
Abstract
We test the hypothesis that endothelial cells adopt an inflammatory phenotype in functionally intact aged human subjects with radiographic evidence of white matter hyperintensity (WMH) suggestive of small cerebrovascular disease. Components of all three complement effector pathways and regulatory proteins were quantified in extracts of plasma endothelial-derived exosomes (EDE) of 11 subjects (age 70–82) with and 15 without evidence of WMH on MRI. Group differences and associations with plasma markers of immune activation (IL6, ICAM1), cognition and neuroimaging were calculated via regression modelling. EDE complement factors within the alternative and classical pathways were found to be higher and regulatory proteins lower in subjects with WMH. EDE levels of some complement components demonstrated significant associations with cognitive slowing and elevated systolic blood pressure. The inhibitor of the membrane attack complex, CD46, showed a significant positive association with cerebral grey matter volume. Plasma inflammatory markers, IL6 and ICAM1, were positively associated with EDE levels of several complement components. These findings provide the first in vivo evidence of the association of endothelial cell inflammation with white matter disease, age-associated cognitive changes, and brain degeneration in functionally normal older individuals. Future endothelial biomarker development may permit recognition of early or preclinical stages of vascular contributions to cognitive impairment and dementia.
Similar content being viewed by others

Introduction
Cerebrovascular disease and the associated blood–brain barrier (BBB) dysfunction are intimately associated with immune activation and among the most common age-associated, inflammation-mediated, degenerative brain changes. Vascular pathways are emerging as an important contributor to neurodegenerative disorders1,2,3,4. Importantly, immuno-vascular dysregulation can cause pathological systemic-brain cross-talk5 in early disease states, prior to frank brain degeneration and clinical manifestations such as mild cognitive impairment6,7. More recently, molecular pathways are emerging to suggest a feed-forward degenerative-inflammatory phenomenon between endothelial cells, innate immune activation, and degenerative myelin debris8. Therefore, identification of molecular biomarkers of immuno-vascular disease in preclinical states has important therapeutic implications for extension of health span, treatment of vascular cognitive impairment and associated neurodegenerative processes, such as Alzheimer’s disease9. However, detection of early or preclinical cellular dysfunctions has been challenging.
In light of the inaccessibility of brain cells, neuroimaging techniques have been developed to capture indirect consequences of cerebrovascular disease (CVD), such as white matter hyperintensities (WMH) on T2/Flair (fluid-attenuated inversion recovery) imaging. However, imaging alone does not suffice, as the underlying molecular etiology of radiographic white matter changes10 can be diverse in aging and across neurodegenerative disorders. Exosomal molecular cargo have emerged as promising biomarkers of disease processes within numerous organ systems, including the brain11,12,13,14,15,16. Exosomes are plasma membrane- and endosomal-derived vesicles, that are released by most cell types and contain cargo molecules from their cell of origin including proteins, mRNAs, microRNA and lipids17,18. Cerebral cell-derived exosomes range in diameter from 30 to 220 nm and are capable of crossing the blood–brain barrier19. Analyses of exosome-derived molecules isolated from bodily fluids are providing an unprecedented ability to non-invasively investigate molecular changes in specific cells in vivo, with great impact on diagnostics of diseases affecting inaccessible organs20.
In the aging brain, WMH on MRI can frequently be seen preclinically and presumed secondary to immuno-vascular effects, involving injury of endothelial cells (ECs) at the blood–brain barrier (BBB) and increased CNS immune activation and reactive gliosis21. Employing a precision medicine approach in functionally-intact subjects, we investigated concentrations of complement factors of innate immunity derived from endothelial-derived exosomes (EDEs) isolated from plasma to provide the first in vivo test of the hypothesis that endothelial cells take on an inflammatory phenotype in association with radiographic evidence of WMH.
Complement factors, essential for innate immunity, form a collection of more than thirty soluble proteins that work with leukocytes to protect the host from pathogens. Complement factors are divided into three distinct but highly connected pathways: the classical, the alternative, and the lectin pathways, as well as complement regulatory proteins (Supplemental Fig. S1). When dysregulated, complement activation can cause robust destruction of host cells. Complement factors also increase cytokine and chemokine production, amplifying inflammation and leading to activation and recruitment of immune cells22,23. Interactions between inflammatory cytokines and EC complement proteins may enable inflammatory homeostasis or exacerbate a dysfunctional state. Mammalian ECs in culture have been shown to produce many complement proteins including C1, C4, C3, factor B, and factors I and H24.
