
Abstract
Lung squamous cell carcinoma (LUSC) possesses a poor prognosis even for stages I–III resected patients. Reliable prognostic biomarkers that can stratify and predict clinical outcomes for stage I–III resected LUSC patients are urgently needed. Based on gene expression of LUSC tissue samples from five public datasets, consisting of 687 cases, we developed an immune-related prognostic model (IPM) according to immune genes from ImmPort database. Then, we comprehensively analyzed the immune microenvironment and mutation burden that are significantly associated with this model. According to the IPM, patients were stratified into high- and low-risk groups with markedly distinct survival benefits. We found that patients with high immune risk possessed a higher proportion of immunosuppressive cells such as macrophages M0, and presented higher expression of CD47, CD73, SIRPA, and TIM-3. Moreover, When further stratified based on the tumor mutation burden (TMB) and risk score, patients with high TMB and low immune risk had a remarkable prolonged overall survival compared to patients with low TMB and high immune risk. Finally, a nomogram combing the IPM with clinical factors was established to provide a more precise evaluation of prognosis. The proposed immune relevant model is a promising biomarker for predicting overall survival in stage I–III LUSC. Thus, it may shed light on identifying patient subset at high risk of adverse prognosis from an immunological perspective.
Similar content being viewed by others
Introduction
Lung cancer remains the leading cause of cancer-related death worldwide, with an estimated 19% 5-year overall survival rate in the United States1. Lung squamous cell carcinoma (LUSC) is a highly aggressive subtype of non-small cell lung cancer (NSCLC), accounting for approximately 30% of all cases2. For patients with resectable LUSC, including stage I–III, relapse is the most common cause of failure3. Biomarkers based on gene expression signature can reliably estimate patient survival and represent a potentially significant adjunct4. Therefore, identification of patients with high risk for recurrence and death is urgently needed.
Accumulating evidence suggests that molecular feature of the tumor microenvironment (TME) has developed as a promising candidate during cancer formation and progression5,6. Immune evasion has been considered as an emerging hallmark of cancer7,8. Recently, immunotherapy targeting immune checkpoints has made remarkable strides in improving LUSC patient survival9,10,11. Certain predictive biomarkers, such as tumor-infiltrating lymphocytes, mutational or neoantigen burden, and T-cell receptor repertoire, are also correlated with prognosis12,13,14,15,16,17. The prevailing view highlights the concept that LUSC exhibits different compositions and functions of the TME18,19. Thus, the molecular signature characterizing immune infiltration in the TME remains to be further investigated5,20,21,22.
Here, we integrated multiple datasets with gene expression to construct and validate an individualized immune-related prognostic model (IPM) for stage I–III LUSC based on immunological genes. Then, we described immune infiltration and mutation burden that are significantly associated with this signature. Further, a more comprehensive investigation was conducted to enhance the predictive value for LUSC prognosis by combing the IPM with clinical characteristics.
Methods
Data acquisition and study population
The gene expression profile and corresponding clinical data of LUSC were retrospectively collected from five public NSCLC cohorts, including one RNA-Seq dataset from The Cancer Genome Atlas (TCGA) LUSC cohort and four microarray datasets from Gene Expression Omnibus (GEO). Only patients with stage I–III squamous carcinoma and complete clinical data were included. We excluded patients with less than ninety-days follow-up. The overview of study design is shown in Fig. S1 and the study characteristics of the included datasets are described in Table S1.
Gene expression data preprocessing
The TCGA LUSC dataset was selected for the training set, which included 440 LUSC and 49 normal tissue samples. For GEO cohorts, the datasets derived from the same microarray platform were combined as validation datasets to improve the statistical power and then eliminated batch effects using the ComBat function by the sva R package (version: 3.28.0)23. Gene expression values were log-transformed and Z-score standardized before removing batch effects for comparable within the same platform.
