
Abstract
Phosphatidic acid phosphatase (PAP) catalyzes the dephosphorylation of phosphatidic acid (PA) yielding diacylglycerol (DAG), the lipid precursor for triacylglycerol (TAG) biosynthesis. PAP activity has a key role in the regulation of PA flux towards TAG or glycerophospholipid synthesis. In this work we have characterized two Mycobacterium smegmatis genes encoding for functional PAP proteins. Disruption of both genes provoked a sharp reduction in de novo TAG biosynthesis in early growth phase cultures under stress conditions. In vivo labeling experiments demonstrated that TAG biosynthesis was restored in the ∆PAP mutant when bacteria reached exponential growth phase, with a concomitant reduction of phospholipid synthesis. In addition, comparative lipidomic analysis showed that the ∆PAP strain had increased levels of odd chain fatty acids esterified into TAGs, suggesting that the absence of PAP activity triggered other rearrangements of lipid metabolism, like phospholipid recycling, in order to maintain the wild type levels of TAG. Finally, the lipid changes observed in the ∆PAP mutant led to defective biofilm formation. Understanding the interaction between TAG synthesis and the lipid composition of mycobacterial cell envelope is a key step to better understand how lipid homeostasis is regulated during Mycobacterium tuberculosis infection.
Similar content being viewed by others

Introduction
Tuberculosis (TB) is one of the top 10 causes of death worldwide and the leading cause of death from a single infectious agent (ranking above HIV/AIDS). In 2019, about 10 million people developed TB and 1.4 million died1. TB is caused by the bacillus Mycobacterium tuberculosis and can persist for decades within the granuloma in a dormant state referred to as latency. In this state, most bacilli are confined to the granuloma where a specific population of macrophages, known as foamy macrophages, are enriched in lipid droplets2,3. In addition, M. tuberculosis accumulates intracytoplasmic lipid inclusions (ILI) in its own cytoplasm that mainly contain triacylglycerol (TAG)4. Interestingly, TAG accumulation has been reported in several actinomycetes including the Mycobacterium genus5. ILI formation has been observed in slow- and fast-growing mycobacteria such as species from the M. tuberculosis complex6,7,8, M. leprae9, M. avium10, M. marinum11,12, M. abscessus13, M. smegmatis14,15 and M. ratisbonense16. Mycobacteria accumulate TAG in response to several growth-limiting stresses but the physiological role of this response remains unclear17,18.
De novo biosynthesis of TAG occurs by the sequential esterification of glycerol-3-phosphate producing phosphatidic acid (PA). PA is a key molecule for the biosynthesis of membrane glycerophospholipids through the synthesis of the common precursor CDP-diacylglycerol (CDP-DAG)19. However, in oleaginous bacteria PA can also be dephosphorylated by a phosphatidic acid phosphatase (PAP) to yield diacylglycerol (DAG), which is in turn acylated by the wax ester/DAG acyltransferases enzymes (WS/DGAT) to synthetize TAG20 (Supplementary Fig. S1). Thus, DAG formation is the first committed reaction of TAG biosynthesis, suggesting a key role of PAP activity in the regulation of PA flux towards TAG or membrane phospholipid biosynthesis. Remarkably, despite the relevance of PAP activity for the biosynthesis of TAGs, there are no studies addressing the physiological role of PAPs in mycobacteria.
Two families of PAP enzymes, Mg2+-dependent (PAP type 1, PAP1) and Mg2+-independent (PAP type 2, PAP2), have been characterized in higher eukaryotic cells and microorganisms21. The PAP1 family utilizes PA as a unique substrate and is localized in the soluble fraction of the cell22. In contrast, the PAP2 enzymes, also known as lipid phosphate phosphatases (LPPs), can use PA, lysophosphatidic acid (LPA), sphingosine- 1-phosphate and DAG pyrophosphate (DGPP) as substrates and are integral membrane proteins. Although PAP2 enzymes have been found both in eukaryotic and prokaryotic organisms, PAP1 enzymes are highly conserved in eukaryotes but absent in prokaryotes. Moreover, only a few PAP2-like enzymes have been characterized in prokaryotes23,24,25. The first PAP2 enzyme characterized in bacteria was PgpB of E. coli23. PgpB had a broad substrate spectrum like PGP, PA, LPA, DGPP and undecaprenyl pyrophosphate (C55-PP)23,26. Furthermore, Comba et al.25 characterized two PAP2 enzymes in Streptomyces coelicolor, Lppα and Lppβ, that showed significant PAP activity when expressed in E. coli. Analysis of mutant and overexpressing strains of these enzymes in S. coelicolor determined a direct link of these enzymes with TAG biosynthesis in oleaginous bacteria.
In this study we present the genetic and physiological characterization of two PAPs from M. smegmatis, demonstrating that msmeg_0633 (PAPα) and msmeg_0634 (PAPβ) encode PAP enzymes catalyzing the formation of DAG from PA. Furthermore, we demonstrated that the absence of PAP activity triggers changes in lipid metabolism in order to maintain TAG levels, highlighting the relevance of TAG synthesis in theses microorganisms.
Results
Identification of putative PAPs in M. smegmatis
In order to identify PAP enzymes responsible for DAG production in M. smegmatis, we performed a BLAST homology search27 over different actinomycetes genome databases using the SCO1102 amino acid sequence as query. SCO1102, named Lppα, has been previously identified and characterized as a PAP protein in S. coelicolor25. The output of this search indicated the presence of several proteins belonging to the PAP2 superfamily in mycobacteria. In particular, two putative PAPs were identified in M. smegmatis, MSMEG_0634 and MSMEG_0633. On the other hand, M. tuberculosis only revealed one PAP candidate, orthologous to MSMEG_0634. Interestingly, MSMEG_0633 protein presents a diacylglycerol kinase catalytic domain (pfam 00781) besides the PAP2 superfamily domain (pfam 01569).
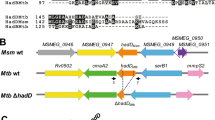
Phylogenetic analysis using curated sequences of PAP2 from several actinomycetes showed that MSMEG_0634 and Rv0308, as well as other putative PAP found in other mycobacteria, clustered with SCO1102 previously characterized in S. coelicolor25. MSMEG_0633 clustered together with other PAP proteins from Streptomyces such as SCO1047 (Fig. 1a).

Bioinformatic analysis of PAPs. (a) Phylogenetic tree of PAP enzymes from different actinomycetes. The maximum likelihood phylogenetic tree was constructed using the MEGA X software. The bootstrap percentage supports (1000 replicates) are indicated in the different branches. (b) Comparative analysis of the genomic context of putative PAP enzymes from mycobacteria and Streptomyces coelicolor. (c) Sequence alignment of PAP2 domains. The key residues for this catalytic activity are highlighted.