At a time when targeted molecular therapies are being developed, a major challenge has been the low specificity of fluid and neuroimaging biomarkers for quantification of endothelial contributions to BBB dysfunction and neurodegeneration. EDE biomarkers allow direct interrogation of endothelial cells and the investigation of associations with risk factors and downstream pathological changes. In this proof of concept study, we investigate levels of complement factor proteins and their regulatory proteins in endothelial cells of functionally normal subjects with WMH by using EDE cargo analyses as a means of performing a “liquid biopsy” of inaccessible endothelial cells in vivo. We then investigate the association of EDE complement factors with an important vascular risk factor, blood pressure, as well as downstream changes to brain structure and function.
Methods
Study participants
We minimized bias, as described25, by prospectively performing consecutive sampling of functionally intact, older study participants from ongoing longitudinal studies of brain aging at the Memory and Aging Center at UCSF (NIH Aging and Cognition study; Larry J. Hillblom foundation study; NIH Chronic Inflammation study) to participate in this study. The inclusion criteria for all subjects were intact daily functioning per an informant (Clinical Dementia Rating = 0), neuropsychological performances within normative standards, and absence of significant clinical neurological disease assessed by history and physical exam. The study participants were selected based on evidence of presence or absence of white matter injury on brain MRI. We selected 11 cerebral small vessel disease (cSVD) cases classified based on global cerebral volume of WMH on T2/FLAIR, corresponding to modified Fazekas score of > = 2. We selected 15 controls who had no significant WMH on brain MRI (Fazekas = 0), or other significant abnormalities, such as focal atrophy. All images were rated for burden of WMH by a board-certified neurologist (FME) in addition to being reviewed by a neuroradiologist to rule out other significant abnormalities. At the time of enrollment in the current study, blood was drawn from each participant for preparation of platelet-poor-plasma according to methods published by Goetzl et al.26. Plasma samples were aliquoted and stored at − 80 °C. All study participants provided informed consent and the study protocols were approved by the UCSF Human Research Protection Program and Institutional Review Board. Research was performed in accordance with the Code of Ethics of the World Medical Association.
Cognition (processing speed)
Participants completed a modified version of the Trail-Making Test, a measure of speeded set-shifting27, which is commonly affected by cSVD. This task requires participants to sequentially alternate between numbers and days of the week as quickly as possible. The outcome variable is the time taken to perform the task (log transformed to normalize the distribution).
Neuroimaging evaluation
MRI acquisition
Subjects were scanned on a Siemens Prisma 3 T scanner at the UCSF Neuroscience Imaging Center. A T1-weighted Magnetization-prepared rapid gradient echo (MP-RAGE) structural scan was acquired in a sagittal orientation, a field-of-view of 256 × 240 × 160 mm with an isotropic voxel resolution of 1 mm3, TR = 2300 ms, TE = 2.9 ms, TI = 900 ms, flip angle = 9°. The T2 fluid attenuated inversion recovery (FLAIR) acquired in the axial orientation, field-of-view = 176 × 256 × 256 mm, resolution 1.00 × 0.98 × 0.98 mm3, TR = 5000 ms, TE = 397 ms, TI = 1800 ms.
MRI processing and analyses
De-identified digital information was transferred from UCSF using secure and HIPAA complaint DICOM server technology. Images were processed by the Imaging of Dementia and Aging (IDeA) lab at UC Davis and full imaging protocol details are reported in prior publications28,29,30,31. In brief, WMH quantification was performed on a combination of FLAIR and 3D T1 images using a modified Bayesian probability structure based on a previously published method of histogram fitting. Prior probability maps for WMH were created from more than 700 individuals with semi-automatic detection of WMH followed by manual editing. Likelihood estimates of the native image were calculated through histogram segmentation and thresholding. All segmentation was initially performed in standard space resulting in probability likelihood values of WMH at each voxel in the white matter. These probabilities were then thresholded at 3.5 SD above the mean to create a binary WMH mask. Further segmentation was based on a modified Bayesian approach that combines image likelihood estimates, spatial priors, and tissue class constraints. The segmented WMH masks were then back-transformed on to native space for tissue volume calculation. Volumes were log-transformed to normalize population variance.