Development and validation of the IPM
We developed a prognostic signature from immune-related genes (IRGs) derived from the ImmPort database24. First, identification of differentially expressed IRGs between LUSC and normal tissue samples was performed using the limma R package (version: 3.36.5)25 with thresholds of false discovery rate (FDR) < 0.05 and log2 |fold change|> 1. Next, we employed univariate Cox analysis to screen IRGs associated with survival using the survival R package (version: 3.1–8)26. Finally, we established the IPM according to the expression value multiplied by the regression coefficient, which was determined by a stepwise variable-selecting procedure in the multivariable Cox regression model. The risk score formula was as follows: \(Risk\;score = ~\mathop \sum \limits_{{i = 1}}^{n} \beta iXi\), where βi stands for the coefficient of individual gene and \(Xi\) represents gene expression value (Z-score). Then we divided patients into high- or low-risk groups based on the median risk score derived from the training set. Accordingly, the IPM was further calculated in the two validation datasets, respectively.
Estimation of the immune infiltration
To quantify the relative proportion of 22 infiltrating immune cell subtypes in complex tumor tissue, we utilized the CIBERSORT algorithm running with the LM22 signature matrix27. The sum of the fractions for all estimated cell types was set equal to 1 in each tumor sample. Spearman correlations between risk score and the proportion of infiltrating immune cell subtypes were calculated and visualized using the ggstatsplot R package (version: 0.1.4)28. The expression levels were normalized by log2(TPM + 1), where TPM denotes transcripts per million. The Human Protein Atlas database was used to verify the protein function by immunohistochemistry.
Function and pathway enrichment analysis
To obtain the biological understanding of our prognostic model, enrichment analysis of the component IRGs was performed with DAVID (version 6.8)29. We visualized the significant biological processes and pathways using the GOplot (Version: 1.0.2)30 and ggalluvial (Version: 0.11.1)31 R package, respectively.
Immunohistochemical scores
Immunohistochemistry staining was semi-quantitatively evaluated based on the percentage of positive cells and stain intensity. The percentage of positive cells was categorized as 0 (< 5%), 1 (5–25%), 2 (26–50%), 3 (51–75%), 4 (76–100%). Stain intensity was scored as 0 (negative), 1 (weak), 2 (moderate), 3 (strong). The immunohistochemical staining score was calculated by multiplying the two scores.
Mutation analysis
In this study, tumor mutation burden (TMB) was defined as the count of nonsynonymous coding mutations per megabase (Mb). Mutation annotation format was downloaded from the GDC data portal and visualized using the maftools R package (version: 1.8.10)32. Patients were stratified into high TMB (> 4 mutations/Mb) and low TMB (≤ 4 mutations/Mb) groups based on the previous research to define the TMB threshold for personalized immunotherapy in NSCLC33. To determine how the immune-related pathways differ between the two groups, gene set enrichment analysis (GSEA) was performed using Java GSEA software (version 4.0.3)34. The normalized enrichment score (NES) and nominal p value were primary statistics for evaluating the enrichment results.
Construction and evaluation of a predictive nomogram
We combined IPM with clinical variables (including TMB, gender, age, stage) in univariate and multivariate Cox regression analyses. To assess the survival probability of 1-, 3-, 5- year for patients with LUSC, a nomogram was formulated with the rms R package (version: 5.1–4)35 on the basis of multivariate analysis results. In addition, the concordance index (C-index) and ROC curve were utilized to evaluate the prediction accuracy of the nomogram and individual prognostic factors. The calibration curve was performed by comparing the prediction of nomogram with the actual observation after bias correction.
Statistical analysis
Kaplan–Meier survival analysis was performed by survminer R package (version: 0.4.6)36, and differences in overall survival between groups were compared using the log-rank method. The receiver operating characteristic (ROC) curve was performed to verify the predictive ability of the IPM by timeROC R package (version: 0.3)37. All statistical analyses were performed using R (V3.5.1), GraphPad Prism 7.0, or SPSS 24.0. All statistical significance level was accepted at a two-sided p value < 0.05 in this study.
