
Abstract
Explorations of stable lead-free perovskites have currently achieved substantial interest to overcome the instability and avoid toxicity related issue faced with the lead-based perovskites. In this study, we have comprehensively studied the stability, nature and origin of electronic, transport and optical properties of inorganic halide double perovskites, which could provide a better understanding of their possible potential applications. The density functional theory is used to investigate the different physical properties of these materials. The stability of these cubic materials is validated by optimizing the structure, tolerance factor, mechanical stability test. The materials are small band gap semiconductors with outshining optoelectronic performance. Due to high optical absorption, high conductivity and low reflectivity they have great potential to be used for optoelectronic application purpose. Because of small band gap we have also investigated the variation of various transport parameters with chemical potential. The semiconducting nature of materials results in ZT close to unity predicting its excellent application in thermoelectric technology.
Similar content being viewed by others
Introduction
Due to increasing energy demands, there has been an increase in the consumption of fossil fuels which sufficiently contributes to the pollution levels and cause global warming. So, an eco-friendly power source is needed to meet the energy crises. To over-come this issue, solar cells made up of silicon proved to be an ideal solution. However, these silicon-based solar cells not only have complicated production procedure but also have less power conversion efficiency. So, there is a sustained research interest toward alternative photovoltaic (PV) materials produced with cost-competitive, facile, and environmentally friendly technologies. In this field perovskite solar cells have gained much progress during the last few decades increasing the efficiency from 3.8% in 2009 to 22.7% in 2017 at the lab-scale1,2,3,4,5. Hybrid halide perovskites with a general formula ABX3 where A is a monovalent cation such as methylammonium (MA+), formamidinium (FA+), and B is a divalent cation such as Pb2+ and X are halides such as Cl, Br, I are the most commonly studied materials for optoelectronic applications. But there are many issues associated with the commercialization of lead based solar cells particularly instability and toxicity6,7. Owing the thermodynamic and environmental stability Cesium halide perovskites proved to be saviors to overcome the less-stability issue of organic cations such MA+, FA+ against the environmental condition. Since lead is a very toxic heavy element has hazardous effects not only on the environment but also on human beings. Therefore, it is very important to reduce or eliminate lead from PV devices8,9. So, keeping this in view research is going on to replace the lead from perovskites by some other non-toxic elements. The replacement for lead must fulfill certain criteria in order to match the excellent performance of the lead-based perovskites. The alternative to lead must be low-cost, easily recycled, should exhibit excellent optoelectronic properties. In addition to being competitive with currently established PV technologies, they should also satisfy some commercial necessities like flexibility, long-term stability, and scalability10,11. The issue is being addressed by the replacement of lead-based perovskites with environment friendly lead-free halide-based perovskites. The success arises because these semi-conducting halide perovskites possess outstanding optoelectronic properties such as high optical absorption coefficient, bandgap that can be tuned, long carrier recombination lifetimes, high carrier mobility, small electron/hole effective masses, and high molar extinction coefficient. Among double halide perovskites, Bi and Sb-based families have drawn a remarkable interest. Cs2MBiCl6 (M = Ag, Cu, Na) and Cs2MBiCl6 (M = K, Rb and Cs), Cs2NaBX6 (B = Sb, Bi; X = Cl, Br, I) have shown excellent stability and good optoelectronic application with smaller and larger band gaps correspondingly12,13. The nanocrystals of Cs2CuMCl6 (M = Bi, Sb) as well as other related family members have been experimentally reported14,15,16,17,18,19,20,21.
Besides the PV, thermoelectric technology has also achieved significant interest during last few decades. Thermoelectric efficiency of materials is given by dimensionless figure of merit (ZT) given by; \(ZT = \frac{{S^{2} \sigma T}}{{\kappa _{e} + \kappa _{l} }}\), where S is Seebeck coefficient, σ is electrical conductivity, T is absolute temperature, κe is electronic thermal conductivity and κl is lattice thermal conductivity22,23. A material with ZT ~ 1.0 is considered a good thermoelectric material24. Such a high ZT value may be obtained when the power factor (PF) is high and thermal conductivity is low. Despite ultra-low thermal conductivity arising because of occupation of cations in the octahedral structure along with high charge mobility, it is quite surprising that these halide double perovskites have been mostly studied for optoelectronic applications. There are only small experimental studies conducted to study thermoelectric efficiency, but the interest towards the thermoelectric response of halide perovskites is now increasing. Theoretical calculations have claimed that the halide and hybrid perovskites could achieve ZT value equal to one. A variety of perovskites have attained figure of merit equal to one like in SrTiO3 (La substituted)25, MASnI3 (ZT ~ 1.0)26, CsSnCl3 (ZT ~ 1.0)27, Cs2AgBiX6 (X = Cl, Br) (ZT ~ 1.0)28. Fatima Aslam et al.29 studied Cs2InAgX6 (X = Cl, Br, I) suggesting that these materials are exhibiting tunable direct energy band gaps that can be employed in practical devices for energy harvesting applications. Motivated by their small band gap and unmatchable desirable properties, we have tried to explore these two materials for optoelectronic application and extended our study to unravel their thermoelectric properties which they equally justify.
Computational methods
The first principle method with the help of Wien2k simulation code30 is used to calculate the electronic structure, optical and transport properties of the materials. The ground state properties are calculated by solving Kohn–Sham equation properly. For the said purpose, different approximation methods like generalized gradient approximation (GGA), onsite coulomb interaction (GGA + U), modified Becke-Johson (mBJ) are utilized to approximate the only unknown term exchange–correlation potential in the state-of-art formulism31,32. Besides these methods we have also considered spin orbit coupling effect for the present set of materials. The unit cell volume is divided into muffin tin spheres where wave function shows atomic like character and interstitial space wherein plane wave basis set is employed. The extension of the basis set is controlled via RMT kmax = 7 and lmax = 10 conditions, where RMT is smallest muffin tin radii and kmax represent maximum value of k. To obtain the convergence of results the unit cell in the k-space is divided into a dense mesh of 1000-k points for integration over the Brillouin zone. As the thermoelectric parameters are sensitive to k point sampling therefore a high dense of 150,000 k points is utilized to calculate the same. The iterations for charge convergence between successive cycles converge up to 0.0001 e and energy up to 0.0001 Ry to obtain better results. The thermoelectric properties are determined under the approximation of constant relaxation time (τ) with the help of BoltzTraP code33. With the help of Gibbs2 code34 we have evaluated some of the thermodynamic parameters like Debye temperature and Grüneisen parameter.
