer picture of the evolutionary relationships in the genus and of other biological factors that might impact phylogenetic reconstructions in Mikania.
Material and methods
Sampling, DNA preparation, sequencing, plastome assembly, and annotation
Whole genomic DNA extraction, Illumina libraries preparation, and NGS sequencing of the 19 Mikania and three outgroup species from other Eupatorieae genera (Table 1) follow33. Sequencing was performed using Illumina HiSeq 2500 Genome Analyzer (Illumina, San Diego, California, USA) in paired-end mode. We assembled all plastomes using Fast-Plast 1.2.852, with the following software: (i) Trimmomatic 0.3253 to remove adaptors and trim low quality reads using the parameters SLIDINGWINDOW:10:20 and MINLEN:40; (ii) Bowtie2 2.1.054 with default parameters to select only chloroplast-like reads using the plastome of Mikania micrantha (NC031833.122 as reference; (iii) SPAdes 3.1.055 to assemble the selected reads into contigs with k-mers of 57 and 87, using the “only-assembler” option; (iv) afin (http://bitbucket.org/benine/afin/) to assemble the contigs from the previous step with the complete reads dataset with the following parameters -l 150,50,50, -f 0.1, -d 100,—× 100, -p 20,15,10, and -i 2,1,1. We evaluated plastome coverage in Jellyfish 2.1.356. We annotated the sequences using Geneious 9.1.557, DOGMA58, and BLAST59,60, with start and stop codons checked manually. We used OGDRAW61 to prepare the graphical representation for the resulting plastome. Finally, we analyzed the boundaries between the plastome regions (i.e., LSC/IRb/SSC/IRa) using Geneious and IRscope62 (https://irscope.shinyapps.io/irapp/).
Comparative analyses of the assembled Mikania plastomes
We conducted comparative analyses within 20 Mikania plastomes (i.e., 19 sequenced here plus M. micrantha, NC031833.1) and among Mikania and the three outgroup taxa plastomes assembled in this study (i.e., Ageratina fastigiata, Litothamnus nitidus, and Stevia collina). We used MAFFT 763 with the FFT-NS-2 method64 to perform the alignment of the complete plastome sequences, with one copy of the IRs manually excluded to avoid data duplication. To search variable regions, we used mVISTA65 with Shuffle-LAGAN66 with the previously annotated M. decora plastome as reference, plus two Mikania species and the three outgroup taxa sequenced here. Based on the phylogeny recovered in this study, we selected one species from each of the three main recovered clades of Mikania (i.e., M. decora, M. decumbens, and M. sylvatica). We calculated nucleotide variability values (π) within 20 Mikania plastomes. We used DnaSP 6.1067 to conduct a sliding window analysis with a 200 bp step size and 800 bp window length. The resulting π values were plotted using R68. We analyzed the variable sites using MEGA 769 in the alignments of the 20 Mikania complete plastomes and of 79 protein-coding genes (Supplementary Table S1) extracted from these genomes. Each gene was extracted from the complete plastome alignment and separately re-aligned in Geneious with the ClustalW plugin70 considering codon positions.
Analyses of repeated regions
We searched for microsatellites or Simple Sequence Repeats (SSRs; i.e., tandemly arranged repeats of short DNA motifs of 1–6 bp in length) and repeated elements using MISA71 and REPuter72, respectively, in the plastomes of the 19 Mikania species and three other Eupatorieae representatives sequenced here. We analyzed SSRs with motifs between 1 and 6 nucleotides and a minimum number of repetition units as follows: 10 for mono-, 5 for di-, and 4 for trinucleotide, and 3 for tetra-, penta-, and hexanucleotide SSRs. We identified repeated elements ≥ 30 bp (forward, palindrome, reverse, and complement) using ≥ 90% of sequence identity and hamming distance = 3.