Results
Construction of the IPM
A total of 687 patients with stage I–III LUSC (502 men [73.1%], 185 women [26.9%]; mean age ± SD, 66.7 ± 8.5) were included in this study. For the training set, 465 IRGs were differentially expressed between LUSC and normal tissue samples among 1792 IRGs from the ImmPort database (Fig. 1A). Sixty-one IRGs significantly associated with overall survival were identified after the univariable Cox analysis (Table S2). We developed the IPM according to the combined effect of twenty-four IRGs extracted from the stepwise multivariate Cox regression model (Fig. S2) (Table S3). On the basis of the median immune risk score in the training set, patients were stratified into high- and low-risk groups. The gene expression information and risk distribution are shown in Fig. 1B.

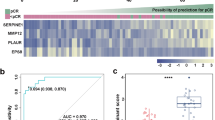
Prognostic analysis of the IPM. (A) Volcano plot of differentially expressed IRGs between tumor and normal tissues. Twenty-four candidates selected for the construction of the IPM are indicated. Gene categories associated with the immune process are shown in different colors. (B) Distribution of risk score and gene expression normalized by Z-score. Kaplan–Meier plots and time-dependent ROC curves according to the IPM in the training set (C,F), validation set 1 (D,G) and validation set 2 (E,F). (C–E) Patients in the high-risk group suffered shorter overall survival (p < 0.001, log-rank test). (F–H) Time-dependent ROC curve validation of the prognostic value of the IPM.
Validation of the IPM
As shown in Fig. 1C, patients with high risk score indicated a significantly worse prognosis than those with low risk score. We performed time-dependent ROC curves to assess the prognostic accuracy of the IPM. The area under curve (AUC) of the prognostic model for overall survival was 0.73 at 1-year, 0.76 at 3-years, 0.69 at 5-years, respectively, in the training set (Fig. 1F). To validate the prognostic power of the IPM, its performance was further evaluated in two independent validation sets. Applying the same algorithm and cutoff value, the IPM divided patients into two groups with high- and low-risk. Similarly, worse prognoses were also shown to correlate with higher risk scores for patients in two validation sets (Fig. 1D,E). The AUC was 0.67, 0.61, 0.60 and 0.73, 0.62, 0.65 in two validation sets, respectively, which showed similar results in predictive ability (Fig. 1G,H). To eliminate the effects of subsequent treatment, we further explored the prognostic performance of the IPM in progression-free survival for LUSC patients. Consistently, patients with high risk displayed worse progression-free survival (Fig. S3).
Immune landscape between the high- and low-risk LUSC patients
Using CIBERSORT algorithm, we evaluated the differences in infiltrating immune cells between high- and low-risk LUSC patients. Figure 2A summarizes the results obtained from 440 LUSC patients. Apparently, the proportion of immune cells in LUSC varies between and within groups (Fig. 2A). Thus, variations in immune cell proportions probably depicted the intrinsic feature which identifies individual differences. Besides, the percentages of different immune cell subpopulations were moderately correlated (Fig. 2B).

Characterization of immune infiltration in high- and low-risk LUSC patients. (A) The relative proportion of 22 infiltrated immune cells in LUSC. (B) Correlation analysis among each immune cell proportions. (C) Violin plots of distribution of significantly different cell subtypes. Significances were measured using the unpaired t-test. (D) Principal components analysis based on the above significantly different immune cells between high- and low-risk LUSC patients.
The high-risk LUSC patients had significantly higher proportions of macrophages M0, dendritic cells activated, T cells CD4 memory resting, neutrophils, and dendritic cells activated, whereas T cells CD8 and plasma cells proportions were relatively lower (Fig. 2C). Similar results were observed for the correlation between risk score and immune cells (Fig. S4). Indeed, the samples of high- and low-risk patients were distinct group‐bias clustering by principal components analysis based on the above significantly different cell subpopulations (Fig. 2D). Taken together, these results surmised that the heterogeneity of immune infiltration in LUSC may serve as a decisive factor that regulates the progress of immunotherapy of cancer.