Structural properties
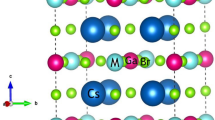
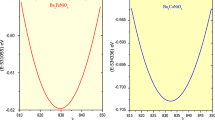
The structural stability of the materials can be determined by various factors like optimizing crystal structure by utilizing the Birch Murnaghan equation of state35, Goldschmidt’s rule from the effective ionic radii or bond length36. The structural stability, the band structure and carrier transport performance of materials and their specific application to a large extent is predominately determined by the combination of cations and anions. The band profile of halide double perovskites with general formula A2B(I)B′(III)X6 are predominantly decided by B (I)-, B(III) and X-site atoms. Fig. S1a (Supplementary Information) shows a possible combination of different cations from the periodic table for the possible formation of halide double perovskites37. The correct combination of these cations leads to the excellent properties of these materials. In the present case, the structural optimization of the titled halide double in ferromagnetic (FM) and non-magnetic (NM) perovskites is done by utilizing the Birch-Murnaghan equation of state which justifies the cubic stability with the non-magnetic (NM) ground state as the stable state as shown in Fig. 1a,b. Also, the cubic stability is determined from the tolerance factor. The ground state parameters of these materials are quoted in Table 1 which agrees well with the previously reported results14,15. The pictorial representation of the titled perovskite in which cesium is enclosed by a cage of 12 halide atoms (Cl) while as d block element Cu and p group elements (Sb/Bi) lie in octahedral of halide atoms having coordination 6 to these atoms is shown in Fig. S1b (Supplementary Information).

(a,b) Energy versus volume optimization curve of Cs2CuMCl6 (M = Sb, Bi) in both spin-polarized and non-polarized states.
Second-order elastic constants and mechanical stability
The elastic constants and thereby mechanical behavior of these considered double halide perovskites are predicted with the help of the Cubic Elastic package38. Equilibrium cubic structure is deformed by applying small strains to predict second-order elastic constants. The cubic structure would be mechanically stable only if the deformed structures are at higher energy compared to the cubic phase. This leads to a limiting condition \(C_{{11}} - C_{{12}} > 0,C_{{11}} > 0,C_{{44}} > 0,C_{{11}} + 2C_{{12}} > 0,C_{{12}} < B < C_{{11}}\)39 for elastic constants to be followed where C11 longitudinal elastic constant indicates elasticity along the axis of unit cell and C12 and C44 are shear elastic constants define elasticity in shape.
The elastic constants help to predict the response of any material to applied stresses. The second-order elastic constants (SOECs) in the present work along with the already reported values have been evaluated utilizing the energy-strain approach in the framework of GGA-PBE given in Table 2. All three elastic constants are non-negative and follow the Born stability criteria condition40. Thereby, advocate the mechanical stability of the materials. From the SOECs, the universal anisotropic factor (AU)41 is deduced. The deviation of AU from unity signifies titled halide double perovskites are highly anisotropic. The anisotropy mainly originates because of the large difference in longitudinal and shear elastic constants. Using SOEC we have estimated mechanical parameters like Young’s (Y), shear (G), and bulk moduli (B), Poisson’s ratio (σ) using the mathematical relations reported elsewhere42. Further, by employing Elate: Elastic tensor analyzer43, we have analyzed the angular dependence of elastic modulus. The results reflect that Young’s modulus and shear modulus are highly anisotropic shown in Fig. S2a,b, while bulk modulus is isotropic as happens in cubic crystals. In addition to 3D graphical representation of directional elastic properties a quantitative analysis by reporting the minimal and maximal values of each modulus is reported in Table S1. Moreover, by executing the Reuss-Vogoit-Hill scheme44 we have defined the average values of different elastic moduli. The obtained results are summarized in Table S2. The C11-values for both double perovskites are greater than the other two shear elastic constants (C12 and C44) and also B being greater than G reflects that these materials show more resistance for volumetric deformation compared to the shape deformation. The Pugh’s ratio (B/G), Poisson’s ratio (σ), and Cauchy pressure (CP = C12 – C44)45,46,47 are greater than their index values of 1.75, 0.26, and 0, respectively as can be seen from Table 2. These values thus signify Cs2CuSbCl6 and Cs2CuBiCl6 double perovskites are ductile. So, these materials can be used to design tools of varying shapes.
Additionally, we have simplified the ultrasonic wave velocities of the titled double perovskites using SOECs and the density of the materials48. In cubic structure pure longitudinal (Vl) and two transverse (VT1 and VT2) modes only happen along [100], [110] and, [111] direction. The magnitude of the sound wave velocity is obtained through the following equation, \(V = \sqrt {\frac{{C_{{eff}} }}{\rho }}\)49 where, the Ceff for different modes along different directions are defined in Table S3. These wave velocities in turn are used to estimate the average Debye velocity (mean sound velocity VD) using relation \(V_{D} = \left\{ {\frac{1}{3}\left( {\frac{1}{{V_{L} }} + \frac{1}{{V_{{T1}} }} + \frac{1}{{V_{{T2}} }}} \right)} \right\}^{{ - \frac{1}{3}}}\)50. The calculated values of Debye velocity or mean sound velocity are presented in Table 2. Moreover, we have tallied the Debye temperature (θD) of the Cs2CuSbCl6 and Cs2CuBiCl6 perovskites by using the Debye average velocity VD in with equation, \(\theta _{D} = \frac{h}{k}\left\{ {\frac{{3n}}{{4\pi }}\frac{{NA\rho }}{M}} \right\}^{{\frac{1}{2}}} V_{D}\)51. The obtained values of ultrasonic sound velocities and Debye temperature furthermore authenticate the anisotropic nature of the materials. Moreover, the high value of Debye temperature signifies these materials are stable at extreme temperatures and could be used for the fabrication of the devices.