Phylogenetic reconstruction
We reconstructed phylogenetic relationships among 20 Mikania plastomes (i.e., 19 sequenced here plus M. micrantha; NC031833.1) and three species from other Eupatorieae genera assembled in this study plus Helianthus annuus (NC007977; Heliantheae) as outgroup. Three concatenated matrices were assembled: one containing the whole plastome sequence with one IR removed (total), one containing only the CDS regions of all 79 protein-coding genes (coding) and one containing all intergenic regions and introns (non-coding). All matrices were aligned using MAFFT 763 using the FFT-NS-2 method64. Maximum likelihood reconstructions were carried out in in RAxML 8.2.973 using the GTR + G model with node support assessed by rapid bootstrap (-f a) using 1000 non-parametric bootstrap pseudo-replicates. The multispecies pseudocoalescent model from Astral III51 was used to obtain species trees from individual gene trees. Three datasets were used in these analyses: one containing only each individual CDS region from all 79 protein-coding genes (coding), one containing intergenic regions longer than 300 bp (non-coding), and one combining both datasets (total). Character evolution models for each gene matrix were calculated with PartitionFinder v.1.1.074,75,76, evaluating the GTR + G and GTR + G + I models in the RAxML version with rcluster search option and unlinked branch lengths, using the corrected Akaike Information Criterion to choose between models. Unrooted gene trees were obtained in RAxML 8.2.9, using the rapid bootstrap mode and 100 pseudo-replicates. Branch support was calculated using local posterior probabilities (LPP)51.
Gene tree discordance
Discordance between the species tree and gene trees, expressed as the proportion of gene trees presenting each of the clades found in the species tree, was calculated using phyparts with the thorough conflict analysis options (-a 1)77. All species and gene trees were rooted using Helianthus annuus as outgroup using the function pxrr in the package phyx78. The proportion of gene trees in agreement with the species tree in each node, as well as the proportion of uninformative gene trees or those supporting alternative topologies, were plotted as pie charts at each node of the tree using the phypartspiecharts.py script79.
Topological comparisons
The adjusted Robinson-Foulds (RF) distance was used to calculate the distance among the six topologies. The RF distance was calculated between all pairs of rooted trees using PAUP* v4.0a80 and adjusted by the number of nodes in the trees (RFadj = RF/(2n − 6)), resulting in values ranging from 0 to 1. A multidimensional scaling approach was used to observe the level of similarity among the topologies, using the “cmdscale” command in the R package “stats”, and subsequently plotted.
Data archiving statement
The complete plastome sequence data of the 19 Mikania plastomes and that of Ageratina fastigiata, Litothamnus nitidus, and Stevia collina are available in GenBank (NCBI) with the accession numbers MT793834–MT793855.
References
Jansen, R. K. & Ruhlman, T. A. Plastid genomes of seed plants. In Genomics of Chloroplasts and Mitochondria: Advances in Photosynthesis and Respiration (Including Bioenergy and Related Processes) (eds Bock, R. & Knoop, V.) 103–126 (Springer, 2012).
Daniell, H., Lin, C. S., Yu, M. & Chang, W. J. Chloroplast genomes: Diversity, evolution and applications in genetic engineering. Genome Biol. 17, 134. https://doi.org/10.1186/s13059-016-1004-2 (2016).
Green, B. R. Chloroplast genomes of photosynthetic eukaryotes. Plant J. 66, 34–44 (2011).
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Müller, K. F. & Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. https://doi.org/10.1007/s11103-011-9762-4 (2011).
Reginato, M., Neubig, K. M., Majure, L. C. & Michelangeli, F. The first complete plastid genomes of Melastomataceae are highly structurally conserved. PeerJ 4, e2715. https://doi.org/10.7717/peerj.2715 (2016).
Chumley, T. W. et al. The complete chloroplast genome sequence of Pelargonium x hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 23, 2175–2190. https://doi.org/10.1093/molbev/msl089 (2006).