Tumor-associated macrophage (TAM) therapy aimed at novel TAM targets for modulation of TME showed promising preclinical results38. We observed the relationship between risk score and emerging biomarkers for immune checkpoints. Our results showed that the expression of critical TAM targets (CD47, CD73, SIRPA, and TIM-3), which positively correlated with immune risk score, were significantly upregulated in the high-risk group, portending that the worse prognosis of high-risk LUSC patients is partly because of the immunosuppressive microenvironment (Fig. 3A,B) (Table S4). Additionally, immunohistochemistry staining results validated from the Human Protein Atlas database revealed the CD47, CD73, SIRPA, and TIM-3 protein to be upregulated in lung cancer tissues compared to normal lung tissues (Fig. S5).

Function and pathway enrichment of the immune prognostic model. (A) Violin plots of the expression of immune checkpoints between high- and low-risk groups. Significances were measured using the unpaired t-test. (B) Correlation of the expression of immune checkpoints with risk scores. (C) Circular visualization of the GO function enrichment of biological processes for the IRGs. (D) Sankey plot of the KEGG pathway enrichment for the IRGs.
Function and pathway enrichment of the IPM
To further investigate the underlying biological effects of the IPM, we performed GO and KEGG enrichment analyses to reveal the function and pathway of the twenty-four survival-related IRGs (Fig. 3C,D) (Table S4). Based on these results, the IRGs were mainly enriched in immune response, migration of immune cells, and immune signaling pathway (Fig. 3C,D) (Table S5). These results depicted the hyperactive immune reaction in the LUSC patients with high immune risk.
Relationship between mutational signature and the IPM
To date, higher TMB has been validated prospectively and emerged as a critical biomarker for patient selection in NSCLC12. Previously, we reported a similar founding that TMB was significantly correlated with immunotherapeutic outcome16. To further illuminate the mechanism underlying immune infiltration and survival in LUSC, we associated TMB and the aforesaid immune-related gene signature. We visualized the somatic mutation data utilizing the maftools R package. Here we depicted a waterfall distribution of mutation information of genes in patients from the TCGA dataset, where annotations with various colors indicate the different patterns of the mutation (Fig. 4A). We calculated the count of nonsynonymous mutations per Mb and further divided patients into two groups with high- and low-TMB levels using 4.1 mutations/Mb as the cutoff value. GSEA analysis revealed that low TMB patients were prominent enriched in immune-related biological processes: Leukocyte migration (NES = 1.87, p = 0.002), Cellular defense response (NES = 1.86, p = 0.032), Inflammatory response (NES = 1.80, p = 0.014), Immune response (NES = 1.76, p = 0.045) (Fig. 4B) (Table S6). Conversely, none of the immune-related biological processes were enriched in high TMB patients (Table S7).

Relationship between mutational signature and the IPM. (A) Stacked plots of mutational frequency in individual tumors (histogram, top), mutations in the top 20 genes (tile plot, middle), their mutational counts (histogram, right) and mutational types (bottom). (B) GSEA enrichment plots of immune-related phenotype in low TMB patients compared with that in high TMB patients. (C) Correlation analyses between survival time and risk score based on TMB level. (D) Risk score distribution in high- and low-TMB groups. Data are presented as mean ± SEM. (E) Kaplan–Meier curves showing favorable survival in patients with high TMB. (F) Kaplan–Meier curves showing significantly prolonged survival in patients with high TMB and low risk compared with patients with low TMB and high risk.
Additionally, correlation analyses exhibited that risk score was pronouncedly negatively correlated with overall survival in patients with high TMB and low TMB (Fig. 4C). Consistently, a significant difference was observed between the two groups (Fig. 4D). Similar to the IPM, we found that patients with low levels of TMB had poor survival (Fig. 4E). Combining the risk score of IPM with TMB level, patients with low levels of TMB and high immune risk suffered significantly shorter survival compared to those with high TMB and low immune risk (Fig. 4F).