Using the Microbial Genome Database (MBGD) tool we analyzed the genetic environment of the coding genes for these enzymes and found that they are highly conserved in several mycobacteria such as M. avium, M. ulcerans, M. bovis and M. marinum as well as M. smegmatis and M. tuberculosis. However, this synteny was not conserved in S. coelicolor (Fig. 1b). Detailed analysis of the protein sequences found in M. tuberculosis and M. smegmatis indicated that they all conserved the key amino acids present in the PAP2 catalytic domain (Fig. 1c).
Heterologous expression of M. smegmatis PAPs enzymes results in an increase in DAG levels in E. coli
To initiate the functional characterization of MSMEG_0633 and MSMEG_0634, an N-terminal His-tag version of each gene was cloned under the control of PBAD promoter in the pBAD33 vector28. Plasmids pAC35 and pAC6 were introduced by transformation in the BL21 derivative strain MPS11, a dgk mutant heterologous expressing the DAG:acyltransferase (DGAT) SCO0958 from S. coelicolor under the control of T7 promoter29. Transformed cells were grown to mid-log phase and then cultivated for 16 h at 23 °C after induction with L-arabinose and/or IPTG. We analyzed the lipid profile of these recombinant strains by metabolic labeling with [14C]-acetate. As shown in Fig. 2a,b, we found that the expression of MSMEG_0633 and MSMEG_0634 increased the intracellular levels of DAG by 3 and fourfold, respectively compared with the same strain grown without the addition of the inducer (Fig. 2). The free fatty acid content of GG-02 and GG-03 strains expressing MSMEG_0634 and MSMEG_0633, respectively, also increased compared with the non-induced strains.

Heterologous expression of MSMEG_0633 and MSMEG_0634 proteins in E. coli. Total lipid extracts from [14C]-acetate labelled cultures of E. coli strains GG-03 (a) or GG-02 (b) were analyzed on silica gel TLC plates and developed in hexane: diethylether:acetic acid (70:30:1, v/v/v). Quantification of the radiolabelling intensity is shown for both TLC in arbitrary units (AU). The results are the mean values and standard deviation of three independent biological replicates. All results were normalized by OD600nm and total protein concentration. TAG, triacylglycerol; FFA, free fatty acids; DAG, Diacylglycerol. (+): Inducer added. (−): Without inducer; AU, arbitrary units. Statistical significance was calculated using two-way ANOVA test followed by Bonferroni correction; ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05.
The sole expression of SCO0958 in the MPS11 strain was sufficient to promote accumulation of significant levels of TAG. However, co-expression of both SCO0958 and MSMEG_0634 enzymes in strain GG-02 increased TAG production by 12-fold (Fig. 2b), suggesting that the expression of these enzymes are sufficient to synthesize de novo TAG using precursors from the glycerophospholipid metabolism of a non-oleaginous host. Altogether, our results suggest that MSMEG_0633 and MSMEG_0634 are central enzymes for DAG generation in M. smegmatis and we named them as msPAPα and msPAPβ.
Construction and analysis of single mutant strains in PAP enzymes
In order to study the physiological role of msPAPα and msPAPβ in storage lipid synthesis we generated two single knockout (KO) mutant strains in the M. smegmatis genes msPAPα and msPAPβ. For this, we constructed two pPR27-derived plasmids, using the upstream and downstream flanking fragments of each gene (Supplementary Fig. S2), and we called them pAC21 and pAC17, respectively. These vectors can be efficiently counterselected on sucrose at 42 °C. The correct double recombination events leading to the deletion of each PAP coding gene were confirmed by genomic PCR (Supplementary Fig. S2) and the resulting mutant strains named ∆PAPα (msmeg_0633 KO) and ∆PAPβ (msmeg_0634 KO), respectively.
To study the effect of each of the PAP protein depletion at different growth stages, the two mutant strains were grown in 7H9 medium and TAG accumulation analyzed by total lipid extraction and fractionation by normal-phase TLC. Both strains showed exponential growth curves comparable with the wild type. However, ∆PAPα presented a longer lag phase and a delayed entry in the exponential phase (Supplementary Fig. S3). None of the single mutants showed reduction in TAG accumulation under these conditions and surprisingly, ∆PAPβ was able to accumulate even more TAG during exponential phase compared with the wild type strain.
PAP depletion results in reduced TAG biosynthesis in early growth phase under stress conditions
To evaluate if a compensation effect was arising in the single mutant strains, we constructed a double mutant strain. Disruption of msPAPα and msPAPβ genes was obtained by a two-step allelic exchange using plasmid pAC20. The correct homologous recombination event was confirmed by PCR (Supplementary Fig. S4) and the resulting strain was named ∆PAP. TAG accumulation was analyzed by total lipid extraction and fractionation by normal-phase TLC. No significant differences in TAG levels were observed when the ∆PAP strain was grown in 7H9 medium (Supplementary Fig. S5). However, under nitrogen-starving conditions, where TAG synthesis is induced, the ∆PAP double mutant presented a longer lag phase as well as a delay in entering into the exponential phase. In addition, de novo TAG biosynthesis was analyzed by metabolic labeling with [14C]-acetate and a significant reduction in TAG content in early exponential growth phase (T1) was observed (Fig. 3). However, during exponential phase, this phenotype was reverted, and the double mutant strain was able to synthetize even more TAG than the wild type strain.

Growth curve and TAG production of M. smegmatis ∆PAP in low ammonium conditions. (a) Growth curves of WT and ∆PAP strains in Sauton medium supplemented with 1 mM NH4Cl. Growth was followed by measuring optical density (OD600nm). (b) Cells from WT and ∆PAP cultures were labelled with [14C]-acetate at the same physiological state: early (T1, OD600nm ~ 0.25), mid-exponential (T2, OD600nm ~ 0.5) and stationary (T3, OD600nm ~ 1.6) phase for 1 h at 37 °C. After organic extraction, 14C-labelled lipids were separated by TLC. Optical density standardization was performed for lipid extraction. (c) Quantification of the radiolabelling intensity of the TLC showed in panel (b) is shown in arbitrary units (AU). The results are the mean values and standard deviation of three independent biological replicates. Solvent system: hexane:diethylether:acetic acid (70:30:1, v/v/v). DAG, Diacylglycerol; TAG, triacylglycerol; WT, M. smegmatis mc2155; AU, arbitrary units. Statistical significance was calculated using two-way ANOVA test followed by Bonferroni correction; ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05. The full-length image corresponding to (b) is shown in Figure S6.
In order to analyze if the total TAG content was affected under a different stress condition, the wild type and ∆PAP strains were grown in 7H9 medium at pH 7 and pH 5. Our results indicated that the ∆PAP strain was impaired in TAG biosynthesis at acidic pH during early exponential phase (T1) compared with the wild type strain (Fig. 4). These observations support the idea that at pH 7, DAG biosynthesis could arise from alternative pathways complementing the absence of PAP activity.