Zhu, A., Guo, W., Gupta, S., Fan, W. & Mower, J. P. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction & loss on substitution rates. New Phytol. 209, 1747–1756. https://doi.org/10.1111/nph.13743 (2016).
Doyle, J. J. Gene trees and species trees: Molecular systematics as one-character taxonomy. Syst. Bot. 17, 144–163 (1992).
Ramsey, A. J. & Mandel, J. R. When one genome is not enough: Organellar heteroplasmy in plants. Annu. Plant Rev. 2, 1–40. https://doi.org/10.1002/9781119312994.apr0616 (2019).
Gonçalves, D. J. P., Simpson, B. B., Ortiz, E. M., Shimizu, G. H. & Jansen, R. K. Incongruence between gene trees and species trees and phylogenetic signal. Mol. Phylogenet. Evol. 138, 219–232. https://doi.org/10.1016/j.ympev.2019.05.022 (2019).
Gonçalves, D. J. P., Jansen, R. K., Ruhlman, T. A. & Mandel, J. R. Under the rug: Abandoning persistent misconceptions that obfuscate organelle evolution. Mol. Phylogenet. Evol. 151, 106903. https://doi.org/10.1016/j.ympev.2020.106903 (2020).
King, R. M. & Robinson, H. The Genera of the Eupatorieae (Asteraceae). Monogr. Syst. Bot. Missouri Bot. Garden 22, 1–581 (1987).
Robinson, H., Schilling, E. & Panero, J. L. Eupatorieae. In Systematics, Evolution, and Biogeography of the Compositae (eds Funk, V. A. et al.) 171–189 (International Association for Plant Taxonomy, 2009).
Funk, V. A., Susanna, A., Stuessy, T. F. & Robinson, H. Classification of compositae. In Systematics, Evolution, and Biogeography of the Compositae (eds Funk, V. A. et al.) 171–189 (International Association for Plant Taxonomy, 2009).
Honório, A. C., Quaresma, A. S. & Oliveira, C. T. Flora do Ceará, Brasil: Mikania (Asteraceae: Eupatorieae). Rodriguésia 70, e02952017. https://doi.org/10.1590/2175-7860201970003 (2019).
Holmes, W. C. A proposed sectional classification for Mikania (Eupatorieae). In Compositae: Systematics. Proceedings of the International Compositae Conference, Kew, 1994 (eds Hind, D. J. N. & Beentje, H. J.) 621–626 (Royal Botanic Gardens, 1996).
Holmes, W. C. Revision of the old world Mikania (Compositae). Bot. Jahrbücher für Syst. 103, 211–246 (1982).
Ritter, M. R. & Miotto, S. T. S. Taxonomia de Mikania Willd. (Asteraceae) no Rio Grande do Sul, Brasil. Hoehnea 32, 309–359 (2005).
Robinson, H. & Holmes, W. C. Compositae-Eupatorieae 190 (3). In Flora of Ecuador Vol. 83 (eds Harling, G. & Andersson, L.) 1–347 (Botanical Institute, Goteborg University, 2008).
Rufatto, L. C., Gower, A., Schwambach, J. & Moura, S. Genus Mikania: Chemical composition and phytotherapeutical activity. Rev. Bras. de Farmacogn. 22, 1384–1403. https://doi.org/10.1590/S0102-695X2012005000099 (2012).
Godoy, S. M. et al. Phylogenetic relationships of Brazilian Mikania species (Asteraceae, Eupatorieae) based on multilocus DNA markers. Bot. J. Linn. Soc. 184, 326–346. https://doi.org/10.1093/botlinnean/box030 (2017).
Huang, L., Wang, Z., Wang, T. & Su, Y.-J. The complete chloroplast genome sequence of Mikania micrantha (Asteraceae), a noxious invasive weed to South China. Mitochondrial DNA B 1, 603–604. https://doi.org/10.1080/23802359.2016.1209090 (2016).