The IPM is an independent prognostic factor
Univariate and multivariate Cox regression analyses were performed to determine whether the IPM acts as an independent prognostic indicator for patients with LUSC. Clinical factors including TMB, age, gender, stage, and IPM were closely related to patient survival (Fig. 5A). After adjusting for other clinical parameters, the IPM remained as an independent significant predictor for LUSC prognosis (HR 1.602, 95% CI 1.301–1.919, p < 0.001) (Fig. 5A). Besides, the expression levels of IRGs were significantly associated with age, gender, and stage (Fig. S6). Furthermore, C-index values were calculated to compare the prognostic performance of each characteristic. C-index estimates the prediction concordance between predicted and actual survival, which ranges from 0.5 (random estimation) to 1 (perfect discrimination)39. The IPM showed a higher mean C-index (0.670) than other clinical characteristics (0.524 to 0.549) (Fig. 5B). In summary, these data confirmed that the IPM possessed valuable predictive capacity independent of other clinical factors.

Nomogram development and validation based on the IPM. (A) Univariate and multivariate Cox regression analysis of the prognostic value of clinical features and the IPM in TCGA LUSC cohort. Red squares indicate adverse outcomes, and green squares indicate favorable outcomes. (B) C-index showing the prognostic performance of clinical features and the IPM. (C) Nomogram integrating clinical characteristics with the IPM for predicting 1-,3-, 5-year survival probability. (D) Calibration plot for internal validation of the nomogram. (E) Time-dependent ROC curves for evaluating the predictive efficacy of the nomogram.
Nomogram development and validation based on the IPM
Next, we established a nomogram that integrated clinical risk factors with the IPM to quantify the survival probability in LUSC patients (Fig. 5C). Consistent with our previous finding, the nomogram illustrated the IPM as the prevailing contribution to prognosis compared with conventional clinical characteristics. Each variable was allocated a score on the point scale. By adding together the total score, the probability of survival was available by drawing a line vertically down to the survival axis. With the same 1000 random splits, the C-index of the nomogram was 0.735 (95% CI 0.691–0.779), which showed good concordance between prediction and observation (Fig. 5D). In addition, the ROC curve was delineated to compare nomogram predictive efficacy with other prognostic factors. Likewise, the nomogram exhibited the largest AUC (Fig. 5E). The AUC of the nomogram for overall survival was 0.79 at 1-year, 0.81 at 3-years, 0.75 at 5-years, respectively. In conclusion, the nomogram displayed better predictive power in both short- and long-term survival in LUSC patients relative to individual prognostic factors.
Discussion
Patients with early-stage and local advanced LUSC are at considerable risk of relapse and death, even after complete surgical excision. With the great advances in immunotherapy, current cancer-related researches focus on the essential role of the immune system. Reliable prognostic biomarkers based on immune characteristics are demanded to predict the risk of cancer. Previous studies have proposed immune-related signatures for survival stratification in NSCLC18,40,41,42,43. However, the further discrimination of LUSC patients by local immune features remains poorly understood. To our knowledge, this study firstly systematic reports the attributes and clinical significance of the IPM in patients with stage I–III LUSC.
In this study, we established a prognostic model based on twenty-four IRGs and validated it in multiple public datasets to describe the immune status of LUSC patients and explore their prognostic potential. The IPM stratified patients into groups with markedly different survival benefits. We could identify patients with LUSC who probably carried a high risk of poor prognosis. Further, integrated analysis combing the IPM with clinical factors was performed to provide a more precise evaluation of overall survival in LUSC.