Enrichment of plasma EDEs and extraction of cargo proteins
Platelet-poor plasma was prepared from 6 ml of venous blood and stored in 0.5 ml aliquots at − 80 °C as previously described26 and EDE were enriched as previously published26. Briefly, after depletion of platelets, EDE exosomes were enriched by sequential immunoprecipitation with two biotinylated monoclonal antibodies to CD31 (MEM-05, Thermo Fisher Scientific) and then CD146 (Novus Biologicals, Littleton, CO, USA) prior to lysis of exosomes for quantification of cargo proteins via ELISA. Sequential rounds of immunoprecipitation aimed to enhance selectivity. We used Nanosight NS300 instrument (Malvern Instruments, Malvern, UK) in combination with an EV membrane label, ExoGlow (System Biosciences, Palo Alto, CA) to confirm that the particle sizes of total exosomal extracts were within range for exosomes (30–220 nm).
Nanoparticle tracking analysis
NanoSight NS300, with the Blue488 laser and sCMOS camera, was used in combination with ExoGlow (System Biosciences, Palo Alto, CA) to quantify the total EV population (5 videos, 30 s each) and confirm that size distribution is within range for exosomes (30–220 nm) (Supplemental Fig. S2). After capture, the videos were analyzed using the NTA 3.3 Dev Build 3.3.104 software, with a detection threshold of 4 and screen gain of 10. As expected, the non-fluorescent NTA captured non EV vesicles with a wider size range (peak ~ 100 nm), while fluorescent NTA with EV membrane dye demonstrated a peak concentration of around 30-50 nm and lower overall concentration of particles, likely due to increased specificity for EVs.
Enzyme-linked immunosorbent assay quantification of proteins
The following ELISA kits were used for the quantification of exosomal proteins: tetraspannin exosome marker CD81 (Cusabio; American Research Products, Waltham, MA, USA), complement fragment C4b (Cusabio Technology, College Park, MD), complement receptor 1, and decay accelerating factor (CR1, DAF; ARP American Research Products, Waltham, MA), factor I (Cloud-Clone Corp, Katy, TX), complement fragment C3d and CD46 (LifeSpan Biosciences, Seattle, WA), complement fragment C3b, Factor B, C1q portion of the C1 complement complex (Abcam, Cambridge, MA), Bb fragment of complement factor B (Quidel-Microvue, San Diego, CA), terminal complement complex C5b-C9 (Elabscience, Bethesda, MD), CD59 and mannose-binding lectin (MBL; Ray Biotech, Norcross, GA), and complement factor D (ThermoFisher-Invitrogen, LaFayette, CO). The mean value for all determinations of CD81 in each assay group was set at 1.00, and relative values of CD81 for each sample were used to normalize their recovery. CD81 was used as surrogate measures of exosome concentration against which each protein was normalized.
Quantification of plasma analytes
Plasma cytokine concentrations were measured by high-performance electrochemiluminiscence (HPE) using the multiplex V-PLEX Human Proinflammatory (IL-6) and Human Vascular Injury (ICAM1) assays. We selected IL-6 and ICAM1 to represent global levels of systematic inflammation and immune activation. The multiplex arrays were analyzed with a MESO QuickPlex SQ 120 imager (MSD, Rockville, MD) and Discovery Workbench v4.0 software. Concentrations were obtained in duplicate per each sample in accordance with the manufacturer’s protocol.
Statistical analyses
Statistical analyses were performed using JMP Pro and PRISM. Significantly skewed variables were log transformed to normalize distributions. For comparison of demographic and EDE proteins of interest between cases and controls, Chi-squared tests (for categorical variables) and Student’s t-tests (for continuous measures) were conducted. Linear models were additionally conducted to determine group EDE differences, adjusting for age, and discriminant analyses were employed for ROC analyses to determine group classification accuracy of EDE biomarkers. Finally, linear models examined associations between EDE levels with plasma inflammatory markers, blood pressure, cognitive speed, and structural brain MRI outcomes, adjusting for age and TIV, as appropriate. We also performed correction for multiple comparisons using the Benjamini–Hochberg False Discovery Rate (FDR)32. For illustration of results, all p-values were rounded to one non-zero digit beyond the decimal.
