
Abstract
Over half of the alpine meadows in the Qinghai-Tibet Plateau (QTP) are degraded due to human activities. Soil degradation from overgrazing is the most direct cause of grassland degradation. It is thus important to synthesize the effects of multiple soil degradation indicators on the belowground biomass of plants and soil microorganisms in the degraded QTP. We studied the diversities and structures of soil bacterial and fungal communities using soil bacterial 16S rRNA and the fungal ITS gene under four degradation gradients, D1: lightly degraded, D2: moderately degraded, D3: highly degraded, and a non-degraded control site (CK). The bacterial Shannon diversity in D3 was significantly lower than that in D1 (p < 0.001), and the bacterial richness index in D3 was significantly lower than that in D1 (p < 0.001). There was no difference in soil fungal diversity among the different degradation levels; however, soil fungal richness decreased significantly from CK to D3. The phyla Actinobacteria, Acidobacteria and the genus Mortierella were differed significantly under the four degradation gradients. Plant litter mass and root C/N ratio were important factors associated with bacterial and fungal diversity and richness. These results indicated that alpine meadow degradation can lead to variations in both microbial diversity and the potential functioning of micro-organisms in the QTP.
Similar content being viewed by others


Introduction
Grassland degradation can produce highly negative environmental impacts, such as desertification, dust storms, and soil erosion1. Alpine meadows, which account for 38% of the grassland area in the Qinghai-Tibet Plateau (QTP), are a principal component of the natural ecosystem2. However, over half of alpine meadows are degraded, largely due to human activities3, such as overgrazing4,5. Overgrazing has reduced above- and below-ground biomass6,7 and decreased diversity and structure in the plant community8,9. It has also reported that overgrazing has resulted in severe soil degradation due to 3.02 Pg of carbon being lost over the last 30 years in the QTP10.
Soil degradation caused by overgrazing is the most direct cause of grassland degradation11. Soil properties that have been proposed as indicators of soil quality include organic carbon, pH, water storage, bulk density, electrical conductivity, and nutrient availability12,13. More than one indicator is typically required for assessing the effects of tillage management systems14 as well as grassland ecosystems. Therefore, it is important to synthesize the effects of multiple soil degradation indicators on belowground plant biomass and soil micro-organisms in the degraded QTP.
The changes in soil nutrient availability and plant diversity caused by alpine meadow degradation can alter the microbial community and its diversity15. Soil water availability16, pH17,18, bulk density19, and plant composition and biomass20 are key factors responsible for changes in microbial communities. Therefore, the changes in soil physicochemical properties and plant attributes during the process of grassland degradation inevitably change the diversity and composition of the soil microbial community6,21. A previous study confirmed that Actinobacteria, Proteobacteria, and Acidobacteria were well adapted to the soil conditions in the degraded alpine steppes at Qinghai-Tibetan Plateau15. Li, et al.6 found no significant difference in bacterial species composition in moderately degraded alpine meadows; however, a shift from Sordariomycetes to Dothideomycetes within Ascomycota was found with increasing degradation level. The microbial alpha diversity might be higher in severely degraded alpine meadows6, but many studies have found no difference in microbial alpha diversity among different degraded steppes15. Given the inconsistent response of soil micro-organisms to grassland degradation22, understanding how both bacterial and fungal community composition and diversity respond to degradation, and clarifying the key soil factors related to degradation, could provide a basis for the health evaluation and management of alpine meadows in the QTP6. There are few studies on the diversity of bacterial and fungal communities in degraded alpine meadows in the QTP, and it is unclear if soil degradation is also related to the composition and structure of soil fungi.
Long-term overgrazing has produced different levels of soil degradation in the alpine meadow of northeastern Qinghai Province, China. In this study, we used high-throughput sequencing of soil bacterial 16S rRNA and the ITS gene region of fungi to study the diversity and structure of soil bacterial and fungal communities under four different degradation conditions. To better understand the potential functional contributions of the observed soil bacteria in the degraded alpine meadow, we targeted human disease functional classes by calculating the metabolic pathways of the 16S rRNA gene sequences in the KEGG (Kyoto Encyclopedia of Genes and Genomes) database23. We hypothesized that (1) long-term overgrazing caused shifts in both plant and soil properties, and (2) soil degradation significantly altered bacterial and fungal diversity. The goals of this study were to (1) identify the plant root and soil properties under different degradation conditions, (2) evaluate the response of soil bacterial and fungal communities to a degraded meadow, and (3) assess the relationships between soil degradation, plant root properties, and soil microbial diversities.
Results
Plant root property responses to grassland degradation
The plant RC and RC/N ratios significantly increased as the degradation level increased (p < 0.05, Table 1), while litter mass, plant RB, and RN significantly decreased (Table 1; p < 0.05). Additionally, the soil pH, EC, and BD values significantly increased as the degradation level increased (p < 0.05, Table S1), while the contents of SWC, TC, TN, and the C/N ratio significantly decreased (p < 0.05).