The immune system can both restrain or facilitate malignant progression, which proceeds in three phases denominated as elimination, equilibrium, and escape according to the hypothesis of cancer immunoediting44. During the escape phase, tumor develops the capacity to circumvent immune recognition which may result from the immunosuppressive state within the TME44,45. At the clinical level, evading immune destruction involves in the elevation of immunosuppressive cells (e.g., TAMs and Treg cells), the expression of immune checkpoint molecules (e.g., CTLA-4 and PD-L1), and the reduction of mutation-derived tumor neoantigens46,47,48. Release from immunosuppression will open the door for immunotherapy that directly stimulate the antitumor immune response. Here, we identified specific populations of immune cells between high- and low-risk patients as well as possible mechanisms to enhance the antitumoral activity. Generally, high-risk LUSC patients possessed a higher proportion of immune suppressive cells such as macrophages M0, while a lower proportion of immune effector cells such as T cells CD8 and plasma cells. These results also increased the reliability of the predictive value of the IPM.
Moreover, the expression of critical TAM targets (CD47, CD73, SIRPA, and TIM-3) was significantly upregulated in the high-risk group. As previously described, TAMs induce immune suppression through various approaches, including negatively modulating the activation of T cells and NK cells49, expressing immune checkpoint ligands that directly constrain T cell functions50, and releasing chemokines to recruit Treg cells in the TME51. Notably, CD47 expression on tumors shuts down macrophages by binding to SIRPA to avoid phagocytosis, termed do not eat me signal52. CD73, a newly admitted immune checkpoint mediator, is highly expressed on tumors and multiple cellular components in TME and inhibits the antitumor immune response47. The interaction between TIM-3 with immune cell signaling components facilitates the suppression of antitumor immunity53. Therefore, in this context, the risk score identified the immune checkpoints compatible with the power of tumor-infiltrating immune cells, indicating that the poor clinical outcome of patients with high risk may as a consequence of the immunosuppressive environment. Based on the above findings, immune dysregulation may be responsible for the differential prognoses between patient groups identified by our prognostic model.
A minority of tumor-specific mutations can lead to the formation of neoantigens that are recognized and targeted by the immune system, and TMB can act as an effective estimation of tumor neoantigen load54. In NSCLC patients receving immunotherapy, high TMB presents a strong correlation with improved response and durable benefit14. In this study, we demonstrated that the high-risk group showed lower TMB, which correlate with worse survival. When further stratified according to the TMB levels and risk score of IPM, patients with high TMB and low risk had a remarkable improved overall survival, underlying the intense immune response triggered by somatic mutations.
By integrating clinical risk factors with the IPM, we established a nomogram that displayed better survival predictive power and potential utility in guiding clinical use. For patients with early-stage LUSC, neoadjuvant and adjuvant immune checkpoint inhibitor therapies showed promising results in survival benefit55. Revealing the status of patients’ immune response using standardized immune assays is becoming an essential requirement to guide the optimal therapeutic intervention56. The development of novel immunotherapy strategies should focus on the tumor-infiltrating immune cell network instead of targeting a single kind of immune cell or immune-related gene. The IPM-based nomogram may have the potential to estimate prognosis and provide insight into guiding individualized clinical management for LUSC patients after surgery.
Some limitations of our study need to be acknowledged include its retrospective nature, and transcriptomic analysis cannot provide a complete molecular picture of the immune environment. In addition, further biological experiments are required for the rigorous validation of the IPM in LUSC.
In summary, the proposed IPM is a promising biomarker for the prediction and stratification of stage I–III LUSC patients. It was proven to act as an independent prognostic factor and reflect the overall intensity of the immune response. This work demonstrates an immune-related model associated with stage I–III LUSC and may contribute to the individualized management. Further, prospective studies are warranted to verify the predictive efficacy of IPM for LUSC.
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2020. CA 70(1), 7–30 (2020).
Travis, W. D. Pathology of lung cancer. Clin. Chest Med. 32(4), 669–692 (2011).
Uramoto, H. & Tanaka, F. Recurrence after surgery in patients with NSCLC. Transl. Lung Cancer Res. 3(4), 242–249 (2014).