Electronic properties
The applications of any material are profoundly characterized by the electronic properties which include band structure and distribution of electrons in these bands52. Herein, with the assistance of density functional theory, we have evaluated the electronic properties of inorganic halide double perovskites. The band structure calculated via non-spin polarized calculations on employing different approximation methods are provided in Figs. 2 and 3. It is clear that the Fermi level is unoccupied indicating the semiconducting nature for Cs2CuMCl6 (M = Sb, Bi). Also, the valence band maxima (VBM) and conduction band minima (CBM) are located at different symmetric points resulting in indirect band gap. The band gap calculated for Cs2CuSbCl6 and Cs2CuBiCl6 by GGA are ∼ 0.61 eV and ∼ 0.89 eV for Sb and Bi-based perovskites, respectively. On adding Hubbard potential and spin orbit coupling (SOC) potential to GGA, almost no change in the band gap is observed. However, on assisting mBJ potential to GGA, indirect band gaps increase to ∼ 1.00 eV for Cs2CuSbCl6 and ∼ 1.20 eV for Cs2CuBiCl6. The energy bands mostly populated of Cl-p and Cu-d stated are pushed away from Fermi level by mBJ-potential result in broadening of band gap. Since the mBJ potential effectively enhance the band gap, so we have further added the SOC and mBJ potentials altogether to GGA. By GGA + mBJ + SOC the band gaps turn out to be ~ 1.50 eV and ~ 2.0 eV for Sb and Bi-based materials, respectively, consistent with the experimental reported results. The mBJ + SOC effect removes the degeneracy of some states, thereby magnify the bandgap. The bandgap values obtained through different approximation in comparison with the experimental and theoretical reported values are summarized in the Table 3. The valence band (VB) is populated by the filled states, empty states enter to conduction band (CB), and partially filled states crossover Fermi level. The oxidation state and the number of remaining valence electrons in the respective oxidation states pre-determines which states would enter to the valence band or conduction band or occupy the Fermi level. In the entitled double perovskites, Cs and Cu have oxidation state + 1, Cl has − 1 oxidation state while the main block elements Sb and Bi are in + 3 oxidation state. In Cs2+1Cu+1 M+3Cl6–1(M = Sb, Bi) oxidation configuration charge of constituents is balanced; moreover, the constituents have only paired electrons.

Band structure of Cs2CuSbCl6 calculated by GGA, GGA + U and GGA + mBJ and GGA + mBJ + SOC methods.

Band structure of Cs2CuBiCl6 calculated by GGA, GGA + U and GGA + mBJ and GGA + mBJ + SOC methods.
The qualitative description of the valence band and conduction band and the energy states associated with them is illustrated with the help of the density of states. The valence state Cs-s, Cu-d, Sb/Bi-p and Cl-p contribution toward the band composition obtained by GGA + mBJ is presented in Fig. 4. Among all the states, most interested states are d-states of Cu which are in the vicinity of Fermi level. The p-states of Cl gets electrons from cations mostly compose the VB. The Cs-s and Sb/Bi-p states are nowhere in the vicinity of the Fermi level; therefore, these states play a passive role in characterizing the electronic properties in these perovskites. The Cu-d-states in the octahedral field split into triplet d-t2g and doublet d-eg states, d-t2g states being at lower energy. The d-t2g can intake a maximum of six electrons (3↑ and 3↓) while as d-eg state is filled by 4-electrons (2↑ and 2↓). Therefore, the electron filling in d-orbitals of Cu1+ is 3t2g (↑), 3t2g (↓), 2eg (↑), and 2eg (↓). All the d-orbitals are filled for both spin channels therefore form the VB. Moreover, the crystal field splitting energy for the configuration is zero. The p-states of Cl gets electrons from cations are filled and happen to be in VB. Therefore, in Cs2CuMCl6 (M = Sb, Bi) the distribution of energy states is; Cu-d and Cl-p are completely filled lie in valence band below Fermi level and while as Sb/Bi-p and Cs-s are empty in the conduction band a small band at Fermi level. The constituent atoms have no unpaired electrons resulting in the non-magnetic character of these materials which is also confirmed from the structural optimization. The obtained semi-conducting nature along with small band gap values signifies that they can outshine in optoelectronic and thermoelectric applications. The partial densities of states obtained by GGA + mBJ + SOC are plotted in Fig. S3 and discussed in the Supplementary Information.

Partial density of states of Cs2CuMCl6 (M = Sb, Bi) calculated by GGA + mBJ scheme.
Thermophysical properties
The variation of transport parameters like carrier concentration, Seebeck coefficient, conductivity, etc. with chemical potential and temperature is remarkable. So, to illustrate the chemical potential dependence of transport coefficients at different temperatures we have used constant relaxation time approximation under BoltzTraP code33. The magnitude of thermoelectrical parameters in semiconductors is mainly characterized by band structure, as the central contribution is from band gap, carrier type, carrier concentration, and carrier effective mass63. The transport behavior is directly linked with the energy bands within the Fermi level. In the Electronic Properties Section, it is found that by incorporating mBJ and mBJ + SOC potentials to GGA, the bandgap changes effectively. The bandgap predominately decides which carries (electrons and holes) take part in the transport phenomenon. So, the transport properties of both conductivities and Seebeck coefficient extensively depend on the value of the bandgap. The high Seebeck coefficient is shown by the insulators, while metals have the least Seebeck coefficient64. So, the narrower the band gap lesser would be the Seebeck coefficient and vice-versa. Conductivity on the other side is proportional to the effective charge carries. In Cs2CuMCl6 (M = Sb, Bi) perovskites, GGA + mBJ and GGA + mBJ + SOC sophistically improves the bandgap. So, to understand the role of bandgap underestimation on the transport properties, we have computed the transport properties by both approximations. The SOC results are presented in the Supplementary Information.