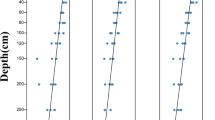
In particular, the degree of soil degradation, obtained from PCA axis 1, also significantly increased as the degradation level increased (p < 0.05, Table 1). The non-linear regression revealed a negative correlation between soil degradation and plant root biomass (R2 = 0.59, p < 0.01); however, the non-linear regression showed a positive correlation between soil degradation and root C/N ratios (R2 = 0.80, p < 0.001) (Fig. 1).

The correlation between soil degradation and plant root biomass and root C/N ratios.
Soil microbial property responses to grassland degradation
The bacterial Shannon diversity index in D3 was significantly lower than that in the CK (Fig. 2a; p < 0.001), and the bacterial Chao richness index in D3 was significantly lower than that in D1 (Fig. 2b; p < 0.001); however, the bacterial Simpson index in D3 was significantly higher than that in CK, D1, and D2 (Fig. 2c; p < 0.001). There was no difference in the soil fungal Shannon and Simpson indices among the four different degradation levels (Fig. 2d, f); however, the soil fungal Chao richness index decreased significantly from CK to D3 (Fig. 2e).

Bacterial and fungal diversity indices (a, d Shannon index; b, e Chao richness index; and c, fSimpson) calculated from 16S rRNA and ITS gene sequence data at the OTU level. *p < 0.05, **p < 0.01, and ***p < 0.001 based on Tukey’s honestly significant difference (HSD) tests.
Actinobacteria, Proteobacteria, Acidobacteria, and Chloroflexi were the main microflora at all four degradation levels, accounting for about 80% of the total bacterial abundance (Fig. 3a). At the genus level, o_Actinomarinales and f_Geminicoccaceae were the main microflora in D3 (Fig. S1a). The NMDS and PERMANOVA tests confirmed that the communities in D1, D2, and D3 were significantly different from those in the CK (R2 = 0.8047, p = 0.001; Fig. 3b). Ascomycota was the main microflora at all four degradation levels, accounting for about 60% of the total fungal abundance (Fig. 3c), and the genera Mortierella, Pseudeurotium, and o_Tremellales were the main microflora in the CK (Fig. S1b). The PERMANOVA tests showed that the communities in D1, D2, and D3 did not differ from those in CK (R2 = 0.0061, p = 0.466; Fig. 3d).

Relative abundances of the soil bacterial (a) and fungal (c) phyla in non-degraded (CK), lightly degraded (D1), moderately degraded (D2), and highly degraded (D3) alpine meadows, and nonmetric multidimensional scaling (NMDS) ordinations based on the mean abundance values of the soil bacterial (b) and fungal (d) phyla at the four different degradation levels.
Relationships between the shifts in soil properties, and microbial community and diversity
In the RDA biplots, a combination of soil variables explained 74.1% and 75.6% of the bacterial and fungal community variance, respectively (Fig. 4). According to the Monte Carlo permutation test, plant root C, soil pH, EC, and soil TC significantly influenced the bacterial communities (Table S2), while only root biomass and soil TN significantly influenced the fungal communities (Table S2). The Spearman’s correlation heatmap showed that the abundance of Bacteroidetes was significantly positively correlated with soil EC (p < 0.001), pH (p < 0.01), root C/N (p < 0.01), and root C (p < 0.01), but negatively correlated with RB (p < 0.001), SWC (p < 0.001), RN (p < 0.01), and LM (p < 0.01) (Fig. 5a). The abundance of Mucoromycota was significantly positively correlated with soil RB (p < 0.001), SWC (p < 0.001), RN (p < 0.001), and LM (p < 0.001), but negatively correlated with root C/N (p < 0.001), root C (p < 0.001), pH (p < 0.01), and EC (p < 0.01) (Fig. 5b).

Redundancy analysis (RDA) showing the impact of plant litter mass, root properties, and soil physiochemical properties (pH, EC, BD, SWC, salt content, and C/N) on bacterial (a) and fungal (b) community structure. The significance of the effect of each property was tested using Monte Carlo permutation tests (permutations = 499).

Spearman’s heatmap analyses between the relative abundances of specific bacterial (a) and fungal (b) genera and soil property parameters. EC (electrical conductivity), BD (bulk density), SWC (soil water content), TC (total carbon), TN (total nitrogen), C/N (soil total carbon/nitrogen), RB (root biomass), RC (root carbon), RN (root nitrogen), LM (litter mass), and RC/N (root carbon/nitrogen). *, **, and *** indicate the significance along the paths at the level of p < 0.05, p < 0.01 and p < 0.001, respectively.
