
Abstract
Trichoderma genus fungi present great potential for the production of carbohydrate-active enzymes (CAZYmes), including glycoside hydrolase (GH) family members. From a renewability perspective, CAZYmes can be biotechnologically exploited to convert plant biomass into free sugars for the production of advanced biofuels and other high-value chemicals. GH54 is an attractive enzyme family for biotechnological applications because many GH54 enzymes are bifunctional. Thus, GH54 enzymes are interesting targets in the search for new enzymes for use in industrial processes such as plant biomass conversion. Herein, a novel metal-dependent GH54 arabinofuranosidase (ThABF) from the cellulolytic fungus Trichoderma harzianum was identified and biochemically characterized. Initial in silico searches were performed to identify the GH54 sequence. Next, the gene was cloned and heterologously overexpressed in Escherichia coli. The recombinant protein was purified, and the enzyme’s biochemical and biophysical properties were assessed. GH54 members show wide functional diversity and specifically remove plant cell substitutions including arabinose and galactose in the presence of a metallic cofactor. Plant cell wall substitution has a major impact on lignocellulosic substrate conversion into high-value chemicals. These results expand the known functional diversity of the GH54 family, showing the potential of a novel arabinofuranosidase for plant biomass degradation.
Similar content being viewed by others
Introduction
Arabinofuranosidases (ABFs) (EC 3.2.1.55) are enzymes that are capable of cleaving residues of l-arabinofuranosyl present in various oligosaccharides and in polysaccharides such as hemicellulose; for this reason, they are interesting targets for biotechnological applications1. These enzymes are members of a group of glycoside hydrolases necessary for the degradation of polymeric substrates, such as arabinan, arabinoxylan and other polysaccharides that constitute the walls of plant cells2. ABFs are grouped into glycoside hydrolase (GH) families 3, 43, 51, 54 and 62, where GH54 including some enzymes described as bifunctional. However, there are few studies focusing on the functional specificity of this last family3.
Ravanal et al.4 characterized an α-l-arabinofuranosidase from Penicillium purpurogenum from the GH54 family, demonstrating that this family contains enzymes capable of acting synergistically with others and carrying out hydrolysis of different substrates in addition to arabinose and xylose. Thus, based on these characteristics presented by some enzymes in this group, further studies are needed to better understand their enzymatic mechanisms, especially with regard to their application for the conversion of complex substrates such as polysaccharides into bioproducts.
Vegetable biomass is a reservoir of sugars organized in complex regular structures represented by cellulose, hemicellulose and lignin5. Such structures need to be deconstructed for the release of sugar-free monomers, which can then be converted into high-added-value chemicals such as second-generation ethanol6. This process requires the enzymatic conversion of biomass, and hemicellulose is a biopolymer that requires a large complex of enzymes for its conversion due to its structural composition7.
For the enzymes to access the main chain of hemicellulose, it is necessary to remove side-chain substitutions, as they are one of the main reasons for the recalcitrance of plant biomass8,9. Thus, the search for enzymes that target hemicellulose substitutions is a promising approach for improving the saccharification of lignocellulolytic material10. It is of interest to apply ABFs for this purpose because in addition, removing l-arabinose, some of these enzymes can act on other sugars involved in substitutions1.
These enzymes are particularly important in the genus Trichoderma, where they play a crucial physiological role11. This genus is composed of heterotrophic filamentous ascomycetous fungi that are capable of growing on different substrates and under different environmental conditions. There are several studies that have addressed the versatility of this genus, which range from applications in sugar and alcohol industries to the development of biofungicides12. Trichoderma reesei is of particular prominence within the group because of its recognized potential to produce several hydrolytic enzymes; however, other species of the genus, especially Trichoderma harzianum, have been shown to be as efficient as T. reesei in this regard13.
Therefore, in this study, we conducted the in silico bioprospecting of a new GH54 from T. harzianum. For this purpose, several approaches were used, such as RNA-seq, in addition to sequence analysis. The enzyme was also characterized biochemically with different substrates and metallic salts to determine their influence on the enzyme.
Results
Obtaining the sequence of ThABF and in silico characterization
Through database searches, a sequence from Trichoderma koningii (AAA81024.1) was selected and used in the steps detailed in the methodology. With the resulting BLAST sequence, the T. harzianum CBS 226.95 genome was mapped. T. harzianum IOC-3844 RNA-seq reads and the target protein sequence (MT439956) were obtained. With this target protein sequence, a BLAST search was performed against the CAZy database, where our data suggest that ThABF is more similar to the GH54 family and that this family has several carbohydrate-binding models (CBMs; 1, 2, 6, 13 and 42), as shown in Supplementary Table S1.