The variation of carrier concentration with chemical potential at different temperatures is presented in Fig. S4. The carrier concentration graph gives us an idea about the nature of transport carriers which describes electronic properties which in turn affects other transport parameters. The sign of the carrier concentration designates the nature of charge carriers; the negative sign indicates that electrons are majority carriers while as positive carrier concentration means holes are majority carriers65. The sharp variation in carrier concentration corresponds to the presence of bandgap/pseudo-gap in the band structure. As the temperature increase electron gain more thermal energy hence carrier concentration increases with an increase in temperature.
The Seebeck coefficient (S) as a function of chemical potential (μ-EF) at different temperatures (300 K, 600 K, 800 K) is plotted in Fig. 5a. For the entire region of chemical potential, the Seebeck coefficient displays prominent peaks and valleys. There are high-intensity peaks for the positive potential as well as negative potential at 300 K and these values decreases as temperature rise to 600 K and 800 K. The decreasing character is because bound electrons get excited by acquiring thermal energy, generate electron–hole pairs. The most prominent peaks are in the range of 0 to 1 eV as the bands are less dispersive with the forbidden region around Fermi level, thereby fewer charge carriers are around in this range. The maximum value of S is 2000 μV/K for Cs2CuSbCl6 and 1500 μV/K for Cs2CuBiCl6 at 300 K, respectively. On comparing the results of both these materials it can conclude that Cs2CuBiCl6 shows a better Seebeck coefficient because of the presence of a larger bandgap. The obtained results are in decent agreement with the already reported theoretical results of similar other materials, thereby validate our results28,29,36. The magnitude thermopower |S| obtained by GGA + mBJ + SOC illustrated in Fig. S5a is higher than the GGA + mBJ counter-partner; it is because the bandgap significantly increases with the incorporation of SOC.

Variation in transport parameters with chemical potentials at different temperatures viz. (a) Seebeck coefficient; (b) Electrical conductivity; (c) thermal conductivity; (d) figure of merit (ZT). Different colors are used to distinguish the temperature: Black-300 K; Red-600 K; and Blue-900 K.
The graphical variation of electronic conductivity (σ) as a function of chemical potential at temperature range 300 K, 600 K, and 800 K is demonstrated in Figs. 5b and S5b calculated by GGA + mBJ and GGA + mBJ + SOC, respectively. Because of the absence of energy bands around the Fermi level making the area desolate of charge carriers and hence the conductivity vanishes around μ-EF = 0. But below or above the Fermi level the conductivity increases because of the presence of energy states. With the rise in temperature the conductivity in the vicinity of the Fermi level increases, because of band smearing. Certain energy states that were filled at T = 0 K becomes empty because with rise in temperature electrons make the transition from the valence band to the conduction band.
The total lattice thermal conductivity comprises of lattice part arising due to lattice vibrations and electronic part arising due to charge carriers. Here, we tried to evaluate both the components of total thermal conductivity with different chemical potentials at a temperature range 300 K, 600 K and 800 K as shown in Fig. 5c. As, thermal conductivity and electronic conductivity both depend on carrier concentration so, with change in chemical potential, they follow a similar profile of variation. However, the thermal conductivity increases abruptly with temperature compared to electrical conductivity. The results depicted by these agree well with the Wiedemann Franz law which states the proportional relation between them as follows: κ = σLT66. The lattice part of thermal conductivity is calculated with the help of Slack’s equation, \(\kappa _{l} = \frac{{A\theta _{D}^{3} V^{{\frac{1}{3}}} m}}{{^{{\gamma ^{2} }} \overline{N} ^{{\frac{2}{3}}} T}}\)22. The equation suggests that κl is dependent on Debye temperature (θD), volume (V), average molar mass per atom (m), Grüneisen parameter (γ), temperature (T) and number of atoms per unit cell (\(\overline{N}\)).The value of A is calculated as, \(A = \frac{{2.43 \times 10^{8} }}{{1 - \frac{{0.514}}{\gamma } + \frac{{0.228}}{{\gamma ^{2} }}}}\)67. The variation of these interdependent quantities such as Grüneisen parameter and Debye temperature with temperature has been plotted in Fig. S6a,b. Debye temperature is an important parameter that characterizes the thermal vibrations in a solid. It is the maximum temperature above which a solid behaves classically and the constituents exhibit coupled vibrations. The degree or extent of anharmonicity in a crystal is determined by Grüneisen parameter. As the temperature is increased the atoms start vibrating more rigorously which leads to an increase in anharmonic effects. From the plots, we can see that θD decreases while as γ increases with an increase in temperature. Finally, with the help of these interdependent quantities, we can evaluate the lattice thermal conductivity using Slack’s equation as shown in Fig. S6c. It can see that these thermodynamic parameters don’t change much with temperature so it is clear that κl depends mostly on the number of atoms (\(\overline{N}\)).
The most important parameter which scrutinizes the efficiency of thermoelectric materials is the dimensionless figure of merit68. The relation of ZT clearly signifies that it increases with electrical conductivity and Seebeck coefficient while it decreases with increasing thermal conductivity. Figures 5d and S5c (with SOC) display the figure of merit (ZT) values of Cs2CuSbCl6 and Cs2CuBiCl6 as a function of chemical potential at temperature 300 K, 600 K and 800 K. The obtained values of ZT are compared with reported results in the literature, tabulated in Table S4. Both these halide perovskites have prominent peaks with the highest peak having ZT nearly equal to 1. The high value of ZT can be attributed to the semiconducting nature of these materials. We can see from the graph that as temperature increases the magnitude of ZT begins to increase and reaches the value of at high temperature.