The SEM explained 83% and 76% of the variance in soil bacterial diversity and richness, respectively (Fig. 6a), and explained 14% and 80% of the variance in soil fungal diversity and richness, respectively (Fig. 6b). Soil degradation was significantly negatively correlated with plant root biomass (R2 = 0.65, p < 0.01), but positively correlated with root C/N ratios (R2 = 0.87, p < 0.001). Although soil degradation decreased the bacterial diversity, there was no significant correlation between soil degradation and soil fungal diversity. Additionally, plant root biomass was significantly negatively correlated with both soil bacterial diversity (R2 = 0.60, p < 0.01) and soil bacterial richness (R2 = 0.97, p < 0.001), and plant root C/N ratios were significantly negatively correlated with both soil bacterial diversity (R2 = 0.67, p < 0.01) and soil bacterial richness (R2 = 0.60, p < 0.01). However, plant root biomass (R2 = 0.34, p < 0.05) and root C/N ratios (R2 = 0.97, p < 0.001) were significantly positively and negatively correlated with soil fungal richness, respectively.

Structural equation model (SEM) based on the effects of soil degradation on soil microbial diversity and richness (a: bacteria, b: fungi). Continuous and dashed arrows indicate positive and negative relationships, respectively. Black and gray arrows represent significant and insignificant correlations, respectively. The width of the arrows is proportional to the strength of the path coefficients. R2 indicates the proportion of the variance explained and appears above every response variable in the model. The *, **, and ***indicate the significance along the paths at the level of p < 0.05, p < 0.01 and p < 0.001, respectively.
Relationships between soil dergradation and metabolic pathways related to human diseases
We found 10 bacteria related to human diseases (Fig. 7a), and infectious disease: bacterial (F = 20.18, p < 0.001) and neurodegenerative disease (F = 49.2, p < 0.001) were significantly increased from CK to D3. The metabolic pathway of substance dependence was significantly decreased from CK to D3 (F = 23.36, p < 0.001). Infectious disease: parasitic was highest in D2 (F = 22.81, p < 0.001). Correlation analysis also showed that infectious disease: bacterial (Fig. 7b; R2 = 0.67, p < 0.01) and neurodegenerative disease (Fig. 7c; R2 = 0.34, p < 0.05) were significantly positively correlated with soil degradation. However, infectious disease: parasitic showed a negative quadratic relation with soil degradation (Fig. 7d; R2 = 0.49, p < 0.05), and the metabolic pathway of substance dependence was significantly negatively correlated with soil degradation (Fig. 7e; R2 = 0.26, p < 0.05).

The predicted KEGG category abundance (a) of non-degraded (CK), lightly degraded (D1), moderately degraded (D2), and highly degraded (D3) alpine meadows, and the correlation analysis between soil degradation and KEGG categories (b–e).
Discussion
Response of bacterial and fungal properties to different degrees of degradation
Bacterial community structures change significantly as alpine meadow degradation increases due to differences in soil properties and vegetation characteristics among different degraded grasslands6,15. With an increase in degradation, the vegetation coverage has been reduced, and soil nutrient contents appear to exhibit a decreasing trend in the Qinghai-Tibetan Plateau21. Similarly, the belowground vegetation characteristics (root biomass, root C, root N, and root C/N ratios) and soil properties (soil pH, electrical conductivity, bulk density, soil water content, soil total C, soil total N, and soil total C/N ratios) differed significantly in this study, which corroborates previous studies24,25. A decrease in aboveground plant biomass can reduce the litter input to the soil, and restrict the activity and abundance of the soil microbial community21,26. We also found that the soil bacterial Shannon diversity decreased greatly in the highly degraded plots. The decreased vegetation biomass due to grassland degradation is likely responsible for decreased C and N concentrations in the soil27, which, in turn, decreased the soil bacterial Shannon diversity under the highly degraded plots caused by overgrazing. This was in agreement with previous results obtained by Zhang et al28. In addition, the soil bacterial richness appeared to be resistant to soil degradation in this alpine meadow; however, fungal richness decreased significantly in the highly degraded plots compared to the non-degraded plots, implying that the fungal community was more sensitive to degradation than the bacterial community6. These differences are attributed to the higher sensitivity of fungi to soil nutrient limitation29. Degradation significantly reduced the soil organic carbon content30, soil water content31, and soil nitrogen content in the present study. Similar to bacterial and fungal studies in QTP grasslands15, Actinobacteria and Ascomycota were the most abundant bacterial and fungal phyla, respectively, detected in these meadows. In addition, the other abundant phyla included Proteobacteria, Acidobacteria and Chloroflexi. Actinobacteria species have strong metabolic capacity at low temperatures32, and Acidobacteria play important roles in organic matter decomposition and nutrient cycles33,34. In this study, Actinobacteria and Acidobacteria were changed significantly in degraded alpine meadows, and this might affect the decomposition and metabolism of soil organic matter. However, no significant differences were found for Proteobacteria, which play a key role in ecosystems such as in the oxidation of organic and inorganic compounds35. The genus Mortierella from the family of Mortierellaceae includes fast-growing fungi capable of rapidly invading new substrates36 and are often the pioneer colonizers of freshly fallen litter37. In this study, plant aboveground biomass decreased significantly from CK to D3, which in turn decreased the litter quantity and the litter decomposition fungi, such as Mortierella.