Growth curve and TAG production of M. smegmatis ∆PAP at acidic stress. (a) Growth curves of WT and ∆PAP strains in 7H9 medium supplemented with glycerol and tyloxapol (pH 7 and 5.4). Growth was followed by measuring optical density (OD600nm). (b) Lipids from WT and ΔPAP cells were extracted at the same physiological state at early exponential phase (T1, OD600nm ~ 0.25) and then analyzed by TLC. (c) Quantification of the intensity of the spots present in the TLC showed in panel (b). The results are the mean values and standard deviation of three independent biological replicates. All results were normalized by OD600nm and total protein concentration. Solvent system: hexane:diethylether:acetic acid (70:30:1, v/v/v). TAG, triacylglycerol; WT, M. smegmatis mc2155; AU, arbitrary units. Statistical significance was calculated using t-test analysis ; ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05.
Absence of PAP enzymes has a global impact on lipid composition in M. smegmatis
Bacteria produce glycerophospholipids exclusively through the CDP-DAG-dependent pathway30,31, making PA a key metabolic branch point between glycerophospholipid and TAG synthesis (Supplementary Fig. S1). To start evaluating the impact of the deletion of the pap genes on glycerophospholipid biosynthesis and on the whole composition of the cell envelope, cultures of the wild type and ∆PAP strains were grown and labeled with [14C]-acetate at early exponential, exponential, and stationary growing phases in 7H9 medium at pH 7 and pH 5.4. The lipid content was extracted from each strain and analyzed by TLC. The results shown in Fig. 5 indicated that the ΔPAP strain synthesized reduced levels of glycopeptidolipids and phospholipids (PE and CL) during early exponential phase (T1) at both pH. Additionally, an increase in trehalose monomycolate (TMM) synthesis was observed (Fig. 5). At neutral pH, phospholipid and TMM synthesis was similar in the wild type and ∆PAP strains in the stationary phase of growth (T3). In contrast, TMM synthesis was still higher in the mutant strain at pH 5.4, (Fig. 5). These results suggested that in the absence of PAP activity there is a metabolic rearrangement that redirects the carbon flux towards TAG synthesis in detriment of phospholipids synthesis and prompted us to perform a comparative lipidomic analysis between the ΔPAP and the wild type strains using Mass Spectrometry (MS). Briefly, lipids were extracted from exponential phase cultures of both strains grown in 7H9 medium at pH 7 and pH 5.4. As expected, the ΔPAP strain did not present significant changes in TAG and DAG content at exponential growth phase at both pH analyzed. However, detailed analysis of the fatty acyl composition of TAG revealed that the ΔPAP strain presented a relative increase in the amount of odd-chain FA esterified into TAG compared to the wild type strain (Figs. 6a and 7a), although the weighted average of the number of carbons, both at neutral pH and at acidic pH remained constant in the mutant strain (Figs. 6b and 7b). Furthermore, this difference was specifically related with an increase in fatty acids C15, C17 and C19 (Fig. 6c,d and 7c,d). These results indicated that although the total content of TAG is similar in the wild type and ∆PAP strains, the composition is different, suggesting that the precursors used for the synthesis of TAG in the mutant strain could come from an alternative pathway.

Lipid analysis of the envelope of M. smegmatis ΔPAP strain. (a) Cells from WT and ∆PAP cultures grown in 7H9 medium (pH 7 and 5.4) were labelled with [14C]-acetate at the same physiological state at early (T1, OD600nm ~ 0.25), mid-exponential (T2, OD600nm ~ 0.5) and stationary (T3, OD600nm ~ 1.5) phase for 1 h at 37 °C. Cultures were normalized by OD and after organic extraction; 14C-labelled lipids were separated by TLC. (b) and (c) Quantification of the intensity of the spots of the TLC showed in panel (a). The results are the mean values and standard deviation of three independent biological replicates. Solvent system: chloroform:methanol:water (20:4:0,5, v/v/v). GPL, glycopeptidolipids; TDM, trehalose dimycolate; TMM, trehalose monomycolate; PE, phosphatylethanol-amine; CL, cardiolipin; WT, M. smegmatis mc2155; AU, arbitrary units. Statistical significance was calculated using two-way ANOVA test followed by Bonferroni correction; ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05.

TAG composition of M. smegmatis WT and ΔPAP strains grown in 7H9 medium at pH 7. (a) Heatmap of the fatty acyl chain length variations in TAG extracted at exponential phase (T2, OD600nm ~ 0.5) from cultures of the WT and ΔPAP strains grown in 7H9 medium at pH 7. (b) Weighted average of numbers of carbon in TAG’s chain. (c) Relative abundance of TAG with even fatty acid chain. (d) Relative abundance of TAG with odd fatty acid chain. WT, M. smegmatis mc2155 wild type strain. The results are the mean values and standard deviation of three independent biological replicates (WT1, WT2, WT3 and ΔPAP1, ΔPAP2, ΔPAP3).

TAG composition of M. smegmatis WT and ΔPAP strains grown in 7H9 medium at pH 5. (a) Heatmap of fatty acid chain length variations in TAG extracted at exponential growth phase (T2, OD600nm ~ 0.5) from cultures of the WT and ΔPAP strains grown in 7H9 medium at pH 5. (b) Weighted average of numbers of carbon in TAG’s chain. (c) Relative abundance of TAG with even fatty acid chain. (d) Relative abundance of TAG with odd fatty acid chain. WT, M. smegmatis mc2155 wild type strain. The results are the mean values and standard deviation of three independent biological replicates (WT1, WT2, WT3 and ΔPAP1, ΔPAP2, ΔPAP3).
∆PAP mutant forms defective biofilms in liquid–air interfaces
A number of components of the mycobacterial cell wall such as glycopeptidolipids, short chain mycolic acids and monomeromycolyl DAG have been shown to play an important role in formation of pellicle biofilms32,33. Even subtle defects in cell wall components can modify the surface properties of individual cells, alter cell-to-cell interactions, and ultimately give rise to colonies with visibly different morphologies34. In this regard, we analyzed if the changes in the lipid composition that we observed in the ∆PAP mutant strain had impact on biofilm formation. For this, cultures of the wild type and ΔPAP strains were grown in modified M63 medium35 in Petri dishes at 30 °C without shaking. As shown in Fig. 8a, the ∆PAP strain formed defective pellicles. Another way to study biofilm formation is the analysis of macrocolonies. They can present different morphologies, from smooth and opaque to rough and transparent36 depending not only on the growth conditions but also in the composition of the extracellular matrix and the cell wall. In this sense, we analyzed colony morphology of the wild type and ∆PAP mutant strain in 7H9-agar medium supplemented with Congo red. The plates were incubated for one week at 37 °C. As shown in Fig. 8b, in 7H9 medium at pH 7 and under nitrogen-starving conditions, the ∆PAP mutant exhibited smooth colony morphology, being unable to form cords in contrast with the opaque and rough colony morphology of the wild type strain. Interestingly, in the presence of acidic stress (pH 5.4) or low concentration of NH4+, both strains formed smaller colonies than those obtained at pH 7 and the ∆PAP colony was even smaller under acidic stress.