Besides band gap, carrier effective mass and carrier concentration are other key parameters for the semiconductor transport performance. The magnitude of the Seebeck coefficient is directly related to the dispersion of energy levels near the Fermi level. The relation of the Seebeck coefficient with carrier concentration and effective mass is: \(S=\frac{8{\pi }^{2}{\mathrm{k}}_{\mathrm{B}}^{2}}{3e{\hslash }^{2}}{m}^{\mathrm{*}}T{\left(\frac{\pi }{3n}\right)}^{2/3}\)64. The relation makes it clear that Seebeck coefficient is directly proportional to effective mass m*, while the magnitude of ‘S’ decreases with an increase in ‘n’. At absolute temperature, semiconductors don’t have free carriers for conduction. Charge carriers are produced only when electrons in the valence band are provided sufficient energy to jump to the conduction band. The VBM and CBM curvatures along with E-K dispersions define the hole and electron effective masses, respectively. Holes have a negative effective mass in comparison to a positive effective mass of electrons. Smaller effective masses mean greater CBM and VBM hybridization. E-K dispersion curves with parabolic curvature have low effective mass, signifies high mobility thereby affording larger electrical conductivity. While high effective mass is the character of the flat band. The energy band in the vicinity of the Fermi level of the entitled materials are less dispersive signify the presence of highly effective mass, which could account high Seebeck coefficient. The various parameters are determined with the help of transM code69. The comparative variation in σ, S altogether with inverse effective mass within the vicinity of the Fermi level is presented in Fig. S7. The large Seebeck coefficient around the Fermi level high is due to significant effective mass. As we move away from the Fermi level, the effective mass decreases gradually result in a decrease in the Seebeck coefficient, correspondingly the conductivity increases. The variation in the Seebeck coefficient, the electrical conductivity with carrier doping concentration is presented in Fig. 6. In a semiconductor, the doping of either type of carries amplifies the electrical conductivity. But the Seebeck coefficient being proportional to n−2/3 decreases as the doping carrier concentration increases. While the conductivity increases up to a certain value beyond which it decreases. The conductivity increases manifolds with electron doping in comparison to hole doping, it may be because of the high mobility of electrons. The ZT plot conveys optimal hole doping can significantly improve the thermoelectric efficiency.

Variation in Seebeck coefficient, conductivity and ZT with electron and hole concentration. Negative carrier concentration is meant for electrons and positive for holes. Different colors are used to distinguish the temperature: Black-300 K; Red-600 K; and Blue-900 K.
Optical properties
The optical properties of a material are directly linked to the dielectric function of the material. These properties are being determined by investigating their visible light energy harvest. This is normally done by calculating the bandgap and the absorption coefficients70. Ideally, direct low band gap semiconductor materials possess promising optoelectronic applications like photo-absorbers for solar-cell71. The optical properties of a material depend on frequency and they are interconnected with each other if we are capable of calculating one e.g., dielectric function we can extort all other properties easily. The optical properties such as the absorption coefficient, refractive index n (ω), reflectivity R (ω) and conductivity function σ (ω) are obtained from the expression of the real part ε (ω) of the dielectric function72.
First, we started with plot of the optical absorption coefficient with photon energy which gives information on the light harvesting capacity of the material. Since, we know that band gap depends inversely upon absorption threshold so materials with higher band gap have narrow absorption in the visible region of electromagnetic spectrum. But the studied materials have smaller band gap showing higher absorption in the visible range as shown in Fig. 7a. These materials show high absorption coefficient ranging from infrared to ultraviolet region and contains entire visible wavelength range. As the photon energy increases the absorption spectrum increases gradually and highest peak occurs at 6.5 eV which corresponds to maximum absorption. This spectrum appeared as a result of electrons exciting from valence to conduction band. The first peak in absorption spectrum for Cs2CuSbCl6 (Cs2CuBiCl6) is about 2 eV (2.5 eV) which arises due to transitions from Cu-d to M-p (M = Sb, Bi) while as second peak occurs at 6 eV (6.5 eV) corresponds to transitions from Cu-d to Cl-p. The late absorption onset was attributed to the indirect band gap. Further, we tried to investigate the optical conductivity of the materials and plotted in Fig. 7b. It follows a similar trend as the absorption spectra as shown in Fig. 7a. Over the entire photo energy range (0–4 eV) conductivity displays high and low peaks with the presence of hump at particular energies. The maximum conductivity is shown at higher energy range. These calculated curves reveal the same trend or same features as observed in case of reported cases14,15. The almost similar nature of band structure leads to similar structure of the optical spectra originates from the top of valence band to the bottom of conduction band. The optical properties determined by the GGA + mBJ + SOC are presented in the supplementary information as displayed in Fig. S8.

The various optical parameters calculated by GGA + mBJ approximation for Cs2CuSbCl6 and Cs2CuBiCl6 where (a) represents optical absorption, (b) represents optical conductivity, (c) represents refractive index, (d) represents extinction coefficient.
An important physical property in optics which provides information about behavior of light inside a material is refractive index. When light is passed through different media, its velocity changes resulting in the variation of refractive of a material. Other important physical quantity which is connected to the light absorption capacity of a material at a particular frequency is called as extinction coefficient. The extinction coefficient is basically complex part of refractive index and represent how electromagnetic wave can propagate in any medium. The variation of refractive index with photon energy is illustrated in Fig. 7c. The static values of refractive index are 2.3 and 2.0 for Cs2CuSbCl6 and Cs2CuBiCl6 respectively. They correspond to the values that can be derived from real part of dielectric function. These obtain a maximum value at around 2 eV. From Fig. 7d we see that the extinction coefficient can be divided into three main absorption peaks which are centered on different photon energy range. These various peaks arise because of electronic transitions from one level to another. These all together properties coveys that these inorganic halide double perovskites would be a potential lead-free alternative for optoelectronic device fabrication.
Spectroscopic limited maximum efficiency: the maximum possible efficiency of a solar absorbing material in PV’s is determined theoretically by Shockley-Queisser limit (SQ) which gives a direct relation between band gap of a material and its maximum power efficiency. The spectroscopic limited maximum efficiency (SLME) is the recently introduced technique which goes beyond the SQ limit to calculate the maximum efficiency of a photovoltaic material by taking into account the absorption coefficient and thickness of material. The SLME takes into account the absorption coefficient as well as radiative/non-radiative recombination losses considering both direct as well as indirect band gap which plays an important role in designing highly efficient photovoltaic device as compared to SQ limit73,74. This approach is based on Fermi golden rule. Considering this approach, we have calculated the effect on efficiency of these materials as a function of the thickness of the absorber layers as shown in Fig. 8. Here, we varied the thickness of material from 0 to 1.4 µm and calculated the efficiency of material. Since we know that Cs2CuSbCl6 perovskite has narrow band gap and high absorption coefficient as compared to Cs2CuBiCl6 perovskite therefore attains higher power conversion efficiency of about 30% while as Cs2CuBiCl6 has efficiency about 19%.