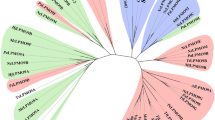
Phylogenetic analysis of ThABF against sequences of the GH54 family (represented mainly by fungal and bacterial sequences) revealed great diversity between the sequences (Fig. 1a). The sequences used in the analysis are shown in Supplementary Table S2. The phylogenetic tree was divided into five groups that best explained this diversity: a Fungi group, containing only fungal enzymes, and Bacterium I to IV groups, containing bacterial enzymes. The predominant taxonomic groups were as follows: Fungi (all Ascomycetes), Bacteria I (Acidobacteria, Verrucomicrobia, Proteobacteria and Actinobacteria), Bacteria II (Actinobacteria and Proteobacteria), Bacteria III (Actinobacteria and Proteobacteria) and Bacteria IV (all Proteobacteria) (Supplementary Table S2).

Phylogenetic analysis and sequence domains of the GH54 family. (a) Phylogenetic tree of the sequences. The star indicates the studied protein. Below the tree is a legend with the bootstrap values divided into ≤ 60%, 61–80 and 81–100. (b) Differences in the composition of domains in the sequences distributed in the groups. The identification of the groups is the same for A and B.
The analysis of the domains of the sequences in the phylogenetic tree (Supplementary Table S2) showed that all members of Fungi contained ArabFuran-catal (α-l-arabinofuranosidase B catalytic) and AbfB (α-l-arabinofuranosidase B) domains, in addition to CBM 42 (Fig. 1b). In Bacterium I, the presence of CBMs 13 and 42 was observed, in addition to sequences with the following combinations of domains: ArabFuran-catal and AbfB; ArabFuran-catal, RICIN (ricin B lectin domain), LTD (lamin tail domain), NPCBM_assoc (NEW3 domain of α-galactosidase) and RPT1; and ArabFuran-catal and RICIN. All Bacterium II sequences exhibited only CBM 42 and ArabFuran-catal and AbfB domains.
The Bacterium III group sequences only CBM 13 and the following combinations of domains: ArabFuran-catal and two RPT1 domains; ArabFuran-catal and RICIN; and ArabFuran-catal and two RICIN domains. Bacterium IV was the smallest group; its sequences included no CBMs and were the most distinct from those of the other groups. The Bacterium IV members included only the ArabFuran-catal domain.
To analyze the similarity of the ThABF sequence with some arabifuranosidases characterized as GH54 members in Trichoderma, alignment was performed (Fig. 2a). It was found that the target sequence was most similar to a sequence from T. virens, with a percent identity of 92.38%, followed by sequences from T. reesei (88.38%) and T. koniingi (87.78%). The structure of the ThABF protein was also predicted, and the model that was used was an Aspergillus kawachii α-l-arabinofuranosidase B with a C-score of 0.95, a TM-score of 0.84 ± 0.08 and an RMSD of 5.3 ± 3.4 Å (Fig. 2b).

Alignment and structure prediction of ThABF. (a) Alignment of ThABF with other Trichoderma sequences belonging to the GH54 family. (b) Prediction of the ThABF structure by I-TASSER.
Production of recombinant protein and evaluation of the folding components
With the sequence obtained from ThABF in silico, cloning and expression were performed. The enzyme was present in the insoluble fraction (Fig. 3a), and it was necessary to use the refolding approach described in the methodology, through which a protein of 53.44 kDa was purified (Fig. 3b). The recombinant ThABF sequence was used to evaluate the secondary and tertiary folding components of the enzyme since it was obtained through refolding. On the basis of the obtained circular dichroism profile, we identified rich α helices in the ThABF structure (Fig. 3c). In the size exclusion chromatography (SEC), the protein was eluted in a single peak, presenting the characteristics of a monomer in solution (Fig. 3d). Thus, these analyses demonstrated that it was possible to obtain a protein with folding components through the refolding method used.

SDS-PAGE in 12% gels showing purification results and graphs of secondary and tertiary folding analyses. (a) Gel containing the soluble (1) and insoluble (2) fractions; M indicates the marker. (b) Gel containing purified ThABF, with as size of approximately 53.44 kDa (1), and a marker (M). Both gels (a) and (b) were stained for 30 min in a dye solution with Coomassie blue. The whole gels from the ThABF purification and from the soluble and insoluble fractions are shown in Supplementary Figs. S1 and S2. (c) Analysis of secondary folding components of the recombinant enzyme by circular dichroism. (d) Analytical size exclusion chromatography of ThABF.