Analysis of biofilms formed in air–liquid interface by the WT and ΔPAP mutant strains. (a) WT and ∆PAP strains were incubated in Petri dishes containing a modified version of the M63 medium without shaking at 30 °C for 5 days. (b) Colonies of WT and ΔPAP strains grown in 7H9 medium (pH 7 and pH 5.4) and Sauton 1 mM NH4Cl during one week at 37° C. The images were acquired with the Olympus MVX10 magnifying glass (0,65X). WT, M. smegmatis mc2155 wild type strain. The results are representative of three independent biological experiments.
Discussion
In this work, we describe the identification and physiological characterization of the first PAP enzymes involved in TAG biosynthesis in mycobacteria. By means of a comprehensive bioinformatic analysis of the M. smegmatis genome, we were able to identify two candidate genes encoding for putative PAP: msmeg_0633 and msmeg_0634. The functional role of these proteins was analyzed through the heterologous expression in E. coli. We observed increased levels of intracellular DAG of up to three and fourfold for MSMEG_0633 and MSMEG_0634, respectively (Fig. 2). Moreover, co-expression of the S. coelicolor DGAT enzyme SCO0958 together with MSMEG_0633 in E. coli reconstituted the complete pathway for de novo TAG biosynthesis in a heterologous host. Altogether, these results indicated that MSMEG_0633 and MSMEG_0634 are phosphatase enzymes able to catalyze in vivo the formation of DAG from intracellular PA or other analogous lipid of the host cell and we named them msPAPα and msPAPβ.
We found that none of the two pap genes were essential for survival of M. smegmatis and disruption of either PAPα or PAPβ caused no alteration in the intracellular levels of DAG/TAG. These results suggested that there may be some redundancy in the activity of both enzymes, and that one could complement the deficiency of the other, similar to what had been previously reported in S. coelicolor25. In this bacterium, growth and DAG/TAG synthesis of the single PAP mutants was indistinguishable from the wild type strain. However, a double mutant strain in the PAP enzymes from S. coelicolor showed reduced levels of TAG accumulation in exponential phase, although upon reaching the stationary phase, the levels were restored to that of the wild type strain25. Moreover, the effect on gene redundancy had previously been observed in other enzymes that participate in the TAG biosynthesis pathway. This is the case of the Tgs enzymes in M. tuberculosis, where a mutant in the tgs1 gene, whose protein product provides the highest TAG synthase activity, led to a drastic decrease in the synthesis of TAG under conditions of hypoxia, acidic stress and NO. However, traces of TAG synthesis were observed in all these conditions37.
We found that M. smegmatis mc2155 is able to synthesize large amounts of TAGs during the exponential growth phase when grown in 7H9 liquid medium with glycerol as carbon source. At stationary phase we were not able to detect TAG accumulation. However, when M. smegmatis was grown in nitrogen limiting conditions, de novo synthesis of TAG was maintained during all stages of growth and this was correlated with a higher accumulation of the reserve compound during the stationary phase. Recently, Santucci et al. reported that the availability of nitrogen and carbon are two factors that influence both the production of TAG and its accumulation in the form of ILI in M. smegmatis38. However, they found that both in rich medium and in nitrogen-limiting conditions M. smegmatis accumulate TAG in stationary phase, and that TAG accumulation was dependent on the concentration of glycerol. The discrepancies with our results could be related with the minimum glycerol concentration used, which was fivefold higher than that used in this work.
Interestingly the double mutant strain ∆PAP, synthesized similar levels of DAG/TAG as the wild type strain (Supplementary Fig. 5S). However, growth and TAG accumulation of the double mutant under nitrogen limiting conditions or acidic stress showed several differences both in growth rate and TAG biosynthesis (Figs. 3 and 4). Moreover, the ΔPAP strain synthesized reduced levels of glycopeptidolipids and phospholipids and increased levels of TMM. While TMM is most likely the form under which mycolic acids are exported to the cell wall and outer membrane, glycopeptidolipids are present in the outer leaflet of M. smegmatis and are important in biofilm formation, aggregation, motility and cell wall integrity39. Altogether, the results suggest that under stress conditions other lipid biosynthetic pathways are altered in order to provide DAG for TAG biosynthesis (Fig. 5). Besides PAP enzymes, there are different alternatives for the production of DAG in bacteria. The experience with the E. coli model suggests that DAG accumulation is deleterious to bacterial growth and DAG kinases are key enzymes in lipid metabolism that phosphorylate DAG formed by the turnover of membrane phospholipids. For example, in E. coli, the phospholipid phosphatidylglycerol (PG) is degraded to DAG by the transfer of the glycerol-1-phosphate head group to membrane-derived oligosaccharides (MDO), that function in osmotic homeostasis40,41. In this case, the DAG formed is converted to phosphatidic acid for the resynthesis of PG by the enzyme DgkA42. In gram positive bacteria like B. subtilis; PG is the source of the glycerol-1-phosphate groups needed for lipoteichoic acid (LTA). LTA is a zwitterionic polymer found in the cell wall of many Gram-positive bacteria, consisting of a polyglycerolphosphate (PGP) chain that is linked via a glycolipid anchor to the bacterial membrane43. PG turnover is rapid in these bacteria and the large amount of DAG formed requires a DagK for its efficient reintroduction into the phospholipid biosynthetic pathway44. It is interesting to note that recent literature has highlighted the presence of a PGP-type LTA in various Actinobacteria. However, these bacteria appear to lack LTA synthase enzyme (LtaS) homologues and, therefore, it has been suggested that the polymer is synthesized by an alternative pathway45. Interestingly, although the double mutant strain accumulated similar levels of DAG/TAG compared to the wild type strain, detailed analysis of TAGs indicated that the ΔPAP strain presented a relative increase in the amount of odd-chain TAG compared to the wild type strain. This difference was specifically related to increased levels of C15, C17 and C19 fatty acids in the TAG of the mutant strain (Figs. 6 and 7). Mycobacteria contain high levels of methyl branched fatty acids within the cell envelope46,47, being the most abundant the tuberculostearic acid48. This fatty acid consists of a C19 monomethyl-branched stearic acid and is mainly found in phospholipids, attached by covalent bonds to lipid virulence determinants such as phosphatidylinositolmannosides (PIMs), lipoarabinomannan (LAM), and related cell wall glycolipids49. Altogether, these observations support an active role for the PAP enzymes during stress and strongly suggest that carbon flux in the ∆PAP mutant is being redirected from the synthesis of phospholipids towards the synthesis of TAG.