Variation of spectroscopic limited maximum efficiency (SLME) as a function of thickness of absorbing layer.
In order to inter-relate the dependency of optical and thermoelectric properties on the band structure, we have comparatively plotted the figure of merit, optical absorbance and band structures altogether, referred in the Fig. S9. The narrow band gap Sb-based perovskite shows more absorption peaks in the visible region compared to Bi-based perovskite, due to its small bandgap. On comparing the magnitude of ZT, low band gap Cs2CuSbCl6 material also have slightly higher ZT-peaks in vicinity of Fermi level in comparison to Cs2CuBiCl6 perovskite. Although Cs2CuBiCl6 show high Seebeck coefficient but still it shows lower ZT peaks. It is because electrical conductivity an important factor for ZT is low for wide bandgap materials. So, narrow band gap materials are most suitable for both applications. If the band gap is reduced to zero, the conductivity may get amplified but Seebeck decreases drastically, so the ZT gets dejected.
Conclusion
In the present study the structural stability along with electronic, elastic, thermoelectric and optical properties of inorganic halide double perovskites have been calculated. Both the materials are stable in cubic structure follow the space symmetry of the Fm-3m space group. The stability in the Fm-3m space group is defined with the help of energy optimization, tolerance factor. Moreover, the positive values of elastic constants authenticate the mechanical stability of the materials. The elastic constants further confirm the ductile and anisotropic nature of the materials. The band structure and density of states reflect the semiconducting character with small indirect ban gap. These materials have excellent optical absorption in the visible range can be used for optoelectronic application purpose. The high Seebeck coefficient with low thermal conductivity is responsible for high figure of merit close to unity.
Data availability
The data would be available from the corresponding author on reasonable request.
References
Kojima, A., Teshima, K., Shirai, Y. & Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for PV cells. J. Am. Chem. Soc. 131, 6050–6051 (2009).
Shao, Y., Yuan, Y. & Huang, J. Correlation of energy disorder and open circuit voltage in hybrid perovskite solar cells. Nat. Energy 1, 15001 (2016).
Shin, S. S. et al. Colloidally prepared La doped BaSnO3 electrodes for efficient, photostable perovskite solar cells. Science 356, 167–171 (2017).
Lee, M. M., Teuscher, J., Miyasaka, T., Murakami, T. N. & Snaith, H. J. Efficient hybrid solar cells based on meso super structured organometal halide perovskites. Science 338, 643–647 (2012).
Liu, M., Johnston, M. B. & Snaith, H. J. Efficient planar heterojunction perovskite solar cells by vapour deposition. Nature 501, 395–398 (2013).
Igbari, F., Wang, Z. & Liao, L. S. Progress of lead-free halide double perovskites. Adv. Energy Mater. 1803150, 1–32 (2019).
Creutz, S. E., Crites, E. N., De Siena, M. C. & Gamelin, D. R. Colloidal nanocrystals of lead-free double-perovskite (elpasolite) semiconductors: Synthesis and anion exchange to access new materials. Nano Lett. 18(2), 1118–1123 (2018).
Bartel, C. J. et al. Inorganic halide double perovskites with optoelectronic properties modulated by sublattice mixing. J. Am. Chem. Soc. 142(11), 5135–5145 (2020).
Jain, A., Voznyy, O. & Sargent, E. H. High-throughput screening of lead-free perovskite-like materials for optoelectronic applications. J. Phys. Chem. C 121, 7183–7187 (2017).
Zhang, Z. et al. Potential applications of halide double perovskite Cs2AgInX6 (X = Cl, Br) in flexible optoelectronics: Unusual effects of uniaxial strains. J. Phys. Chem. Lett. 10, 1120–1125 (2019).
Zelewski, S. J. et al. Revealing the nature of photoluminescence emission in the metal-halide double perovskite Cs2AgBiBr6. J. Mater. Chem. C 7, 8350–8356 (2019).
Wenwu, S., Cai, T., Wang, Z. & Chen, O. The effects of monovalent metal cations on the crystal and electronic structures of Cs2MBiCl6 (M = Ag, Cu, Na, K, Rb, and Cs) perovskites. J. Chem. Phys. 153, 141101 (2020).
Zhao, S., Yamamoto, K., Iikubo, S., Hayase, S. & Ma, T. First-principles study of electronic and optical properties of lead-free double perovskites Cs2NaBX6 ( B = Sb, Bi; X = Cl, Br, I). J. Phys. Chem. Solids 117, 117–121 (2018).
Zhou, W. et al. Lead-free small-bandgap Cs2CuSbCl6 double perovskite nanocrystals. J. Phys. Chem. Lett. 11, 6463–6467 (2020).
Zhao, X.-G. et al. Design of lead-free inorganic halide perovskites for solar cells via cation-transmutation. J. Am. Chem. Soc. 139(7), 2630–2638 (2017).
Feng, H.-J., Deng, W., Yang, K., Huang, J. & Zeng, X. C. Double perovskite Cs2BBiX6 (B = Ag, Cu; X = Br, Cl)/TiO2 heterojunction: An efficient Pb-free perovskite interface for charge extraction. J. Phys. Chem. C 121, 4471–4480 (2017).
Jafar, A. M., Khalaph, K. A. & Mahmood, A. Lead-free perovskite and double perovskite solar cells. Mater. Sci. Eng. 765, 012047 (2020).
Che, X., Wang, C., Li, Z., Hou, Z. & Yin, W.-J. Bayesian optimization based on a unified figure of merit for accelerated materials screening: A case study of halide perovskites. Sci. China Mater. 63(6), 1024–1035 (2020).
Roknuzzaman, M. et al. Electronic and optical properties of lead-free hybrid double perovskites for PV and optoelectronic applications. Sci. Rep. 9, 718 (2019).