Results
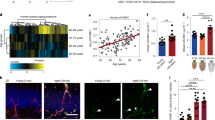
Demographics (Table 1, Fig. 1)

Demographic, cognitive and vascular risk group comparisons between subjects with WMH and without. Demographics include age, gender, and education; vascular risk factors include systolic and diastolic blood pressure, as well as clinical labs: hemoglobin A1C, insulin, homeostatic model assessment of insulin resistance, total cholesterol, low density lipoprotein, ratio of low to high density lipoprotein, and triglycerides; neuroimaing: white matter hyperintensity, global white matter volumes, global grey matter volumes, and mean of bilateral hippocampal volumes. For volumetric neuroimaging measures, total intracranial volumes were regressed out and residual used in models. Dependent variables were log transformed as needed to approximate normal distributions. All significance levels are from ANCOVA for continuous variables controlling for age, and Chi-square for gender. The graphs show plots of raw values, however the significance of statistical models are for ANCOVA models. The dissociation between statistical models built with age-adjusted versus un-adjusted values is most notable for WMV in this figure. SBP systolic blood pressure; DBP diastolic blood pressure; HgbA1c hemoglobin A1C; HOMA-IR homeostatic model assessment of insulin resistance; LDL low density lipoprotein; HDL high density lipoprotein; MMSE mini-mental status examination; WMH white matter hyperintensity; WMV white matter volume; GMV grey matter volume. Stars depict p-value significance levels from Student's t tests: ****; ≤ 0.001 = ***; ≤ 0.01 = **; ≤ 0.05 = *; ≥ 0.05 = ns.
Numerical values are summarized in Table 1. The mean age for subjects with WMH was significantly higher than controls (p = 0.0003). We therefore controlled for age in all our analyses. There were no significant differences with respect to sex and educational attainment between groups. The global measure of cognitive function, mini-mental status exam (MMSE), did not significantly differ between groups (p = 0.60). With respect to vascular risk factors, subjects with WMH had on average higher systolic blood pressures (p = 0.0005). No significant difference was noted for diastolic blood pressure (p = 0.70). Diagnosis of hypercholesterolemia (p = 0.90), or hyperglycemia (p = 0.30) did not differ between groups, nor were any significant differences found in laboratory values for fasting lipid panel (triglycerides p = 0.40; HDL p = 0.40; and LDL p = 0.10), blood insulin concentration (p = 0.90), hemoglobin-A1C (p = 0.40), and the homeostatic model assessment of insulin resistance (HOMA-IR) (p = 0.60)—a surrogate for assessing β-cell function.
Group differences (Table 2, Fig. 2)


Group comparison of EDE cargo in group of subjects with WMH ( +) versus without (−). Complement factors are grouped by pathway. C3b and C5b-9 grouped under alternative pathway are, as depicted in Supplemental Fig. S1, shared across classical, and alternative pathways. Stars depict significance levels from ANCOVA models, controlling for age, detailed in Table 1: ≤ 0.0001 = ****; ≤ 0.001 = ***; ≤ 0.01 = **; ≤ 0.05 = *; ≥ 0.05 = ns.
Numerical values are summarized in Table 2. Results are consistent with the hypothesis of endothelial inflammatory changes in subjects with WMH. No group differences were noted with respect to EV concentrations or EV size distributions (Supplemental Fig. S2). Levels of EDE complement effector proteins in the classical and alternative pathways were significantly elevated and complement regulatory proteins were significantly lower in subjects with WMH. After normalizing for EDE concentrations, we found the following factors to have significantly higher levels in subjects with WMH with large effect sizes: C1q (p = 0.002), C3b (p < 0.0001), Factor B (p < 0.0001), Bb (p < 0.0001), and C5b-9 (p = 0.0002). EDE levels of the complement regulatory proteins Factor I (p = 0.02), and CD59 (p < 0.0001) were significantly lower in subjects with WMH. Results remained significant when adjusting for age in a regression model. The following complement factors and regulatory proteins did not reach statistical significance between groups: Factor D (p = 0.40), C4b (p = 0.60), CR1 (p = 0.60), DAF (p = 0.10), CD46 (p = 0.90), and lectin pathway MBL (p = 0.50).
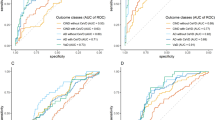
Individual markers had AUC values ranging 0.74 to 0.99 (Table 2). The greatest group effect sizes were found for C3b, Bb and CD59. For these EDE complement proteins, the likelihood ratio of having significant WMH in comparison to none or mild WMH was found to be 13.64 at sensitivity of 91% (95% CI 59–100) and specificity of 93% (95% CI 68–100). The associated optimal diagnostic accuracy cut off was > 4.7 ng/mL for C3b, > 28.609 ng/mL for Bb, and < 0.225 ng/mL for CD59.