The ability of environmental mycobacteria to form biofilm has been widely described in many mycobacteria species including M. avium50, M. fortuitum51, M. marinum52 and M. smegmatis32. It was also observed in subsequent studies that some mycobacteria can develop structures not only on surfaces, but also at the air–liquid interface33,53. This phenomenon can be explained by the composition of the extracellular matrix of the biofilm and the unique characteristic of the mycobacterial cell wall, especially the presence of a large number of lipids. Furthermore, it has been observed that M. smegmatis is able to form well organized colonies which have been described as a type of biofilm. These colonies are composed of cells encapsulated by a large amount of exopolysaccharides54. In both cases, the components of the cell wall play a central role both in the initial formation of the biofilm32 as well as in its maturation55. Additionally, high amounts of DAG and TAG have been found in the outer membrane of M. smegmatis56 but it is unknown if they play a role in the formation of these structures. In this work we found that the ∆PAP strain formed a defective biofilm and exhibited smooth colony morphology in contrast with the opaque and rough colony morphology of the wild type strain (Fig. 8). In mycobacteria, mycolic acids can also be found as mycolyl-diacylglycerols (MDAG), in which a mycolic acid (or mycolate) is esterified to a glycerol moiety. Based on our results, one possible explanation is that upon PAP depletion, the DAG necessary for TAG biosynthesis could arise from the recycling of MDAG. Chen et al. showed that reduction of MDAG biosynthesis resulted in the accumulation of TAGs, suggesting a metabolic relationship between these two molecules57. MDAG play a role in colony morphology and biofilm formation of mycobacteria. Loss of hydrophobic MDAGs is consistent with a smooth colony phenotype on solid media and defect in pellicle formation in liquid media.
It is known that during infection M. tuberculosis faces variable environments. For instance, the pH is highly dynamic and can fluctuate depending on the activation state of macrophages58 or within the granulomas, with a mean value of pH 5.559. Deb et al. observed that when acidic stress is combined with other types of stress such as hypoxia or low availability of nutrients, M. tuberculosis accumulates large amounts of TAG6. However, the effect of acidic stress has not been described either in the ability to form macrocolonies or in the synthesis of TAG in M. smegmatis. In this study we found that both, the wild type and ∆PAP mutant strains formed smaller colonies when they were grown under stress conditions such as limiting nitrogen concentration or acidic pH (Fig. 8).
The identification of msPAPα and msPAPβ completes the minimal set of enzymes together with the redundant set of DGATs previously characterized13,17,37,60,61 required for de novo TAG biosynthesis in mycobacteria. ILI formation in M. tuberculosis has been described as an adaptation strategy promoting survival during periods of non-replicating persistence in vitro but also in vivo, mainly regulated by the dormancy regulon18,62. Overall, our results provide new elements for the study of TAG biosynthesis and regulation in mycobacteria.
Methods
Bacterial strains, culture, and transformation conditions
The strains and plasmids used in this study are described in Supplementary Tables S1 and S2. The E. coli strain DH5α63 was used for routine sub-cloning and was transformed according to Sambrook et al.64. E. coli strains were grown in Luria–Bertani (LB) medium at 37 °C and supplemented when needed with the following antibiotics: 100 µg ml−1 amplicillin (Ap), 50 µg ml−1 kanamycin (Km), 20 µg ml−1 chloramphenicol (Cm) or 100 µg ml−1 hygromycin (Hyg). M. smegmatis mc2155 is an electroporation-efficient mutant of mc2665. Liquid cultures of M. smegmatis mc2155 (WT), ∆PAPα and ∆PAPβ, and ∆PAP were grown at 37 °C in Middlebrok 7H9 or modified Sauton medium containing (g l−1) K2HPO4, 0.5; MgSO4, 0.5; ferric citrate, 0.05 g; ZnSO4, 0.001; citric acid, 2, supplemented with 0.2% glycerol and 0.03% Tyloxapol. When required, gentamicin 20 mg ml−1 was added to the medium.
Cultures were started with 1/200 dilutions from pre-made cultures with an initial OD of 1; except for the experiment shown in Fig. 4 where 1/100 dilutions were used for the cultures grown at pH 5.4.
For biofilm cultures grown on liquid medium, 10 ml of biofilm medium (a modified version of M6335) was inoculated with 10 ml of a saturated culture in a 60 × 15 mm2 polystyrene Petri dish and incubated at 30 °C (unless otherwise stated) without disturbance.
DNA manipulation, plasmid construction and mutant generation
Isolation of plasmid DNA, restriction enzyme digestion and agarose gel electrophoresis were carried out by conventional methods64. Genomic DNA of M. smegmatis was obtained as described previously66.
pAC6
msmeg_0634 was PCR-amplified from genomic DNA of M. smegmatis mc2155 using the oligonucleotides ACA-003 to introduce an NdeI site at the translational start codon of msmeg_0634 gene, and ACA-004 to introduce an EcoRI site at the end of the ORF. To generate a msmeg_0634 His tag fusion gene, the PCR product was digested with NdeI and EcoRI and cloned into NdeI/EcoRI cleaved pET-28a(+), yielding pAC5. Finally, pAC5 was digested with XbaI and HindIII and cloned into XbaI/HindIII cleaved pBAD33, yielding pAC6.
pAC17
For the construction of the M. smegmatis ∆PAPβ mutant strain, the upstream region of msmeg_0634 gene was amplified with the primers ACA-005 and ACA-006. The 866 bp PCR product was cloned into pCR BluntII TOPO (Invitrogen) yielding pAC2 plasmid. The downstream region of msmeg_0634 gene was amplified with the primers ACA-007 and ACA-008. The 1210 bp PCR product was cloned into pCR BluntII TOPO (Invitrogen) yielding plasmid pAC3. Plasmids pAC2 and pAC3 were digested with XbaI/NdeI or NdeI/SpeI, respectively, and the fragments obtained were cloned into SpeI cleaved pPR27 plasmid, yielding pAC17.
pAC20
For the construction of the M. smegmatis ∆PAP mutant strain the upstream region of msmeg_0633 gene was amplified with the primers ACA-013 and ACA-014. The 942 bp PCR product was cloned into pGEM-T Easy (Promega) and digested with NdeI/SpeI. The fragment was inserted into NdeI/SpeI cleaved pAC2, yielding pAC19. Finally, the pAC19 was digested with SpeI and the ∆PAP fragment was inserted into SpeI cleaved pPR27 plasmid, yielding pAC20.