Meyer, E., Mutukwa, D., Zingwe, N. & Taziwa, R. Lead-free halide double perovskites: A review of the structural, optical, and stability properties as well as their viability to replace lead halide perovskites. Metals 8(667), 1–6 (2018).
Volonakis, G. et al. Lead-free halide double perovskites via heterovalent substitution of noble metals. J. Phys. Chem. Lett. 7(7), 1254–1259 (2016).
Bhat, T. M. & Gupta, D. C. Magneto-electronic, thermal, and thermoelectric properties of some Co-based quaternary alloys. J. Phys. Chem. Solids 112, 190–199 (2018).
Huang, H. H., Fan, X., Singh, D. J. & Zheng, W. T. Thermoelectric properties of monolayer GeAsSe and SnSbTe. J. Mater. Chem. C 8(28), 9763–9774 (2020).
Bhandari, S. R. et al. Electronic, magnetic, optical and thermoelectric properties of Ca2Cr1−xNixOsO6 double perovskites. RSC Adv. 10, 16179–16186 (2020).
Lu, Z., Zhang, H., Lei, W., Sinclairand, D. C. & Reaney, I. M. Reaney high-figure-of-merit thermoelectric La-doped A-site-deficient SrTiO3 ceramic. Chem. Mater. 28(3), 925–935 (2016).
Singh, R., Kottokkaran, R., Dalal, V. L. & Balasubramanian, G. Engineering band gap and electronic transport in organic–inorganic halide perovskites by superlattices. Nanoscale 9, 8600–8607 (2017).
Yu, L. et al. Thermoelectric property analysis of CsSnX3materials (X= I, Br, Cl). In 2015 21st International Workshop on Thermal Investigations of ICs and Systems (THERMINIC) 1–5 (IEEE, 2015).
Guechi, N., Bouhemadou, A., Omran, S. B., Bourzami, A. & Louail, L. Elastic, optoelectronic and thermoelectric properties of the lead-free halide semiconductors Cs2AgBiX6 (X= Cl, Br): Ab initio investigation. J. Electron. Mater. 47(2), 1533–1545 (2018).
Aslam, F., Sabir, B. & Hassan, M. Structural, electronic, optical, thermoelectric, and transport properties of indium-based double perovskite halides Cs2InAgX6 (X= Cl, Br, I) for energy applications. J. Appl. Phys. A 127(2), 1–12 (2021).
Schwarz, K., Blaha, P. & Madsen, G. K. H. Electronic structure calculations of solids using the WIEN2k package for material sciences. Comput. Phys. Commun. 147(1–2), 71–76 (2002).
Perdew, P. J., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett 77(18), 3865–3868 (1996).
Rezkallah, T. et al. Investigation of the electronic and magnetic properties of Mn doped ZnO using the FP-LAPW method. Chin. J. Phys. 55(4), 1432–1440 (2017).
Madsen, G. K. H. & Singh, D. J. BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 175(1), 67–71 (2006).
Roza, A. O. D. L., Pérez, D. A. & Luaña, V. Gibbs2: A new version of the quasi-harmonic model code. II. Models for solid-state thermodynamics, features and implementation. Comput. Phys. Commun. 182, 2232–2248 (2011).
Katsura, T. & Tange, Y. A simple derivation of the Birch-Murnaghan equations of state (EOSs) and comparison with EOSs derived from other definitions of finite strain. Minerals 9(12), 745 (2019).
Mir, S. A. & Gupta, D. C. Analysis of cage structured halide double perovskites Cs2NaMCl6 (M = Ti, V) by spin polarized calculations. J. Alloys Compd. 854, 1–10 (2020).
Hoefler, S. F., Trimmel, G. & Rath, T. Progress on lead-free metal halide perovskites for PV applications: A review. Monatsh Chem. 148, 795–826 (2017).
Jamal, M. Jalali Asadabadi, S., Ahmad, I., & Rahnamaye Aliabad, H. A. Elastic constants of cubic crystals. Comput. Mater. Sci. 95, 592–599 (2014).
Nabi, M. & Gupta, D. C. Small-band gap halide double perovskite for optoelectronic properties. Int J. Energy Res. 45(5), 7222–7234 (2020).
Born, M. On the stability of crystal lattices. I. Math. Proc. Cambridge Philos. Soc. 36, 160. https://doi.org/10.1017/s0305004100017138 (1940).
Ranganathan, I. S. & Starzewski, M. O. Universal elastic anisotropy index. Phys. Rev. Lett. 101, 055504 (2008).
Luan, X. et al. The mechanical properties and elastic anisotropies of cubic Ni3Al from first principles calculations. Curr. Comput.-Aided Drug Des. 8, 307 (2018).
Gaillac, R., Pullumbi, P. & Coudert, F.-X. ELATE: An open source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 28, 275201 (2016).
Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. 65, 349–354 (2002).
Pugh, S. F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 45, 823–843 (2009).
Huang, B., Duan, Y. H., Hu, W. C., Sun, Y. & Chen, S. Structural, anisotropic elastic and thermal properties of Mb (M=Ti, Zr and Hf) monoborides. Ceram. Int. 41, 6831–6843 (2015).
Zhang, M. J., Zhang, Y., Xu, W. K. & Ji, V. Young’s modulus surface and poisson’s ratio curve forcubic metals. J. Phys. Chem. Solids 68, 503–510 (2007).
Bhat, T. M. & Gupta, D. C. Transport, structural and mechanical properties of quaternary FeVTiAl alloy. J. Electron. Mater. 45(11), 6012–6018 (2016).
Khandy, S. A. & Gupta, D. C. Study of ferromagnetism, spin-polarization, thermoelectrics and thermodynamics of layered perovskite Ba2FeMnO6 under pressure and temperature Phys. Status Solidi B 1800625, 1–7 (2019).
Zahedi, E. & Xiao, B. DFT study of structural, elastic properties and thermodynamic parameters of Bi2S3 under hydrostatic pressures. Comput. Mater. Sci. 101, 301–312 (2015).