Relationships between the shifts in soil properties and microbial diversity and richness
The composition of soil microbes can change with plant type changes due to the influence of vegetation growth on soil properties and the alteration of microclimates affecting microbial environments38,39. In terrestrial regions, pH is the most vital factor affecting the microbial community and diversity in soils40,41. In the present study, the Monte Carlo permutation test suggested that plant root C, soil pH, EC, and soil TC significantly influenced the bacterial communities, while only root biomass and soil TN significantly influenced the fungal communities. In addition, soil pH increased significantly in degraded alpine meadows. Thus, the development of microbial communities could be limited by the high pH caused by grassland degradation27. As previously reported42,43, Bacteroidetes was significantly positively correlated with pH, and this was confirmed in the present study. This result showed that Bacteroidetes soil bacteria were highly resistant to land degradation because of their positive correlation with soil pH. Interestingly, root biomass decreased significantly as the degradation level increased. A decrease in root biomass can limit the litter input to the soil, thus restricting the abundance of Basidiomycota and Mucoromycota under land degradation37.
One goal of this research was to evaluate the relationships between soil degradation, plant root characteristics, and bacterial and fungal diversity. Changes in plant diversity and soil properties due to alpine meadow degradation can result in variations in the bacterial community structure and composition15. Although soil degradation decreased the bacterial diversity, there was no significant correlation between soil degradation and soil fungal diversity in the SEM, implying that fungal diversity is less influenced by degradation than bacterial diversity. Plant root biomass was significantly negatively and positively correlated with soil bacterial and fungal richness, respectively. Interestingly, plant root C/N ratios showed consistent negative relationships with both soil bacterial and soil fungal richness.
Relationships between soil degradation and metabolic pathways related to human diseases
As the degradation degree increases, microbes might produce specialized proteins to survive in the degraded alpine steppe environment15. Without metagenomic data, the functional capacities of microorganisms cannot be effectively analyzed. Recently, Aßhauer et al.23 suggested that Tax4Fun analyses are useful in supplementing 16S rRNA analyses as an inexpensive alternative to PICRUSt analyses. The potential KEGG Orthologue (KO) functional profiles of microbial communities have also been predicted by 16S rRNA gene sequences using PICRUSt44. A recent study based on Tax4Fun analyses showed that most bacteria involved in human diseases increase as alpine steppe degradation increases15. In this work, we found that infectious disease: bacterial and neurodegenerative disease were significantly positively correlated with soil degradation. Alpine meadows and steppes are the two main livestock grazing regions in the QTP, and overgrazing has caused severe environmental degradation in this area, causing a loss of soil nutrient and plant biodiversity. Infections, including by Salmonella, are reportedly suffered by approximately 1.4 million people in the United States as a result of the consumption of contaminated meat45. People who live in degraded meadows in the QTP could suffer negative health effects when using livestock without prior examination and treatment15, and thus human health might be impacted by livestock grazing on degraded soils.
Conclusions
This study demonstrates that the plant and soil properties, and the rhizosphere microbiomes under field conditions varied with degradation intensity. The soil pH, EC, and BD values significantly increased as the degradation level increased. The abundances of Bacteroidetes and Gemmatimonadetes were significantly positively correlated with soil EC, pH, root C/N, and root C, whereas Basidiomycota and Mucoromycota were significantly negatively correlated with soil pH and EC. Soil degradation caused significant shifts in bacterial diversity and fungal richness, but did not significantly alter bacterial richness and fungal diversity in alpine meadow of the QTP. Plant litter mass and root C/N ratio proved to be the important factors controlling the shifts in bacterial and fungal diversity and richness. This study provides an improved understanding of the adapatation of the microbial community to soil degradation caused by overgrazing.
Materials and methods
Study area
The field experiment was conducted in the Haibei prefecture (36° 55′ N, 100° 57′ E, 3040 m el.), Qinghai Province, northeast of Tibetan Plateau (Fig. 8a). The mean annual temperature and mean annual rainfall at this site are − 0.45 °C and 400 mm, respectively. This area is dominated by meadow vegetation, including Stipa capillata, Poa pratensis, and Kobresia spp. The study site was grazed by 20 yaks (Bos grunniens) in a year-long continuous grazing system (5 ha), and the grazing intensity was four yaks ha–1 year –1, which is a high grazing intensity according to Wang, et al.46. We selected four sites with different degradation levels to represent the degradation intensity (Fig. 8b): non-degraded (CK), lightly degraded (D1), moderately degraded (D2), and highly degraded (D3). The plant total coverage was 88.25% (CK), 64.25% (D1), 52.5% (D2), and 28.5% (D3) (Fig. 8c). Details of the soil physiochemical properties are presented in Table S1.