Ramaswamy, S. et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc. Natl. Acad. Sci. U.S.A. 98(26), 15149–15154 (2001).
Gentles, A. J. et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 21(8), 938–945 (2015).
Angell, H. & Galon, J. From the immune contexture to the Immunoscore: The role of prognostic and predictive immune markers in cancer. Curr. Opin. Immunol. 25(2), 261–267 (2013).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: The next generation. Cell 144(5), 646–674 (2011).
Munn, D. H. & Bronte, V. Immune suppressive mechanisms in the tumor microenvironment. Curr. Opin. Immunol. 39, 1–6 (2016).
Govindan, R. et al. Phase III trial of ipilimumab combined with paclitaxel and carboplatin in advanced squamous non-small-cell lung cancer. J. Clin. Oncol. 35(30), 3449–3457 (2017).
Rizvi, N. A. et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): A phase 2, single-arm trial. Lancet Oncol. 16(3), 257–265 (2015).
Topalian, S. L. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366(26), 2443–2454 (2012).
Hellmann, M. D. et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 378(22), 2093–2104 (2018).
Tumeh, P. C. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515(7528), 568–571 (2014).
Rizvi, N. A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (New York, N.Y) 348(6230), 124–128 (2015).
Han, J. et al. TCR repertoire diversity of peripheral PD-1(+)CD8(+) T cells predicts clinical outcomes after immunotherapy in patients with non-small cell lung cancer. Cancer Immunol. Res. 8(1), 146–154 (2020).
Zhang, K. et al. Identification of deleterious NOTCH mutation as novel predictor to efficacious immunotherapy in NSCLC. Clin. Cancer Res. 26(14), 3649–3661 (2020).
Liu, Y. Y. et al. Characteristics and prognostic significance of profiling the peripheral blood T-cell receptor repertoire in patients with advanced lung cancer. Int. J. Cancer 145(5), 1423–1431 (2019).
Mazzaschi, G. et al. Low PD-1 Expression In Cytotoxic CD8(+) tumor-infiltrating lymphocytes confers an immune-privileged tissue microenvironment in NSCLC with a prognostic and predictive value. Clin. Cancer Res. 24(2), 407–419 (2018).
Coussens, L. M., Zitvogel, L. & Palucka, A. K. Neutralizing tumor-promoting chronic inflammation: A magic bullet?. Science (New York, NY) 339(6117), 286–291 (2013).
Bindea, G. et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39(4), 782–795 (2013).
Rooney, M. S., Shukla, S. A., Wu, C. J., Getz, G. & Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160(1–2), 48–61 (2015).
Suzuki, K. et al. Prognostic immune markers in non-small cell lung cancer. Clin. Cancer Res. 17(16), 5247–5256 (2011).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics (Oxford, England) 28(6), 882–883 (2012).
Bhattacharya, S. et al. ImmPort: Disseminating data to the public for the future of immunology. Immunol. Res. 58(2–3), 234–239 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7), e47 (2015).
Therneau, T. M. A Package for Survival Analysis in S. version 2.38. https://CRAN.R-project.org/package=survival. (2015).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12(5), 453–457 (2015).
I P. ggstatsplot: 'ggplot2' Based Plots with Statistical Details. https://doi.org/10.5281/zenodo.2074621 (2018).
da Huang, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4(1), 44–57 (2009).
Walter, W., Sánchez-Cabo, F. & Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics (Oxford, England) 31(17), 2912–2914 (2015).
Brunson, J. C. ggalluvial: Alluvial Plots in 'ggplot2'. https://CRAN.R-project.org/package=ggalluvial. (2019).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28(11), 1747–1756 (2018).
Devarakonda, S. et al. Tumor mutation burden as a biomarker in resected non-small-cell lung cancer. J. Clin. Oncol. 36(30), 2995–3006 (2018).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102(43), 15545–15550 (2005).
Jr FEH. rms: Regression Modeling Strategies. https://CRAN.R-project.org/package=rms (2019).