Relation with secreted plasma cytokine and chemokine markers (Table 3)
Significant positive associations were found between systemic inflammatory cytokine IL6 and EDE C3b (β = 0.44; p = 0.03), C5b-9 (β = 0.43; p = 0.03), Bb (β = 0.40; p = 0.05), and Factor B (β = 0.42; p = 0.04), and negative association with CD59 (β = − 0.63; p = 0.0009). We also found a significant association between the endothelial and macrophage expressed adhesion molecule, ICAM1, and EDE C1q (β = 0.39; p = 0.05), (C3b (β = 0.41; p = 0.04), C5b-9 (β = 0.44; p = 0.03), Bb (β = 0.51; p = 0.009), and Factor B (β = 0.47; p = 0.02), and negative association with CD59 (β = − 0.59; p = 0.002).
Relation with blood pressure (Table 4)
Significant positive associations were found between an important vascular risk factor, systolic blood pressure, and C1q (β = 0.46; p = 0.05) and Bb (β = 0.58; p = 0.02) of the classical and alternative pathways, respectively. A significant negative association was found between systolic blood pressure and CD59 (β = − 0.62; p = 0.02), a complement regulatory protein.
Relation with brain imaging (Table 4)
Levels of C3b and C5b-9 involved in both the classical and alternative pathways, and Factor B and Bb of the alternative pathway, were significantly positively associated with global burden of WMH (p-value range 0.001–0.05 detailed in Table 4). DAF levels were significantly inversely associated with global volume of WMH (R2 = 0.57, β = − 42, p = 0.02). CD46, a key regulatory protein of the end membrane attack complex (Supplemental Fig. S1), was significantly associated with total grey matter volumes, controlling for total intracranial volume and age (R2 = 0.45, β = 0.43, p = 0.03).
Relation with cognitive function (Table 4)
Higher levels of EDE complement proteins C1q (β = 0.59, p = 0.05) and Bb (β = 0.71, p = 0.005) were associated with significantly lower cognitive function as measured by set-shifting performance speed, controlling for age in all analyses.
Discussion
We had previously shown high sensitivity and specificity of EDE BBB markers for vascular-mediated brain injury (WMH)25. In this study, we extended our previous work into EDE markers of innate inflammation to investigate the hypothesis that endothelial inflammation is involved in white matter injury in functionally normal elders with evidence of WMH on brain imaging. To this end, EDE concentrations of two complement factors in the classical (C1q, and C4b), two in classical and alternative (C3b, and C5b-9), three in the alternative (Factor B, Bb, and Factor D), and one in the lectin pathway (MBL), along with five regulatory proteins (CD59, Factor I, CR1, DAF, and CD46) were compared between groups of subjects with and without evidence of WMH on brain imaging. Overall, our results suggest significant activation of alternative and classical complement pathways in tandem with decreased levels of protective complement regulatory proteins in subjects with WMH.
We demonstrated significant differences for C1q, C3b, C5b-9, Factor B, and Bb, as well as CD59 and Factor I regulatory proteins. All were large effect sizes, particularly for proteins within the alternative pathways Bb, Factor B, and C3b. The largest group differences were found for factors C3b and Bb, such that discriminant function of subjects with WMH versus without reached an AUC of 0.99 for either factor alone. These findings implicate both the classical and alternative complement pathways, and the resulting opsonizing C3b and the C5b-9 membrane attack complex, in inflammatory endothelial contributions to WMH. The lack of clear difference in EDE MBL levels in subjects with and without WMH suggests that the lectin pathway may not be an important contributor to cerebral endothelial inflammation in functionally normal subjects with WMH. Taken together, the presented findings support involvement of endothelial inflammation in WMH and associations with slowed processing speed in aging. Complement factors involved in innate immune dysfunction are emerging as important molecular contributors to neuronal dysfunction in Alzheimer’s disease23,33,34,35. A prior study has shown activation of alternative and classical complement pathways in astrocytes in AD via astrocytic-derived exosomal analyses36. Therefore, the group differences noted in this study likely increase risk of future cognitive decline and Alzheimer’s disease.