pAC21
For the construction of the M. smegmatis ∆PAPα mutant the upstream region of msmeg_0633 gene was amplified with the primers ACA-013 and ACA-014 and the downstream region with the primers ACA-015 y ACA-016. The 942 pb and 1069 pb PCR products, were cloned into pGEM-T Easy (Promega) yielding pAC15 and pAC16 plasmids, respectively. pAC15 was digested with PstI/NdeI and the 966 pb fragment was cloned into PstI/NdeI cleaved pAC16, yielding pAC18. Finally, pAC18 was digested with SpeI and the ∆ms0633 fragment was inserted into SpeI cleaved pPR27, yielding pAC21.
pAC35
msmeg_0633 was PCR-amplified from genomic DNA of M. smegmatis mc2155 using the primers ACA-011 to introduce an NdeI site at the translational start codon of msmeg_0633 gene, and ACA-012 to introduce an EcoRI site at the end of the ORF. To generate a msmeg_0634 His tag fusion gene, the PCR product was digested with NdeI and EcoRI and cloned into NdeI/EcoRI cleaved pET-28a(+), yielding pAC5. Finally, pAC5 was digested with XbaI and HindIII and cloned into XbaI/HindIII cleaved pBAD33, yielding pAC6.
Construction of ∆PAPα, ∆PAPβ and ∆PAP double mutant strains of M. smegmatis
For the construction of the M. smegmatis mutants ∆PAPα, ∆PAPβ, and ∆PAP the plasmids pAC21, pAC17 or pAC20 were used to transform M. smegmatis cells. Gm-resistant and XylE+ transformants were analyzed by colony PCR and those that presented a legitimate recombination event in the chromosomal copy of the respective gene were selected and grown at 37 °C and plated at 42 °C in the presence of sucrose to promote a second recombination event, resulting in the replacement of the wild type copy of the genes for the truncated versions. The correct double recombination events present in the mutant strains were confirmed by PCR.
Recombinant protein expression in E. coli and lipid analysis
For the expression of heterologous proteins in E. coli, the GG-02 and GG-03 strains derived from MPS11 were grown in LB media at 37 °C until DO600nm 0.6. PAP protein expression was induced by addition of l-arabinose 0.2% and cultures were grown overnight at 23 °C. For SCO0958-MS0633 or SCO0958-MS0634 co-expression experiments, cells at OD600nm 0.6 were induced by l-arabinose 0.2% and IPTG 0.1 mM and grown overnight at 23 °C. For lipid analysis, cultures were incubated for 1 h with 3 µCi of [14C]-acetate and total lipids were extracted as described by Bligh & Dyer67.
The lipid extracts were dried and analyzed by TLC on silica gel 60 F254 plates (0 ± 2 mm, Merck), using the solvent system hexane/diethylether/acetic acid (70:30:1, v/v/v)68. For 14C labeled lipids the radioactivity incorporated into each lipid fraction was quantified using Typhoon FLA 7000 (GE Healthcare) and the corresponding spots were quantified using ImageJ software (version 1.52a).
Lipids analysis
Lipid analysis using thin layer chromatography (TLC)
For lipid extraction, the volume of each sample was normalized by OD600nm, e.g. 10 ml of a culture of OD600nm 0.5 was collected by centrifugation and lipids were extracted as described by Bligh & Dyer67. The lipid extracts were dried and resuspended in 50 µl of chloroform and 10 µl were analyzed by TLC on silica gel 60 F254 plates (0 ± 2 mm, Merck). For TAG analysis, the solvent system used was hexane/ diethylether/acetic acid (70: 30: 1, v/v/v) and for phospholipid and cell wall component analyses the solvent system was chloroform/methanol/water (20:4:0.5, v/v/v). Lipid fractions were visualized by Cu-phosphoric staining and identified by comparing to the mobility of known standards. For 14C labeled lipids, the radioactivity incorporated into each lipid fraction was quantified using Typhoon ™ FLA 7000 laser scanner (GE Healthcare) and the corresponding spots were quantified using ImageJ software (version 1.52a).
LC–MS-MS lipid analysis
Lipids were extracted from 2 mg of lyophilized cells using a biphasic separation with MTBE, methanol and water69. Briefly, 500 µl of ice-cold methanol were added to the cells and the mixture was vortexed for 20 s. Then, 2000 µl of methyl tert-butyl ether (MTBE) were added and the mixture was incubated under agitation for 30 min at 4 °C. After addition of 500 µl of water, samples were vortexed for 1 min and centrifuged at 14,000×g for 10 min at room temperature. The upper phase containing the lipids was collected and dried down under nitrogen. The dry extracts were reconstituted with 300 μL of 9:1 methanol:toluene with 10 mM of ammonium acetate and centrifuged at 14,000×g for 5 min before analysis by MS. Water extracted using the same protocol was used as a blank control.
Flow injection analysis (FIA) was performed at 7 μl min−1 using a Nexera X2 UHPLC system (Shimadzu) and 1:1 dichloromethane:methanol with 10 mM of ammonium acetate as running solution. Lipids were analyzed using a QTRAP 5500 mass spectrometer (Sciex).
Lipid molecular species were relatively quantified by MRM in positive and negative ionization mode. Lipid classes were quantified using the following ion forms: [M+NH4] + DAG, MAG and TAG, [M−H]− for phospholipids and FFA. Source and gas setting were as follow: Curtain Gas = 17, CAD Gas = Medium, Ion Spray Voltage = 4.1 kV in positive mode and − 2.5 kV in negative mode, Temperature = 200 °C, Nebulizing Gas = 17 and Heater Gas = 25.
Statistical analysis
The statistical analyses were made using GraphPad Prism software. Data are reported as arithmetic means of the results obtained from three independent experiments ± standard deviations. Statistical significance was calculated using t-test or ANOVA test followed by Bonferroni correction as described in the figure legends. Statistical significance was accepted at p < 0.05.
Curve fitting was done using the following equation and parameters:
Parameters: t1/2 is the time that gives an OD at half way between OD0 (initial OD) and ODf (final OD).
References
Global Tuberculosis Report 2020. Licence: CC BY-NC-SA 3.0 IGO (World Health Organization, 2020).
Russell, D. G., Cardona, P., Kim, M. & Allain, S. Foamy macrophages and the progression of the human TB granuloma. Nat. Immunol. 10, 943–948 (2010).
Peyron, P. et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 4, 1–14 (2008).
Neyrolles, O. Mycobacteria and the greasy macrophage: Getting fat and frustrated. Infect. Immun. 82, 472–475 (2014).
Alvarez, H. M. Triacylglycerol and wax ester-accumulating machinery in prokaryotes. Biochimie 120, 28–39 (2016).
Deb, C. et al. A novel in vitro multiple-stress dormancy model for mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS ONE 4, e6077 (2009).