Determination of enzyme specificity and tests with metal ions
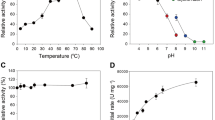
In the first experiment, enzyme activity was tested with the pNPAra substrate because in silico analysis indicated that ThABF would present a relatively high affinity with this substrate. However, the detected activity was not significant, leading to the hypothesis that a cofactor is needed to activate the catalytic site of the enzyme. Thus, we evaluated the effects of different metals on the activity of ThABF (Fig. 4). It was observed that the enzyme requires a metal ion cofactor to exert its catalytic activity, since the control exhibited a profile similar to that of EDTA. CUCl2 had no effect on the enzyme’s activity. MgCl2 had the greatest effect on the enzyme’s activity among the eleven tested metal salts, followed by MnCl2, CoCl2, CaCl2 and NiCl2. Despite the demonstration that ThABF requires a cofactor, its activity against pNPAra was not significantly increased; thus, a panel of chromogenic substrates derived from nitrophenol was tested to obtain information on ThABF enzymatic specificity (Table 1). The enzyme exhibited the greatest activity against pNPG, followed by pNPAp, pNPF and finally pNPAra.

Relative activity of ThABF in the presence of different metallic ions.
Optimization of pH and temperature conditions and enzymatic kinetics
Following the selection of ThABF-specific substrates and ions, the optimization of pH and temperature was performed under different conditions to identify the ideal performance of the enzyme. As shown in Fig. 5a,b, maximum ThABF activity was observed at 55 °C and pH 6.5. The enzyme exhibited greater than 50% activity between temperatures of 45 and 60 °C. However, at lower temperatures, in the range of 20 to 40 °C, its activity was below 40%. ThABF exhibited a wide range of performance levels at pH values from 6.0 to 9.0, and the optimal pH was shown to be 6.5 in sodium phosphate buffer. At pH 4 to 5.5, enzyme activity decreased significantly. Experiments were carried out to examine the kinetics of ThABF with the substrates selected in the specificity tests; the parameters Vmax, Km, Kcat and Kcat/Km were calculated, and the results are shown in Table 2. The curves constructed with the substrates pNPG and pNPAra are shown in Fig. 5c,d.

Biochemical characterization of ThABF. Relative activity of the recombinant protein at temperatures of 20 to 70 °C (a) and in the pH range of 4 to 10 (b). Enzymatic kinetics of TiBgal54A with the substrates pNPG (c) and pNPA (d).
Discussion
Arabinofuranosidases are enzymes that, in addition to acting on l-arabinose, can also degrade other types of sugars and can be applied in various industrial processes1,14. In this work, we bioprospected an arabifuranosidase from the metal-dependent GH54 family from T. harzianum and demonstrated that it showed expanded activity against other substrates. This enzyme can be introduced in enzymatic cocktails to assist in the hydrolysis of hemicellulose, since this biopolymer is present in approximately 50% of the most diverse sources of plant biomass, and its degradation requires highly diverse enzymes due to its structural complexity15,16,17,18.
Through the in silico analyses carried out in this study, great functional diversity was revealed in the GH54 family. Some studies have demonstrated that enzymes of this family exert activity against other types of substrates, in addition to those that are commonly identified, such as β-d-galactofuranoside, α-l-arabinopyranoside and β-d-xylopyranoside4,19. Evidence supporting this conclusion was found in a phylogenetic analysis in which we observed the presence of LTD, RICIN and NPCBM domains (related to the cleavage of simple sugars such as galactose and lactose) in several not yet characterized sequences20,21,22,23. We also identified several types of CBMs related mainly to enzymes that act on hemicellulose from both the phylogenetic and BLAST results. CBMs 42 and 13 are the most prevalent in GH54 sequences, possibly because they are present in some arabinofuranosidases and xylanases24,25.
Through the alignment and prediction of the structure of ThABF, we showed that the target protein identified in this study has the characteristics of an arabinofuranosidase, particularly regarding the high conservation of amino acids in the ArabFuran-catal and AbfB catalytic domains24,26,27. The ArabFuran-catal domain was present in all sequences analyzed in the family. This domain targets several l-arabinose bonds present in hemicellulose, as does AbfB24,26. For the purification of ThABF, it was necessary to use a refolding method adapted to recover the protein from inclusion bodies in its best oligomeric state28. The final protein yield following purification was relatively low, at 0.51 mg/mL, which made it difficult to carry out other types of biochemical tests; however, the method allowed us to characterize the enzyme and study it, as demonstrated in other studies28,29.