Schematic of the field experimental designs in the study area (a), and the distance from the watering point (b) representing the degradation intensity (lightly degraded, D1; moderately degraded, D2; and highly degraded, D3), and the plant total coverage (c). The photographs were taken by Chao Yang at Haibei prefecture (36°55′N, 100°57′E), and the map was created by Chao Yang with ArcGIS 10.2 for Desktop (http://www.esri.com/arcgis/about-arcgis).
Plant root biomass and soil sampling
The field investigation was conducted in late-July 2019, which is the period of maximum annual biomass. Across each of the four degradation intensities (CK, D1, D2, and D3), four replicate points were separated at intervals of 50 m, and the sampling points from D1 to D3 were also separated at intervals of 50 m. In each point, we set up three random quadrats (1 m × 1 m) to measure the vegetation coverage and obtain root and rhizosphere soil samples. In total, there were 16 plots (four degradation intensities × four replicates) and 48 quadrats.
Plant total coverage was calculated by the ratio of the physically shaded area of the observed species in each quadrat to the total area of a quadrat47. The aboveground litter mass (LM) was collected and weighed. The belowground root biomass at 0–20 cm depth was obtained using a 10-cm-diameter root auger in each quadrat after measurement of plant coverage. Additionally, in each quadrat, a mixed rhizosphere soil sample at 0–20 cm depth from three random samples was collected using a 3-cm-diameter soil auger. The constant weight of the aboveground biomass and root biomass (RB) was obtained by oven-drying at 65 °C for 24 h after removal of the attached soil.
Plant root and soil properties analyses
Root carbon (RC), root nitrogen (RN), soil total carbon (TC), and soil total nitrogen (TN) concentrations were measured using a CHNS Element Analyzer (Elementar, Germany). Soil pH and electrical conductivity (EC) were measured using a glass electrode in a 1:2.5 soil: water suspension. Soil bulk density (BD) was calculated using the ring knife method at 0–20 cm depth. A foil sampler with a volume of 100 cm−3 was used to obtain the samples, which were then oven-dried at 105 °C for 24 h. Soil water content (SWC) was measured gravimetrically and expressed as a percentage of soil water to dry soil weight.
DNA extraction and PCR amplification
The bacterial and fungal extraction and determination methods were similar to those used in a previous study19. In brief, the genomic DNA was extracted from each soil sample using a Fast DNA SPIN Kit for soil (MP Biomedicals, CA, USA). We weighed a 0.30 g soil sample from each treatment. Soil DNA integrity was then detected by 0.8% agarose gel electrophoresis. The V3 and V4 regions of the bacterial 16S rRNA gene were amplified using universal primers 338F and 806R, and the fungal ITS gene was amplified by ITS1 primers. The PCR amplification of the 16S rRNA gene and ITS1 region included pre-denaturation at 95 °C for 3 min; 27 cycles at 95 °C for 30 s, annealing at 55 °C for 30 s, elongation at 72 °C for 45 s, and an extension at 72 °C for 10 min.
Processing of MiSeq sequencing data
Purified amplicons were pooled in equimolar ratios and paired-end sequenced on an Illumina MiSeq PE300 platform(Illumina, San Diego,USA) according to the standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China). Processing of the raw data and diversity indices including Shannon and Chao1 richness were implemented in QIIME (ver. 1.3.0)48. The reads were truncated to obtain an average quality score lower than 20; a sliding window over 50 bp and over 3 continuous bases, and the reads with lengths fewer than 300 bp were discarded as well. We then used UPARSE (ver. 7.1) to cluster the high-quality sequences with a 97% identity threshold into operational taxonomic units (OTUs)49. The original sequencing data of the bacteria and fungi were deposited into the NCBI Sequence Read Archive (SRA) database (Accession Number: SRP251789).
Statistical analysis
Significance analyses of litter mass, plant root indices (RB, RC, RN, and RC/N), soil indices (pH, EC, BD, SWC, SOC, TN, C/N, and soil degradation), and microbial diversity indices (Shannon and Chao 1) between four degradation levels were performed using one-way analysis of variance (ANOVA) in PAST (ver. 3.25; Natural History Museum, University of Oslo, Norway). The level of significance was tested at p < 0.05 using Tukey’s pairwise test.