Liu, B. et al. Mikania micrantha genome provides insights into the molecular mechanism of rapid growth. Nat. Commun. 11, 340. https://doi.org/10.1038/s41467-019-13926-4 (2020).
Hind, D. J. N. & Robinson, H. Tribe Eupatorieae Cass. In The Families and Genera of Vascular Plants Vol. 7 (ed. Kubitzki, K.) 510–574 (Springer, 2007).
Nie, X. et al. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 7, e36869. https://doi.org/10.1371/journal.pone.0036869 (2012).
Zhang, Y., Li, L., Yan, T. L. & Liu, Q. Complete chloroplast genome sequences of Praxelis (Eupatorium catarium Veldkamp), an important invasive species. Gene 549, 58–69. https://doi.org/10.1016/j.gene.2014.07.041 (2014).
Qiao, Z.-P., Chen, Z.-X. & Wang, Q.-Z. The complete chloroplast genome of Ageratum conyzoides (Asteraceae). Mitochondrial DNA B 4, 3342–3343. https://doi.org/10.1080/23802359.2019.1673241 (2019).
Mandel, J. R. et al. A fully resolved backbone phylogeny reveals numerous dispersals and explosive diversifications throughout the history of Asteraceae. Proc. Natl. Acad. Sci. U.S.A. 116, 14083–14088. https://doi.org/10.1073/pnas.1903871116 (2019).
Panero, J. L. et al. Resolution of deep nodes yields an improved backbone phylogeny and a new basal lineage to study early evolution of Asteraceae. Mol. Phylogenet. Evol. 80, 43–53. https://doi.org/10.1016/j.ympev.2014.07.012 (2014).
Timme, R. E., Kuehl, J. V., Boore, J. L. & Jansen, R. K. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: Identification of divergent regions and categorization of shared repeats. Am. J. Bot. 94, 302–312. https://doi.org/10.3732/ajb.94.3.302 (2007).
Lin, N. et al. Plastome sequencing of Myripnois dioica and comparison within Asteraceae. Plant Divers. 41, 315–322. https://doi.org/10.1016/j.pld.2019.07.003 (2019).
Kim, G. et al. Comparative chloroplast genome analysis of Artemisia (Asteraceae) in East Asia: Insights into evolutionary divergence and phylogenomic implications. BMC Genomics 21, 415. https://doi.org/10.1186/s12864-020-06812-7 (2020).
Loeuille, B. et al. Extremely low nucleotide diversity among thirty-six new chloroplast genome sequences from Aldama (Heliantheae, Asteraceae) and comparative chloroplast genomics analyses with closely related genera. PeerJ 9, e10886. https://doi.org/10.7717/peerj.10886 (2021).
Kim, K. J. & Lee, H. L. Widespread occurrence of small inversions in the chloroplast genomes of land plants. Mol. Cells 19, 104–113 (2005).
Jansen, R. K. & Palmer, J. D. A chloroplast DNA inversion marks an ancient evolutionary split in the sunflower family (Asteraceae). Proc. Natl. Acad. Sci. U.S.A. 84, 5818–5822. https://doi.org/10.1073/pnas.84.16.5818 (1987).
Jansen, R. K. & Palmer, J. D. Chloroplast DNA from lettuce and Barnadesia (Asteraceae): Structure, gene localization, and characterization of a large inversion. Curr. Genet. 11, 553–564. https://doi.org/10.1007/BF00384619 (1987).
Salih, R. H. M., Majeský, L., Schwarzacher, T., Gornall, R. & Heslop-Harrison, P. Complete chloroplast genomes from apomictic Taraxacum (Asteraceae): Identity and variation between three microspecies. PLoS ONE 12, e0168008. https://doi.org/10.1371/journal.pone.0168008 (2017).
Lahaye, R. et al. DNA barcoding the floras of biodiversity hotspots. Proc. Natl. Acad. Sci. U.S.A. 105, 2923–2928. https://doi.org/10.1073/pnas.0709936105 (2008).