Bouzid, F. et al. Mycobacterium canettii Infection of Adipose Tissues. Front. Cell. Infect. Microbiol. 7, 189 (2017).
Kai, L. L. et al. Triacylglycerol utilization is required for regrowth of in vitro hypoxic nonreplicating Mycobacterium bovis bacillus Calmette-Guerin. J. Bacteriol. 191, 5037–5043 (2009).
Mattos, K. A. et al. Mycobacterium leprae intracellular survival relies on cholesterol accumulation in infected macrophages: A potential target for new drugs for leprosy treatment. Cell. Microbiol. 16, 797–815 (2014).
Caire-Brändli, I. B. et al. Reversible lipid accumulation and associated division arrest of mycobacterium avium in lipoprotein-induced foamy macrophages may resemble key events during latency and reactivation of tuberculosis. Infect. Immun. 82, 476–490 (2014).
Barisch, C. & Soldati, T. Breaking fat! How mycobacteria and other intracellular pathogens manipulate host lipid droplets. Biochimie https://doi.org/10.1016/j.biochi.2017.06.001 (2017).
Barisch, C., Paschke, P., Hagedorn, M., Maniak, M. & Soldati, T. Lipid droplet dynamics at early stages of Mycobacterium marinum infection in Dictyostelium. Cell. Microbiol. 17, 1332–1349 (2015).
Viljoen, A., Blaise, M., de Chastellier, C. & Kremer, L. MAB_3551c encodes the primary triacylglycerol synthase involved in lipid accumulation in Mycobacterium abscessus. Mol. Microbiol. 102, 611–627 (2016).
Garton, N. J., Christensen, H., Minnikin, D. E., Adegbola, R. A. & Barer, M. R. Intracellular lipophilic inclusions of mycobacteria in vitro and in sputum. Microbiology 148, 2951–2958 (2002).
Weir, M. P., Langridge, W. H. & Walker, R. W. Relationships between oleic acid uptake and lipid metabolism in Mycobacterium smegmatis. Am. Rev. Respir. Dis. 106, 450–457 (1972).
Silva, R. A., Grossi, V. & Alvarez, H. M. Biodegradation of phytane (2,6,10,14-tetramethylhexadecane) and accumulation of related isoprenoid wax esters by Mycobacterium ratisbonense strain SD4 under nitrogen-starved conditions. FEMS Microbiol. Lett. 272, 220–228 (2007).
Daniel, J. et al. Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture. J. Bacteriol. 186, 5017–5030 (2004).
Baek, S. H., Li, A. H. & Sassetti, C. M. Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PLoS Biol. 9, e1001065 (2011).
Parsons, J. B. & Rock, C. O. Bacterial lipids: Metabolism and membrane homeostasis. Prog. Lipid Res. 52, 249–276 (2013).
Jaiyanth, D. et al. Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in mycobacterium tuberculosis as it goes into a dormancy-like state in culture. Microbiology 186, 5017–5030 (2004).
Kocsis, M. G. & Weselake, R. J. Phosphatidate phosphatases of mammals, yeast, and higher plants. Lipids 31, 785–802 (1996).
Carman, G. M. & Han, G. S. Roles of phosphatidate phosphatase enzymes in lipid metabolism. Trends Biochem. Sci. 31, 694–699 (2006).
Dillon, D. A. et al. The Escherichia coli pgpB gene encodes for a diacylglycerol pyrophosphate phosphatase activity. J. Biol. Chem. 271, 30548–30553 (1996).
Carman, G. M. Phosphatidate phosphatases and diacylglycerol pyrophosphate phosphatases in Saccharomyces cerevisiae and Escherichia coli. Biochim. Biophys. Acta 1348, 45–55 (1997).
Comba, S., Menendez-Bravo, S., Arabolaza, A. & Gramajo, H. Identification and physiological characterization of phosphatidic acid phosphatase enzymes involved in triacylglycerol biosynthesis in Streptomyces coelicolor. Microb. Cell Fact. 12, 9 (2013).
Lu, Y. H., Guan, Z., Zhao, J. & Raetz, C. R. H. Three phosphatidylglycerol-phosphate phosphatases in the inner membrane of Escherichia coli. J. Biol. Chem. 286, 5506–5518 (2011).
BLAST: Basic Local Alignment Search Tool [http://www.ncbi.nlm.nih.gov/BLAST/].
Guzman, L. M., Belin, D., Carson, M. J. & Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose P(BAD) promoter. J. Bacteriol. 177, 4121–4130 (1995).
Arabolaza, A., Rodriguez, E., Altabe, S., Alvarez, H. & Gramajo, H. Multiple pathways for triacylglycerol biosynthesis in Streptomyces coelicolor. Appl. Environ. Microbiol. 74, 2573–2582 (2008).
Kalscheuer, R. Handbook of hydrocarbon and lipid microbiology. Handb. Hydrocarb. Lipid Microbiol. https://doi.org/10.1007/978-3-540-77587-4 (2010).
Zhang, Y.-M. & Rock, C. O. Membrane lipid homeostasis in bacteria. Nat. Rev. Microbiol. 6, 222–233 (2008).
Recht, J., Martínez, A., Torello, S. & Kolter, R. Genetic analysis of sliding motility in Mycobacterium smegmatis. J. Bacteriol. 182, 4348–4351 (2000).
Sambandan, D. et al. Keto-Mycolic acid-dependent pellicle formation confers tolerance to drug-sensitive Mycobacterium tuberculosis. MBio 4, 1–10 (2013).
Alexander, D. C., Jones, J. R., Tan, T., Chen, J. M. & Liu, J. PimF, a mannosyltransferase of mycobacteria, is involved in the biosynthesis of phos- phatidylinositol mannosides and lipoarabinomannan. J. Biol. Chem. 279, 18824–18833 (2004).
Recht, J. & Kolter, R. Glycopeptidolipid acetylation affects sliding motility and biofilm formation in Mycobacterium smegmatis. J. Bacteriol. 183, 5718–5724 (2001).
Torrelles, J. B. et al. Characterization of virulence, colony morphotype and the glycopeptidolipid of Mycobacterium avium strain 104. Tuberculosis 82, 293–300 (2002).
Sirakova, T. D. et al. Identification of a diacylglycerol acyltransferase gene involved in accumulation of triacylglycerol in Mycobacterium tuberculosis under stress. Microbiology 152, 2717–2725 (2006).
Santucci, P. et al. Nitrogen deprivation induces triacylglycerol accumulation, drug tolerance and hypervirulence in mycobacteria. Sci. Rep. 9, 1–15 (2019).
Batt, S. M., Minnikin, D. E. & Besra, G. S. The thick waxy coat of mycobacteria, a protective layer against antibiotics and the host’s immune system. Biochem. J. 447, 1983–2006 (2020).