Since soil indices within each degradation are often closely correlated, we conducted principal component analyses (PCA) to create multivariate indexes for each degradation level, reducing collinearity among the predictor variables50. Therefore, we used the eigenvalues of PCA axis 1 to characterize soil degradation (Figure S2). Additionally, the correlation between the soil degradation and plant root biomass and root C/N ratios was analyzed using an exponential model in PAST (ver. 3.25).
Nonmetric multidimensional scaling (NMDS) based on Bray–Curtis similarity matrices was performed to identify the total structural changes in soil bacteria and fungi, and significance was tested by a permutational multivariate ANOVA (PERMANOVA) in PAST (ver. 3.25).
The relationship between litter and root properties (LM, RB, RC, RN, and RC/N), soil properties (pH, EC, BD, SWC, and C/N) and soil microbial community (bacteria and fungi) was determined by redundancy analysis (RDA) using CANOCO (ver. 4.5, Plant Research International, Wageningen, Netherlands). The significance of the effect of each variable was defined using Monte Carlo permutation tests (permutations = 999), and the resulting significance level was tested by the F- and p-values. The causal relationships among soil degradation, plant root indices, and soil microbial diversities and richness were quantified by structural equation modeling (SEM) using AMOS (ver. 21.0, IBM, SPSS, Inc., Chicago, IL, USA). A non-significant chi-square (χ2) test and the root mean square error of approximation (RMSEA) were used to define the goodness of fit of the model according to Yang et al.19. We used 16S rRNA gene sequences to calculate the metabolic pathways related to human diseases in the Kyoto Encyclopedia of Genes and Genomes database51 (KEGG, www.kegg.jp/kegg/kegg1.html) using the Tax4fun package in R (ver. 3.5.1). Additionally, the correlation between the soil degradation and the metabolic pathways related to human diseases was analyzed using a linear model in PAST (ver. 3.25).
References
Bryan, B. A. et al. China’s response to a national land-system sustainability emergency. Nature 559, 193–204 (2018).
Zhang, W. J., Xue, X., Peng, F., You, Q. G. & Hao, A. H. Meta-analysis of the effects of grassland degradation on plant and soil properties in the alpine meadows of the Qinghai-Tibetan Plateau. Glob. Ecol. Conserv. 20, e00774 (2019).
Pan, T., Zou, X. T., Liu, Y. J., Wu, S. H. & He, G. M. Contributions of climatic and non-climatic drivers to grassland variations on the Tibetan Plateau. Ecol. Eng. 108, 307–317 (2017).
Shen, H. H., Wang, S. P. & Tang, Y. H. Grazing alters warming effects on leaf photosynthesis and respiration in Gentiana straminea, an alpine forb species. J. Plant. Ecol. 6, 418–427 (2013).
Li, G. Y., Jiang, C. H., Cheng, T. & Bai, J. Grazing alters the phenology of alpine steppe by changing the surface physical environment on the northeast Qinghai-Tibet Plateau, China. J. Environ. Manage. 248, 109257 (2019).
Li, Y. M. et al. Changes of soil microbial community under different degraded gradients of alpine meadow. Agric. Ecosyst. Environ. 222, 213–222 (2016).
Guo, N. et al. Changes in vegetation parameters and soil nutrients along degradation and recovery successions on alpine grasslands of the Tibetan plateau. Agric. Ecosyst. Environ. 284, 106593 (2019).
Lin, L. et al. Predicting parameters of degradation succession processes of Tibetan Kobresia grasslands. Solid Earth 6, 1237–1246 (2015).
Li, H. D. et al. Assessing revegetation effectiveness on an extremely degraded grassland, southern Qinghai-Tibetan Plateau, using terrestrial LiDAR and field data. Agric. Ecosyst. Environ. 282, 13–22 (2019).
Wang, G. X., Qian, J., Cheng, G. D. & Lai, Y. M. Soil organic carbon pool of grassland soils on the Qinghai-Tibetan Plateau and its global implication. Sci. Total Environ. 291, 207–217. https://doi.org/10.1016/s0048-9697(01)01100-7 (2002).
Yuan, Z. Q. et al. Responses of soil organic carbon and nutrient stocks to human-induced grassland degradation in a Tibetan alpine meadow. CATENA 178, 40–48 (2019).
Askari, M. S. & Holden, N. M. Quantitative soil quality indexing of temperate arable management systems. Soil Till Res. 150, 57–67 (2015).
Lima, A. C. R., Brussaard, L., Totola, M. R., Hoogmoed, W. B. & de Goede, R. G. M. A functional evaluation of three indicator sets for assessing soil quality. Appl. Soil Ecol. 64, 194–200 (2013).
Masto, R. E., Chhonkar, P. K., Singh, D. & Patra, A. K. Alternative soil quality indices for evaluating the effect of intensive cropping, fertilisation and manuring for 31 years in the semi-arid soils of India. Environ. Monit. Assess 136, 419–435. https://doi.org/10.1007/s10661-007-9697-z (2008).