In addition to the investigation of group differences in levels of EDE complement markers, we investigated the associations of complement factors with critical vascular risk factors, such as blood pressure, systemic inflammation (plasma IL6 levels), and immune activation (plasma ICAM1 levels), as well as downstream consequences of cerebral vascular disease, such as white matter injury (as a continuous variable), grey matter atrophy, and slowed processing speed. Bb, a key factor in the alternative pathway emerged as significantly associated with both upstream and downstream vascular risk and injury, respectively. Interestingly, a potent inhibitor of the membrane attack complex, CD46, showed a significant positive association with cerebral grey matter volume. CD59 also demonstrated an inverse association with systolic blood pressure. Prior studies have shown changes in EC levels of CD59 in hypoxic states and some microvascular disorders, such as diabetic vasculopathy. It is possible that upregulations of CD59 represent a reactive protective mechanism by endothelial cells. Overall, interactions between inflammatory cytokines and EC complement proteins may enable inflammatory homeostasis or exacerbate a dysfunctional state. Complement binding to ECs results in increased expression of adhesive proteins and production of inflammatory mediators including ICAM1 and IL6, that we measured in this study. Together these changes result in increased vascular permeability and transmigration of PMN leukocytes, resulting in white matter injury and radiographic evidence of WMH.
Since this is the first study to investigate complement activation in endothelial cells of subjects with only radiographic evidence of WMH, we sought to quantify complement factors across all pathways. Overall, abnormal EC-derived exosome cargo levels of complement proteins reflect the nature and extent of dysfunctional complement system homeostasis in ECs and is the basis for the present analytical approach. ECs normally respond to the continuous challenges of inflammatory signals through diverse mechanisms that optimize host defenses and minimize vascular injury. EC exosomes are components of a constitutive system to dispose of cellular “waste” or potentially damaging endogenous inflammatory mediators, in addition to presenting some of them as stimuli to protective blood leukocytes. Complement-mediated injury of ECs in a broad range of vascular diseases is attributable to failures of inflammatory homeostasis that include both enhanced activation of complement effector pathways and diminished complement regulatory elements. Components of activated complement pathways, including C1q, C4b, C3b, or membrane attack complex (MAC) C5b-9 and the anaphylatoxins C5a and C3a are capable of evoking numerous EC inflammatory responses. In addition to representing a means of disposing of unwanted metabolites and proteins, exosomes released from ECs can provide a means of cell–cell communication. Therefore, C3b and C5b-9 packaged in EDEs could potentially get delivered to neurons and display their surface membranes, initiating microglial cytotoxic attacks and neuronal destruction. Moreover, enhanced production of classical and alternative complement factors could be amplified by the relative deficiencies of complement regulatory proteins, also noted in this study (see Supplemental Fig. S1). The main source of complement factors in states of health is the liver. However, it is becoming clear that other cells can also express complement factors. In culture, mammalian ECs have been shown to produce C1, C4, C3, factor B, and factors I among other factors. Of note, although EDE complement factors could be produced by endothelial cells, they could also be sourced elsewhere and simply up taken by ECs. More work remains to be done to confirm with confidence that these complement factors are cargo as opposed to attached to the surface. Indeed, single-cell RNA sequencing in addition to protease digestion studies could clarify sources of these factors. Last, it should be noted that a limitation of our cross-sectional, non-interventional study, is with respect to causal inference. Indeed, complement activation can be an instigator of disease, as well as a consequence of injury, thereby acting as a propagator or amplifier of endothelial injury.
In summary, cerebral vascular inflammation is emerging as an important contributor to brain injury and cognitive decline. In addition, higher load of WMH is associated with adverse age-associated cognitive outcomes, such as slowed processing speed and increased risk of AD dementia37,38. In the absence of frank neurodegenerative phenotypes, age-associated WMH is frequently presumed secondary to cerebral vascular endotheliopathies resulting in increased BBB leakage and activation of CNS immune system, leading to reactive gliosis37,39,40,41,42. In this model, endothelial cells play a central role. However, a direct in vivo interrogation of EC molecular content and testing of specific hypotheses has only just been made possible via EC exosomal isolation techniques. In this proof of concept study, we demonstrate a strong association between endothelial innate inflammation and cerebral white matter injury, a hallmark of small vessel disease, and important contributor to age-associated cognitive slowing, neurodegeneration and dementia. EDE complement factors can provide promising preclinical biomarkers—an avenue that should be pursued further in light of the value of detecting preclinical disease as well as using fluid biomarkers as surrogate outcomes in therapeutic trials targeting endothelial inflammation.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Change history
01 February 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41598-022-05977-3
References
Gravitz, L. Drugs: A tangled web of targets. Nature 475, S9-11 (2011).