In tests with different substrates, we demonstrated that ThABF exerted activity on the substrates pNPG, pNPAp, pNPF and pNPAra. As previously described, some GH54 enzymes exhibit activity against substrates other than those reported in this family; however, there have been only two studies addressing this topic4,19. Such activity may be related to some modification of the active site of these enzymes, and resolution of their structures would provide a better understanding of this enzymatic activity toward different substrates, as only one structure in the family has been resolved to date24. At the time of writing the present report, 19 enzymes had been characterized in this family, among which only 6 have been tested on different types of synthetic substrates4,19,30,31. In addition, as demonstrated via in silico analyses, there are several unstudied proteins of this family harboring domains related to the hydrolysis of galactose, among other types of sugars. This demonstrates the limitation of studies involving enzymes of this family of GHs.
The data obtained in the tests with metallic cofactors showed that the enzyme was dependent on metals. This role of Mg2+ has been reported for other enzymes; there are several hypotheses regarding how magnesium acts on these enzymes, one of which is that it helps maintain the conformation of the active site32. The analysis of the optimal conditions of the enzyme showed that ThABF presents characteristics similar to those of other fungal arabifuranosidases of the GH54 family, making it quite adaptable to changes in the conditions imposed in several types of biotechnological processes, such as the hydrolysis of plant biomass1.
Based on the results presented above, ThABF has the potential to be used in the saccharification of lignocellulosic material, mainly because it acts on different types of sugars that constitute the side chains of hemicellulose33,34. These side chains contribute to the recalcitrancy of plant biomass due to its diverse structural composition2,35. The removal of hemicellulose makes it less recalcitrant, improving the hydrolysis process8,9. For this purpose, enzymes such as arabinofuranosidases that act on these sugars are necessary34.
In conclusion, the present study reports the in silico bioprospection, expression, purification and biochemical characterization of a GH54-dependent metal with expanded substrate specificity that acts in a wide pH range. These characteristics indicate the potential to use this enzyme in industrial bioprocesses, such as the saccharification of plant biomass. For future studies, it would be interesting to test the enzyme on different types of complex substrates, in addition to solving its structure to investigate the structural basis of the substrate specificity of ThABF.
Methods
In silico data mining, RNA-Seq read mapping and phylogenetic analyses of ThABF
A search for sequences of bifunctional Trichoderma enzymes that act on hemicellulose was carried out in the NCBI nonredundant protein36, UniProt and CAZy databases. With the selected sequence, a BLASTP search was performed versus the genome of T. harzianum CBS 226.95 (TaxID: 983964) in the NCBI database. The most similar sequence was used to map the RNA-Seq reads of the T. harzianum IOC-3844 data generated in the work of Horta et al.37 and, thus, obtain the sequence of the specific protein of the target lineage. For mapping, CLC Genomics Workbench software (CLC bio—v4.0; Finlandsgade, Dk) was used. Analyses of physical–chemical parameters were also performed with ProtParam; the presence of signal peptides was evaluated on the SignalP-5.0 server; domain prediction was conducted with SMART; and protein structure prediction was conducted with I-TASSER.
A BLASTP search of the sequence resulting from mapping was performed versus the CAZy database, and different enzymes of the GH54 family were then selected to perform phylogenetic analysis. The sequences were aligned using ClustalW38 implemented in Molecular Evolutionary Genetics Analysis software, version 7.039. Gaps were removed manually, and incomplete or difficult-to-align sequences were excluded from the analysis. Phylogenetic analyses were performed with MEGA7 using maximum likelihood (ML)40 inference based on the Jones–Taylor–Thornton (JTT) model with 1000 bootstrap41 repetitions for each analysis. The tree, drawn to scale, was obtained automatically by applying the neighbor-joining BioNJ algorithms to a matrix of distances in pairs estimated using the JTT model and the topology with the highest log probability values. The trees were visualized and edited using the program Figtree42.
Heterologous production and refolding of ThABF
The ThABF gene was cloned into the pET-28a (+) vector using the standard gene cloning protocol of Sambrook43 with the aid of the forward primer TAAGAATTCGGGCCCTGTGA and the reverse primer TGGTCGACTTAAGCAAACTGG. The recombinant plasmids were transformed into Escherichia coli Rosetta (Novagen, Darmstadt, Germany) for protein overexpression.