Zhou, H. et al. Changes in the soil microbial communities of alpine steppe at Qinghai-Tibetan Plateau under different degradation levels. Sci. Total Environ. 651, 2281–2291 (2019).
Yang, C., Zhang, F. G., Liu, N., Hu, J. & Zhang, Y. J. Changes in soil bacterial communities in response to the fairy ring fungus Agaricus gennadii in the temperate steppes of China. Pedobiologia 69, 34–40 (2018).
Li, J. J. & Yang, C. Inconsistent response of soil bacterial and fungal communities in aggregates to litter decomposition during short-term incubation. Peerj 7, e8078 (2019).
Yang, C., Li, J. J., Liu, N. & Zhang, Y. J. Effects of fairy ring fungi on plants and soil in the alpine and temperate grasslands of China. Plant Soil 441, 499–510 (2019).
Yang, C., Liu, N. & Zhang, Y. J. Soil aggregates regulate the impact of soil bacterial and fungal communities on soil respiration. Geoderma 337, 444–452 (2019).
Wardle, D. A. et al. Ecological linkages between aboveground and belowground biota. Science 304, 1629–1633 (2004).
Wu, G.-L., Ren, G.-H., Dong, Q.-M., Shi, J.-J. & Wang, Y.-L. Above- and belowground response along degradation gradient in an alpine grassland of the Qinghai-Tibetan Plateau. Clean-Soil Air Water 42, 319–323. https://doi.org/10.1002/clen.201200084 (2014).
Che, R. X. et al. Degraded patch formation significantly changed microbial community composition in alpine meadow soils. Soil Till Res. 195, 104426 (2019).
Aßhauer, K. P., Wemheuer, B., Daniel, R. & Meinicke, P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 31, 2882–2884 (2015).
Harris, R. B. Rangeland degradation on the Qinghai-Tibetan plateau: a review of the evidence of its magnitude and causes. J. Arid Environ. 74, 1–12. https://doi.org/10.1016/j.jaridenv.2009.06.014 (2010).
Ren, G., Shang, Z., Long, R., Hou, Y. & Deng, B. The relationship of vegetation and soil differentiation during the formation of black-soil-type degraded meadows in the headwater of the Qinghai-Tibetan Plateau China. Environ. Earth Sci. 69, 235–245. https://doi.org/10.1007/s12665-012-1951-1 (2013).
Zhang, Y. et al. Diversity of nitrogen-fixing, ammonia-oxidizing, and denitrifying bacteria in biological soil crusts of a revegetation area in Horqin Sandy Land Northeast China. Ecol. Eng. 71, 71–79. https://doi.org/10.1016/j.ecoleng.2014.07.032 (2014).
Wang, Y. et al. Effects of grassland degradation on ecological stoichiometry of soil ecosystems on the Qinghai-Tibet Plateau. Sci. Total Environ. 722, 137910. https://doi.org/10.1016/j.scitotenv.2020.137910 (2020).
Zhang, Y. et al. Soil bacterial and fungal diversity differently correlated with soil biochemistry in alpine grassland ecosystems in response to environmental changes. Sci. Rep. 7, 43077. https://doi.org/10.1038/srep43077 (2017).
Hartmann, M. et al. Resistance and resilience of the forest soil microbiome to logging-associated compaction. ISME J. 8, 226–244. https://doi.org/10.1038/ismej.2013.141 (2014).
Liu, S. B., Zamanian, K., Schleuss, P. M., Zarebanadkouki, M. & Kuzyakov, Y. Degradation of tibetan grasslands: consequences for carbon and nutrient cycles. Agric. Ecosyst. Environ. 252, 93–104 (2018).
He, S. Y. & Richards, K. Impact of meadow degradation on soil water status and pasture managementA case study in tibet. Land Degrad. Dev. 26, 468–479. https://doi.org/10.1002/ldr.2358 (2015).
Yergeau, E., Hogues, H., Whyte, L. G. & Greer, C. W. The functional potential of high Arctic permafrost revealed by metagenomic sequencing, qPCR and microarray analyses. ISME J. 4, 1206–1214. https://doi.org/10.1038/ismej.2010.41 (2010).
Eichorst, S. A. et al. Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments. Environ. Microbiol. 20, 1041–1063 (2018).
Fang, D. X. et al. Microbial community structures and functions of wastewater treatment systems in plateau and cold regions. Bioresour. Technol. 249, 684–693 (2018).
Mukhopadhya, I., Hansen, R., El-Omar, E. M. & Hold, G. L. IBD—what role do proteobacteria play?. Nat. Rev. Gastroenterol. Hepatol. 9, 219–230. https://doi.org/10.1038/nrgastro.2012.14 (2012).
Kjoller, A. H. & Struwe, S. Fungal communities, succession, enzymes, and decomposition (2002).