Kassambara, A., Kosinski, M. & Biecek, P. survminer: Drawing Survival Curves Using 'ggplot2'. https://CRAN.R-project.org/package=survminer. (2019).
Blanche, P., Dartigues, J. F. & Jacqmin-Gadda, H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat. Med. 32(30), 5381–5397 (2013).
Cassetta, L. & Pollard, J. W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 17(12), 887–904 (2018).
Mayr, A. & Schmid, M. Boosting the concordance index for survival data: A unified framework to derive and evaluate biomarker combinations. PLoS ONE 9(1), e84483 (2014).
Seo, J. S. et al. Whole exome and transcriptome analyses integrated with microenvironmental immune signatures of lung squamous cell carcinoma. Cancer Immunol. Res. 6(7), 848–859 (2018).
Li, B., Cui, Y., Diehn, M. & Li, R. Development and validation of an individualized immune prognostic signature in early-stage nonsquamous non-small cell lung cancer. JAMA Oncol. 3(11), 1529–1537 (2017).
Sorich, M. J., Rowland, A., Karapetis, C. S. & Hopkins, A. M. Evaluation of the lung immune prognostic index for prediction of survival and response in patients treated with atezolizumab for NSCLC: Pooled analysis of clinical trials. J. Thorac. Oncol. 14(8), 1440–1446 (2019).
Xu, Z., Yan, Y., Wang, X., Zeng, S. & Gong, Z. Lung immune prognostic index for outcome prediction to immunotherapy in patients With NSCLC. J. Thorac. Oncol. 14(9), e207–e208 (2019).
Schreiber, R. D., Old, L. J. & Smyth, M. J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science (New York, NY) 331(6024), 1565–1570 (2011).
Radoja, S., Rao, T. D., Hillman, D. & Frey, A. B. Mice bearing late-stage tumors have normal functional systemic T cell responses in vitro and in vivo. J. Immunol. 164(5), 2619–2628 (2000).
Schumacher, T. N. & Schreiber, R. D. Neoantigens in cancer immunotherapy. Science (New York, NY) 348(6230), 69–74 (2015).
Allard, B., Longhi, M. S., Robson, S. C. & Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 276(1), 121–144 (2017).
Mahoney, K. M., Rennert, P. D. & Freeman, G. J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 14(8), 561–584 (2015).
Morandi, F. & Pistoia, V. Interactions between HLA-G and HLA-E in physiological and pathological conditions. Front. Immunol. 5, 394 (2014).
Santarpia, M. & Karachaliou, N. Tumor immune microenvironment characterization and response to anti-PD-1 therapy. Cancer Biol. Med. 12(2), 74–78 (2015).
Noy, R. & Pollard, J. W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 41(1), 49–61 (2014).
Jaiswal, S. et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 138(2), 271–285 (2009).
Das, M., Zhu, C. & Kuchroo, V. K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 276(1), 97–111 (2017).
Chan, T. A. et al. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 30(1), 44–56 (2019).
Forde, P. M. et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N. Engl. J. Med. 378(21), 1976–1986 (2018).
Bruni, D., Angell, H. K. & Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 20(11), 662–680 (2020).
Funding
This study was supported by the National Key Research and Development Program of China (2016YFC1303800) and National Natural Science Foundation of China (81773056 & 82073179). The funders had no role in study design, data collection, data analysis, interpretation and writing of the manuscript.
Author information
Authors and Affiliations
Contributions
Y.H. and L.L. contributed to the study design. Q.F.Y., J.W., L.B. performed the data curation and analysis. D.W., C.T., Y.T.L. analyzed and interpreted the results. Q.F.Y. drafted and reviewed the manuscript. D.W. helped revise the manuscript. All authors read and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, QF., Wu, D., Wang, J. et al. Development and validation of an individualized immune prognostic model in stage I–III lung squamous cell carcinoma. Sci Rep 11, 12727 (2021). https://doi.org/10.1038/s41598-021-92115-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-92115-0
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