Buckholtz, N. S. Perspective: in search of biomarkers. Nature 475, S8 (2011).
Ryu, J. K. et al. Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat. Immunol. 19, 1212–1223 (2018).
Merlini, M. et al. Fibrinogen induces microglia-mediated spine elimination and cognitive impairment in an Alzheimer’s disease model. Neuron https://doi.org/10.1016/j.neuron.2019.01.014 (2019).
Sankowski, R., Mader, S. & Valds-Ferrer, S. I. Systemic inflammation and the brain: Novel roles of genetic, molecular, and environmental cues as drivers of neurodegeneration. Front. Cell. Neurosci. 9, 1–20 (2015).
Sweeney, M. D. et al. Vascular dysfunction: The disregarded partner of Alzheimer’s disease. Alzheimer’s Dement. 15, 158–167 (2019).
Gottesman, R. F. et al. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA https://doi.org/10.1001/jama.2017.3090 (2017).
Zhou, T. et al. Microvascular endothelial cells engulf myelin debris and promote macrophage recruitment and fibrosis after neural injury. Nat. Neurosci. https://doi.org/10.1038/s41593-018-0324-9 (2019).
Casserly, I. & Topol, E. Convergence of atherosclerosis and Alzheimer’s disease: inflammation, cholesterol, and misfolded proteins. Lancet 363, 1139–1146 (2004).
Elahi, F. M. et al. Longitudinal white matter change in frontotemporal dementia subtypes and sporadic late onset Alzheimer’s disease. NeuroImage Clin. 16, 595–603 (2017).
Zhao, Z. & Zlokovic, B. V. Remote control of BBB: A tale of exosomes and microRNA. Cell Res. 27, 849–850 (2017).
Goetzl, E. J. et al. Low neural exosomal levels of cellular survival factors in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2, 769–773 (2015).
Goetzl, E. J. et al. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 30, 4141–4148 (2016).
Fiandaca, M. S. et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimer’s Dement. 11, 600-607.e1 (2015).
Goetzl, E. J. et al. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 85, 40–47 (2015).
Mustapic, M. et al. Plasma extracellular vesicles enriched for neuronal origin: A potential window into brain pathologic processes. Front. Neurosci. 11, 1–12 (2017).
Kanninen, K. M., Bister, N., Koistinaho, J. & Malm, T. Exosomes as new diagnostic tools in CNS diseases. Biochim. Biophys. Acta 1862, 403–410 (2016).
Piper, R. C. & Katzmann, D. J. Biogenesis and Function of Multivesicular Bodies. Annu. Rev. Cell Dev. Biol. 23, 519–547 (2007).
Chen, C. C. et al. Elucidation of exosome migration across the blood-brain barrier model in vitro. Cell. Mol. Bioeng. 9, 509–529 (2016).
Armstrong, D. & Wildman, D. E. Extracellular vesicles and the promise of continuous liquid biopsies. J. Pathol. Transl. Med. 52, 1–8 (2018).
Ryu, J. K. et al. Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat. Commun. 6, 1–15 (2015).
Matsuo, K. et al. Complement activation in capillary cerebral amyloid angiopathy. Dement. Geriatr. Cogn. Disord. https://doi.org/10.1159/000486091 (2018).
Veerhuis, R., Nielsen, H. M. & Tenner, A. J. Complement in the brain. Mol. Immunol. 48, 1592–1603 (2011).
Sartain, S. E., Turner, N. A. & Moake, J. L. Brain microvascular endothelial cells exhibit lower activation of the alternative complement pathway than glomerular microvascular endothelial cells. J. Biol. Chem. https://doi.org/10.1074/jbc.RA118.002639 (2018).
Elahi, F. M. et al. “Liquid Biopsy” of white matter hyperintensity in functionally normal elders. Front. Aging Neurosci. 10, 343 (2018).
Goetzl, E. J. et al. Altered cargo proteins of human plasma endothelial cell–derived exosomes in atherosclerotic cerebrovascular disease. FASEB J. 31, 3689–3694 (2017).
Kramer, J. H. et al. Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn. Behav. Neurol. 16, 211–218 (2003).
DeCarli, C., Fletcher, E., Ramey, V., Harvey, D. & Jagust, W. J. Anatomical mapping of white matter hyperintensities (WMH): Exploring the relationships between periventricular WMH, deep WMH, and total WMH burden. Stroke 36, 50–55 (2005).