Poll, C., Brune, T., Begerow, D. & Kandeler, E. Small-scale diversity and succession of fungi in the detritusphere of rye residues. Microbial. Ecol. 59, 130–140. https://doi.org/10.1007/s00248-009-9541-9 (2010).
Jangid, K. et al. Land-use history has a stronger impact on soil microbial community composition than aboveground vegetation and soil properties. Soil Biol. Biochem. 43, 2184–2193. https://doi.org/10.1016/j.soilbio.2011.06.022 (2011).
Cao, C. et al. Soil bacterial community responses to revegetation of moving sand dune in semi-arid grassland. Appl. Microbiol. Biotechnol. 101, 6217–6228. https://doi.org/10.1007/s00253-017-8336-z (2017).
Tripathi, B. M. et al. Tropical soil bacterial communities in Malaysia: pH dominates in the equatorial tropics too. Microbial. Ecol. 64, 474–484. https://doi.org/10.1007/s00248-012-0028-8 (2012).
Chu, H. et al. Bacterial community dissimilarity between the surface and subsurface soils equals horizontal differences over several kilometers in the western Tibetan Plateau. Environ. Microbiol. 18, 1523–1533. https://doi.org/10.1111/1462-2920.13236 (2016).
Wu, X. et al. Bacterial communities in the upper soil layers in the permafrost regions on the Qinghai-Tibetan plateau. Appl. Soil Ecol. 120, 81–88. https://doi.org/10.1016/j.apsoil.2017.08.001 (2017).
Yang, C. et al. Assessing the effect of soil salinization on soil microbial respiration and diversities under incubation conditions. Appl. Soil Ecol. https://doi.org/10.1016/j.apsoil.2020.103671 (2020).
Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814. https://doi.org/10.1038/nbt.2676 (2013).
Mermin, J. et al. Reptiles, amphibians, and human Salmonella infection: a population-based, case-control study. Clin. Infect. Dis. 38, S253–S261. https://doi.org/10.1086/381594 (2004).
Wang, J. et al. Plant community ecological strategy assembly response to yak grazing in an alpine meadow on the eastern Tibetan Plateau. Land Degrad. Dev. 29, 2920–2931. https://doi.org/10.1002/ldr.3050 (2018).
Ji, S., Geng, Y., Li, D. & Wang, G. Plant coverage is more important than species richness in enhancing aboveground biomass in a premature grassland, northern China. Agric. Ecosyst. Environ. 129, 491–496. https://doi.org/10.1016/j.agee.2008.11.002 (2009).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. https://doi.org/10.1038/nmeth.f.303 (2010).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. https://doi.org/10.1038/ismej.2012.8 (2012).
Chen, W. et al. Consistent responses of surface- and subsurface soil fungal diversity to N enrichment are mediated differently by acidification and plant community in a semi-arid grassland. Soil Biol. Biochem. 127, 110–119. https://doi.org/10.1016/j.soilbio.2018.09.020 (2018).
Kanehisa, M. et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484. https://doi.org/10.1093/nar/gkm882 (2008).
Acknowledgements
We thank the staff at the Haibei Demonstration Zone of Plateau Modern Ecological Animal Husbandry Science and Technology in Qinghai Province, China for their help with fieldwork. This study was funded by the Natural Science Foundation of Shandong Province (ZR2020QC188), the China Agricultural Research System (No. CARS-34), the Start Up Funds for High Level Talents of Qingdao Agricultural University (QAU-1120025), and the First Class Grassland Science Discipline Programme in Shandong Province of China.
Author information
Authors and Affiliations
Contributions
C.Y. designed the details of the study. L.D. collected and prepared data that were analyzed by J.L. and J.S.; L.D wrote the first draft of the manuscript with contributions from J.L. Both authors contributed to editing and reviewing the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dong, L., Li, J., Sun, J. et al. Soil degradation influences soil bacterial and fungal community diversity in overgrazed alpine meadows of the Qinghai-Tibet Plateau. Sci Rep 11, 11538 (2021). https://doi.org/10.1038/s41598-021-91182-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-91182-7
This article is cited by
-
Short-term restoration practices change the bacterial community in degraded soil from the Brazilian semiarid
Scientific Reports (2024)
-
Distribution characteristics of soil carbon density and influencing factors in Qinghai–Tibet Plateau region
Environmental Geochemistry and Health (2024)
-
Soil microbial diversity and composition response to degradation of the alpine meadow in the southeastern Qinghai-Tibet Plateau
Environmental Science and Pollution Research (2024)
-
Types of vegetables shape composition, diversity, and co-occurrence networks of soil bacteria and fungi in karst areas of southwest China
BMC Microbiology (2023)
-
Influence of simulated warming on soil nitrogen fractions in a Tibetan alpine meadow
Journal of Soils and Sediments (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