Shaw, J. et al. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 92, 142–166. https://doi.org/10.3732/ajb.92.1.142 (2005).
Shaw, J., Lickey, E. B., Schilling, E. E. & Small, R. L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 94, 275–288. https://doi.org/10.3732/ajb.94.3.275 (2007).
Zhang, X. et al. Plastome phylogenomics of Saussurea (Asteraceae: Cardueae). BMC Plant Biol. 19, 290. https://doi.org/10.1186/s12870-019-1896-6 (2019).
Little, M. C. & Hallick, R. B. Chloroplast rpoA, rpoB & rpoC genes specify at least three components of a chloroplast DNA-dependent RNA polymerase active in tRNA and mRNA transcription. J. Biol. Chem. 263, 14302–14307 (1988).
Krawczyk, K. & Sawicki, J. The uneven rate of the molecular evolution of gene sequences of DNA-dependent RNA polymerase I of the genus Lamium L.. Int. J. Mol. Sci. 14, 11376–11391. https://doi.org/10.3390/ijms140611376 (2013).
Park, S. et al. Contrasting patterns of nucleotide substitution rates provide insight into dynamic evolution of plastid and mitochondrial genomes of Geranium. Genome Biol. Evol. 9, 1766–1780. https://doi.org/10.1093/gbe/evx124 (2017).
Smith, S., Walker-Hale, N., Walker, J. F. & Brown, J. W. Phylogenetic conflicts, combinability, and deep phylogenomics in plants. Syst. Biol. 69, 579–592. https://doi.org/10.1093/sysbio/syz078 (2020).
Walker-Hale, N., Vargas, O. M., Larson, D. A. & Stull, G. W. Characterizing gene tree conflict in plastome-inferred phylogenies. PeerJ 7, e7747. https://doi.org/10.7717/peerj.7747 (2019).
Watson, L. E., Siniscalchi, C. M. & Mandel, J. Phylogenomics of the hyperdiverse daisy tribes: Anthemideae, Astereae, Calenduleae, Gnaphalieae, and Senecioneae. J. Syst. Evol. 58, 841–852. https://doi.org/10.1111/jse.12698 (2020).
Thode, V. A., Lohmann, L. G. & Sanmartín, I. Evaluating character partitioning and molecular models in plastid phylogenomics at low taxonomic levels: A case study using Amphilophium (Bignonieae, Bignoniaceae). J. Syst. Evol. 58, 1071–1089. https://doi.org/10.1111/jse.12579 (2020).
Sayyari, E. & Mirarab, S. Fast coalescent-based computation of local branch support from quartet frequencies. Mol. Biol. Evol. 33, 1654–1668. https://doi.org/10.1093/molbev/msw079 (2016).
Walker, J. F., Brown, J. W. & Smith, S. Analyzing contentious relationships and outlier genes in phylogenomics. Syst. Biol. 67, 916–924. https://doi.org/10.1093/sysbio/syy043 (2018).
Zhang, C., Rabiee, M., Sayyari, E. & Mirarab, S. ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 19, 153. https://doi.org/10.1186/s12859-018-2129-y (2018).
McKain, M. & Afinit Mrmckain/Fast-Plast: Fast-Plast 1.2.6. Zenodo (2017). https://doi.org/10.5281/zenodo.597709.
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. https://doi.org/10.1093/bioinformatics/btu170 (2014).
Langmead, B. & Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comp. Biol. 19, 455–477. https://doi.org/10.1089/cmb.2012.0021 (2012).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. https://doi.org/10.1093/bioinformatics/btr011 (2011).
Kearse, M. et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. https://doi.org/10.1093/bioinformatics/bts199 (2012).
Wyman, S. K., Jansen, R. K. & Boore, J. L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. https://doi.org/10.1093/bioinformatics/bth352 (2004).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Lohse, M., Drechsel, O., Kahlau, S. & Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. https://doi.org/10.1093/nar/gkt289 (2013).