Goldberg, D. E., Rumley, M. K. & Kennedy, E. P. Biosynthesis of membrane-derived oligosaccharides: A periplasmic phosphoglyceroltransferase. Proc. Natl. Acad. Sci. U. S. A. 78, 5513–5517 (1981).
Kennedy, E. P. Osmotic regulation and the biosynthesis of membrane derived oligosaccharides in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 79, 1092–1095 (1982).
Raetz, C. R. H. & Newman, K. F. Diglyceride kinase mutants of Escherichia coli: Inner membrane association of 1,2-diglyceride and its relation to synthesis of membrane-derived oligosaccharides. J. Bacteriol. 137, 860–868 (1979).
Fischer, W. Bacterial phosphoglycolipids and lipoteichoic acids. In Handbook of Lipid Research Vol. 6 123–234 (Plenum Press, 1990).
Jerga, A., Lu, Y.-J., Schujman, G. E., de Mendoza, D. & Rock, C. O. Identification of a soluble diacylglycerol kinase required for lipoteichoic acid production in Bacillus subtilis. J. Biol. Chem. 282, 21738–21745 (2007).
Rahman, O., Dover, L. G. & Sutcliffe, I. C. Lipoteichoic acid biosynthesis: Two steps forwards, one step sideways?. Trends Microbiol. 17, 219–225 (2009).
Kaneda, T. Biosynthesis of branched chain fatty acids. II. Microbial synthesis of branched long chain fatty acids from certain short chain fatty acid substrates. J. Biol. Chem. 238, 1229–1235 (1963).
Campbell, I. & Naworal, J. Composition of the saturated and monounsaturated fatty acids of Mycobacterium phlei. J. Lipid Res. 10, 593–598 (1969).
Carson, J. & Sumrell, G. Certain characteristics of the fatty acids from the lipids of the tubercle bacillus. J. Am. Chem. Soc. 72, 4837 (1950).
Brennan, P. J. The envelope of mycobacteria. Annu. Rev. Biochem. 64, 29–63 (1995).
Carter, G., Wu, M., Drummond, D. C. & Bermudez, L. E. Characterization of biofilm formation by clinical isolates of Mycobacterium avium. J. Med. Microbiol. 52, 747–752 (2003).
Hall-Stoodley, L. & Lappin-Scott, H. Biofilm formation by the rapidly growing mycobacterial species Mycobacterium fortuitum. FEMS Microbiol. Lett. 168, 77–84 (1998).
Hall-Stoodley, L. et al. Direct detection of bacterial biofilms on the middle-ear mucosa of children with chronic otitis media. J. Am. Med. Assoc. 296, 202–211 (2006).
Ojha, A. K. et al. Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol. Microbiol. 69, 164–174 (2008).
Danese, P. N., Pratt, L. A. & Kolter, R. Exopolysaccharide production is required for development of Escherichia coli K-12 biofilm architecture. J. Bacteriol. 182, 3593–3596 (2000).
Ojha, A. et al. GroEL1: A dedicated chaperone involved in mycolic acid biosynthesis during biofilm formation in mycobacteria. Cell 123, 861–873 (2005).
Bansal-Mutalik, R. & Nikaido, H. Mycobacterial outer membrane is a lipid bilayer and the inner membrane is unusually rich in diacyl phosphatidylinositol dimannosides. Proc. Natl. Acad. Sci. USA 111, 4958–4963 (2014).
Chen, J. M. et al. Roles of Lsr2 in colony morphology and biofilm formation of Mycobacterium smegmatis. J. Bacteriol. 188, 633–641 (2006).
Schaible, U. E., Sturgill-Koszycki, S., Schlesinger, P. H. & Russell, D. G. Cytokine activation leads to acidification and increases maturation of Mycobacterium avium-containing phagosomes in murine macrophages. J. Immunol. 160, 1290–1296 (1998).
Kempker, R. R. et al. Lung tissue concentrations of pyrazinamide among patients with drug-resistant pulmonary tuberculosis. Antimicrob. Agents Chemother. 61, 1–11 (2017).
Low, K. L. et al. Lipid droplet-associated proteins are involved in the biosynthesis and hydrolysis of triacylglycerol in Mycobacterium bovis bacillus Calmette-Guérin. J. Biol. Chem. 285, 21662–21670 (2010).
Rastogi, S. et al. The diacylglycerol acyltransferase Rv3371 of Mycobacterium tuberculosis is required for growth arrest and involved in stress-induced cell wall alterations. Tuberculosis 104, 8–19 (2017).
Reed, M. B., Gagneux, S., DeRiemer, K., Small, P. M. & Barry, C. E. The W-Beijing lineage of Mycobacterium tuberculosis overproduces triglycerides and has the DosR dormancy regulon constitutively upregulated. J. Bacteriol. 189, 2583–2589 (2007).
Hanahan, D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166, 557–580 (1983).
Sambrook, J., Fritsch, F. & Maniatis, T. Molecular Cloning: A Laboratory Manual 2nd edn. (Cold Spring Harbor Laboratory Press, New York, 1989).
Snapper, S. B., Melton, R. E., Mustafa, S., Kieser, T. & Junior, W. R. J. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 4, 1911–1919 (1990).
Connell, N. D. Mycobacterium: Isolation, maintenance, transformation, and mutant selection. Methods Cell. Biol. 45, 107–125 (1994).
Bligh, E. G. & Dyer, W. J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 (1959).
Rotering, H. & Raetz, C. R. Appearance of monoglyceride and triglyceride in the cell envelope of Escherichia coli mutants defective in diglyceride kinase. J. Biol. Chem. 258, 8068–8073 (1983).
Matyash, V., Liebisch, G., Kurzchalia, T. V., Shevchenko, A. & Schwudke, D. Lipid extraction by methyl-terf-butyl ether for high-throughput lipidomics. J. Lipid Res. 49, 1137–1146 (2008).
Acknowledgements
We thank Dr. Ana Arabolaza and Dr. Silvia Altabe for helpful discussion of the results. This research was supported by Agencia Nacional de Promoción de la Investigación, el Desarrollo Tecnológico y la Innovación (ANPCyT) grants 2015-0796 and 2018-02539 to GG and 2015-2022 to HG. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
G.G. and H.G. contributed to conception and design of the study. A.C.A., F.S. and M.C. performed the experiments and prepared the figures. All authors contributed to the interpretation of the data. G.G. designed the study, supervised the experimental work, and wrote the final version. All authors contributed to manuscript revision, read and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Crotta Asis, A., Savoretti, F., Cabruja, M. et al. Characterization of key enzymes involved in triacylglycerol biosynthesis in mycobacteria. Sci Rep 11, 13257 (2021). https://doi.org/10.1038/s41598-021-92721-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-92721-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