Brik, M. G. First-principles calculations of electronic, optical and elastic properties of ZnAl2S4 and ZnGa2O4. J. Phys. Chem. Solids 71(10), 1435–1442 (2010).
Markov, M., Hu, X. & Liu, H. C. et al. Semi-metals as potential thermoelectric materials. Sci. Rep. 8, 9876 (2018).
Gill, D., Bhumla, P., Kumar, M. & Bhattacharya, S. High-throughput screening to modulate electronic and optical properties of alloyed Cs2AgBiCl6 for enhanced solar cell efficiency. J. Phys. Materials 4(2), 025005 (2021).
Gill, D., Bhumla, P., Kumar, M. & Bhattacharya S. High-throughput screening for band gap engineering by sublattice mixing of Cs2AgBiCl6 from first-principles. arXiv preprint arXiv:2006.00183. (2020).
McClure, E. T., Ball, M. R., Windl, W. & Woodward, P. M. Cs2AgBiX6 (X= Br, Cl): New visible light absorbing, lead-free halide perovskite semiconductors. Chem. Mater 28(5), 1348–1354 (2016).
Wei, F. et al. Enhanced visible light absorption for lead-free double perovskite Cs2AgSbBr6. Chem. Commun 55(26), 3721–3724 (2019).
Du, K.-Z., Meng, W., Wang, X., Yan, Y. & Mitzi, B. D. Bandgap engineering of lead-free double perovskite Cs2AgBiBr6 through trivalent metal alloying. Angew. Chem. Int. Ed 56(28), 8158–8162 (2017).
Xiao, Z. et al. Intrinsic instability of Cs2In(I)M(III)X6 (M= Bi, Sb; X= halogen) double perovskites: A combined density functional theory and experimental study. J. Am. Chem. Soc. 139(17), 6054–6057 (2017).
Slavney, A. H. et al. Small-band-gap halide double perovskites. Angew. Chem. Int. Ed. 57(39), 12765–12770 (2018).
Saeed, Y. et al. Cs2NaGaBr6: A new lead-free and direct band gap halide double perovskite. RSC Adv. 10(30), 17444–17451 (2020).
Zhou, J. et al. Composition design, optical gap and stability investigations of lead-free halide double perovskite Cs2AgInCl6. J. Mater. Chem. A 5(29), 15031–15037 (2017).
Rajeev Kumar, N. & Radhakrishnan, R. Electronic, optical and mechanical properties of lead-free halide double perovskites using first-principles density functional theory. Mater. Lett. 227, 289–291 (2018).
Yaseen, M. et al. Phase transition and thermoelectric properties of cubic KNbO3 under pressure: DFT approach. J. Mater. Res. Technol. 11, 2106–2113 (2021).
Zhang, X., & Zhao, L.-D. Thermoelectric materials: Energy conversion between heat and electricity. J. Materiomics. 1(2), 92–105 (2015).
Reshak, A. H. Transport properties of Co-based Heusler compounds Co2VAl and Co2VGa: Spin-polarized DFT+U. RSC Adv. 6(59), 54001–54012 (2016).
Kim, W. Strategies for engineering phonon transport in thermoelectrics. J. Mater. Chem. C 3, 10336–10348 (2015).
Nath, P. et al. High throughput combinatorial method for fast and robust prediction of lattice thermal conductivity. Scr. Mater. 129, 88–93 (2017).
Mir, S. A. & Gupta, D. C. Scrutinizing the stability and exploring the dependence of thermoelectric properties on band structure of 3d–3d metal-based double perovskites Ba2FeNiO6 and Ba2CoNiO6. Sci. Rep. 11(1), 10506. https://doi.org/10.1038/s41598-021-90027-7 (2021).
Li, Y., Zhang, L. & Singh, D. J. New stable ternary alkaline-earth metal Pb(II) oxides: Ca/Sr/BaPb2O3 and BaPbO2. Phys. Rev. Mater. 1, 055001 (2017).
Jong, U.-G., Yu, C.-J. & Kye, Y.-H. Computational prediction of structural, electronic, and optical properties and phase stability of double perovskites K2SnX6 (X = I, Br, Cl). RSC Adv. 10, 201–209 (2020).
Fox, M. Optical properties of solids. Am. J. Phys. 70, 1269 (2002).
Nabi, M., Bhat, T. M. & Gupta, D. C. Effect of pressure on electronic, magnetic, thermodynamic, and thermoelectric properties of tantalum-based double perovskites Ba2MTaO6 (M= Mn, Cr). Int. J. Energy Res. 43(9), 4229–4242 (2019).
Yu, L. & Zunger, A. Identification of potential photovoltaic absorbers based on first-principles spectroscopic screening of materials. Phys. Rev. Lett. 108, 6–10 (2012).
Basera, P., Kumar, M., Saini, S. & Bhattacharya, S. Reducing lead toxicity in the methylammonium lead halide MAPbI3: Why Sn substitution should be preferred to Pb vacancy for optimum solar cell efficiency. Phys. Rev. B 101, 054108 (2020).
Acknowledgements
One of the authors, Muskan Nabi wants to acknowledge Chandan Vishwakarma (IIT Delhi) for assistance with SLME calculation and Dr. Tahir M Bhat (Jiwaji University) for his useful discussion and valuable suggestions.
Author information
Authors and Affiliations
Contributions
Both the authors have significant contributions in conducting this research work. M.N. has carried out the calculations and wrote the original manuscript. D.C.G. contributed in analysis and discussion for the results. He also significantly helped in improving the current form of manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nabi, M., Gupta, D.C. Potential lead-free small band gap halide double perovskites Cs2CuMCl6 (M = Sb, Bi) for green technology. Sci Rep 11, 12945 (2021). https://doi.org/10.1038/s41598-021-92443-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-92443-1
This article is cited by
-
Theoretical insight of stabilities and optoelectronic properties of double perovskite Cs2CuIrF6: Ab-initio calculations
Journal of Molecular Modeling (2023)
-
Recent progress of lead-free halide double perovskites for green energy and other applications
Applied Physics A (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