Amiryousefi, A., Hyvönen, J. & Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. https://doi.org/10.1093/bioinformatics/bty220 (2018).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. https://doi.org/10.1093/nar/gkf436 (2002).
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M. & Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. https://doi.org/10.1093/nar/gkh458 (2004).
Brudno, M. et al. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 19, i54–i62. https://doi.org/10.1093/bioinformatics/btg1005 (2003).
Rozas, J. et al. DnaSP v6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 34, 3299–3302. https://doi.org/10.1093/molbev/msx248 (2017).
R Development Core Team. R: A Language and Environment for Statistical Computing (2017). http://www.r-project.org (Accessed 2 January 2020)
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. https://doi.org/10.1093/molbev/msw054 (2016).
Larkin, M. A. et al. Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948. https://doi.org/10.1093/bioinformatics/btm404 (2007).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585. https://doi.org/10.1093/bioinformatics/btx198 (2017).
Kurtz, S. et al. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. https://doi.org/10.1093/nar/29.22.4633 (2001).
Stamatakis, A. RAxML Version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. https://doi.org/10.1093/bioinformatics/btu033 (2014).
Stamatakis, A. RAxML-VI-HPC, maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. https://doi.org/10.1093/bioinformatics/btl446 (2006).
Lanfear, R., Calcott, B., Ho, S. Y. W. & Guindon, S. PartitionFinder, combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29, 1695–1701. https://doi.org/10.1093/molbev/mss020 (2012).
Lanfear, R., Calcott, B., Kainer, D., Mayer, C. & Stamatakis, A. Selecting optimal partitioning schemes for phylogenomic datasets. BMC Evol. Biol. 14, 82. https://doi.org/10.1186/1471-2148-14-82 (2014).
Smith, S. A., Moore, M. J., Brown, J. W. & Yang, Y. Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evol. Biol. 15, 150. https://doi.org/10.1186/s12862-015-0423-0 (2015).
Brown, J. W., Walker, J. F. & Smith, S. A. Phyx: Phylogenetic tools for unix. Bioinformatics 33, 1886–1888. https://doi.org/10.1093/bioinformatics/btx063 (2017).
Johnson, M. (Accessed 25 March 2020); https://github.com/mossmatters/phyloscripts/tree/master/phypartspiecharts (2017).
Swofford, D. L. PAUP*: Phylogenetic Analysis Using Parsimony, version 4.0 b10 (2003).
Acknowledgements
We thank Gustavo Heiden for help during field work. This research was supported by a post-doctoral fellowship from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (PNPD-CAPES) to VAT from the Programa de Pós-Graduação em Botânica da Universidade Federal do Rio Grande do Sul (UFRGS). Additional funds were provided by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Grant ID 305776/2010-0) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, Grant IDs 2011/18385-0 and 2012/12325-9).
Author information
Authors and Affiliations
Contributions
V.A.T., C.T.O., B.L. and C.M.S. designed the study, defined sampling, obtained samples, interpreted the results, and co-wrote the manuscript. V.A.T. assembled Illumina sequences, annotated plastomes, and performed comparative and phylogenetic analyses. C.M.S. conducted phylogenetic analyses. J.R.P. co-wrote the manuscript, supervised, and obtained funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thode, V.A., Oliveira, C.T., Loeuille, B. et al. Comparative analyses of Mikania (Asteraceae: Eupatorieae) plastomes and impact of data partitioning and inference methods on phylogenetic relationships. Sci Rep 11, 13267 (2021). https://doi.org/10.1038/s41598-021-92727-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-92727-6
This article is cited by
-
Comparative chloroplast genomics and insights into the molecular evolution of Tanaecium (Bignonieae, Bignoniaceae)
Scientific Reports (2023)
-
The complete chloroplast genome of Carpesium abrotanoides L. (Asteraceae): structural organization, comparative analysis, mutational hotspots and phylogenetic implications within the tribe Inuleae
Biologia (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