DeCarli, C. et al. Measures of brain morphology and infarction in the framingham heart study: Establishing what is normal. Neurobiol. Aging 26, 491–510 (2005).
Maillard, P. et al. White matter hyperintensity penumbra. Stroke 42, 1917–1922 (2011).
Maillard, P. et al. Coevolution of white matter hyperintensities and cognition in the elderly. Neurology 79, 442–448 (2012).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. https://doi.org/10.2307/2346101 (1995).
Querfurth, H. W. & LaFerla, F. M. Alzheimer’s disease. N. Engl. J. Med. 362, 329–344 (2010).
Kang, S. S. et al. Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J. Exp. Med. https://doi.org/10.1084/jem.20180653 (2018).
Park, L. et al. Brain perivascular macrophages initiate the neurovascular dysfunction of Alzheimer Aβ peptides. Circ. Res. 121, 258–269 (2017).
Goetzl, E. J., Schwartz, J. B., Abner, E. L., Jicha, G. A. & Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 83, 544–552 (2018).
Fazekas, F., Chawluk, J. B., Alavi, A., Hurtig, H. I. & Zimmerman, R. A. Mr Signal abnormalities at 1.5-T in Alzheimer dementia and normal aging. Am. J. Roentgenol. 149, 351–356 (1987).
Filley, C. M. & Fields, R. D. White matter and cognition: Making the connection. J. Neurophysiol. 116, 2093–2104 (2016).
Gupta, N. et al. White matter hyperintensity-associated blood-brain barrier disruption and vascular risk factors. J. Stroke Cerebrovasc. Dis. 27, 466–471 (2018).
Zhang, C. E. et al. Blood–brain barrier leakage is more widespread in patients with cerebral small vessel disease. Neurology https://doi.org/10.1212/WNL.0000000000003556 (2016).
Li, Y. et al. Higher blood–brain barrier permeability is associated with higher white matter hyperintensities burden. J. Neurol. 264, 1474–1481 (2017).
Li, Y. et al. Compromised blood-brain barrier integrity is associated with total magnetic resonance imaging burden of cerebral small vessel disease. Front. Neurol. 9, 221 (2018).
Acknowledgements
This work was funded by the National Institute on Aging and Department of Veterans Affairs IK2CX002180, Larry L. Hillblom Foundation 2019A012SUP and New Vision Research (to F.M.E.). Participant recruitment and data collection was funded by the MarkVCID (UH3NS100608) and Hillblom Aging Network for the Prevention of Age-Associated Cognitive Decline study grants (2140-A-004-NET) (to J.K.) and NIH-NIA ADRC (P50 AG023501) and PPG (P01AG019724) (to B.L.M.).
Author information
Authors and Affiliations
Contributions
All authors reviewed and edited the manuscript. FE and EG designed the study, performed experiments and interpreted the results, FE performed statistical analyses, figure and table preparation, and wrote the manuscript. NB and NF performed nanoparticle tracking assays. For rigor, DH independently ran all statistical analyses and confirmed the results. MA recruited participants, collected biospecimen and MRI scans. CD and PM processed MRI scans. JK and BM provided funding, resources, and critical input. JH, KC, and AS provided critical input.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained errors in the presentation of the author names Fanny M. Elahi, Danielle Harvey, Marie Altendahl, Nivetha Brathaban, Nicole Fernandes, Kaitlin B. Casaletto, Adam M. Staffaroni, Pauline Maillard, Jason D. Hinman, Bruce L. Miller, Charles DeCarli, Joel H. Kramer and Edward J. Goetzl, which were incorrectly given as F. M. Elahi, D. Harvey, M. Altendahl, N. Brathaban, N. Fernandes, K. B. Casaletto, A. M. Stafaroni, P. Maillard, J. D. Hinman, B. L. Miller, C. DeCarli, J. H. Kramer and E. J. Goetzl.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elahi, F.M., Harvey, D., Altendahl, M. et al. Elevated complement mediator levels in endothelial-derived plasma exosomes implicate endothelial innate inflammation in diminished brain function of aging humans. Sci Rep 11, 16198 (2021). https://doi.org/10.1038/s41598-021-91759-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-91759-2
This article is cited by
-
Plasma derived extracellular vesicle biomarkers of microglia activation in an experimental stroke model
Journal of Neuroinflammation (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