Transformed E. coli Rosetta cells were grown to an optical density of approximately 0.8 at 800 nm. To trigger the transcription of the ThABF gene, it was inserted in a culture containing IPTG at 0.4 mM. Then, the cells were incubated overnight at 25 °C, harvested by centrifugation and resuspended in 50 mL of buffer A (250 mM NaCl, 40 mM sodium phosphate, 20 mM imidazole, 3 mM MgCl2, 1 mM EDTA and pH 8.0), 1 mg/mL lysozyme and 1 mM PMSF (phenylmethylsulfonyl fluoride) for 30 min with shaking on ice. The cells were disrupted by sonication, and the soluble fraction was obtained by centrifugation (16,000 rpm, 40 min, 4 °C).
After rupturing the cells and resuspending the inclusion bodies, the refolding protocol of Santos et al.28 was applied. The pellet was resuspended in 15 mL of buffer A containing 1 M urea. Then, sonication and centrifugation (16,000 rpm, 15 min, 4 °C) were performed 6 times. Dialysis was subsequently performed under agitation overnight with buffer A plus 0.4 M arginine, followed by centrifugation (10,000 rpm, 10 min, 4 °C). The concentrations of the purified proteins were determined spectroscopically using the molar extinction coefficient (ε) predicted on the basis of the amino acid sequence, and sample purity was estimated by polyacrylamide gel electrophoresis with 12% sodium dodecyl sulfate (SDS-PAGE).
Analysis of folding components
The far-UV CD spectra of ThABF were collected using a Jasco model J-810 spectropolarimeter from the National Biorenewables Laboratory (LNBR) coupled to a Peltier control system (PFD 425S-Jasco). The CD spectra were generated using the purified recombinant protein at a concentration of approximately 1.024 mg/mL in 10 mM sodium phosphate buffer, pH 8.0. A total of 12 accumulations were recorded within the range of 260 to 208 nm at a rate of 50 nm min−1 using a quartz cuvette with a travel length of 1 mm, and the results were averaged.
The tertiary folding components of the recombinant refolded ThABF were evaluated by analytical SEC using a Superdex 200 10/300 GL column (GE Healthcare, Uppsala, Sweden). The protein sample at a concentration of approximately 0.56 mg/mL was dialyzed against buffer A, and gel filtration was then performed at a flow rate of 0.5 mL min−1. The elution fractions from each chromatographic run were collected and analyzed by 12% SDS-PAGE.
Biochemical characterization
The substrate specificity of ThABF was tested with the following pNP glycosides: α-d-arabinopyranoside (pNPAp), β-d-galactopyranoside (pNPG), α-d-arabinofuranoside (pNPAra), β-d-fucopyranoside (pNPF), α-d-glucopyranoside, α-d-xylopyranoside, α-d-mannopyranoside, α-l-ramnopyranoside, α-l-fucopyranoside, β-d-glucopyranoside, α-d-galactopyranoside, β-d-xylopyranoside, β-d-cellobioside and β-d-mannopyranoside. A 10 mM concentration of pNPs was used in the tests, which were conducted in 100 mM sodium phosphate buffer, pH 6.5, at 55 °C for 10 min. The reactions were stopped with 100 mM CaCO3, and the released pNP was quantified spectrophotometrically at 410 nm.
After selecting the substrates against which ThABF exhibited activity, its activity was tested with different 1 mM concentrations of different metal salts (CaCl2, KCl, NaCl, MgCl2, MnCl2, CoCl2, ZnSO4, CuCl2, NiCl2, FeCl3 and BaCl2) and EDTA. Before these tests, any metal ions that could interfere with the analysis were removed via two dialysis steps performed on the recombinant protein overnight under agitation. First, 15 mM EDTA was added to chelate the metals present in the sample, and the protein was then transferred to a buffer containing 10 mM sodium phosphate. Subsequently, the tested chemicals were individually incubated with the recombinant enzyme; the reactions occurred at 55 °C at pH 6.5 for 10 min with pNPG as the substrate.
After the selection of the ion with the greatest effect on the enzyme, it was used to optimize pH and temperature conditions. The optimal pH was determined by incubating 0.12 mg/mL of purified ThABF with 10 mM pNPG in buffers with pH levels ranging from 3.0 to 10.0 [sodium acetate buffer (100 mM, pH 3.0 to 5.0), sodium phosphate buffer (100 mM, pH 6.0 to 8.0) and Tris-NaCl (100 mM, pH 7.0 to 9.0)]. The mixtures were incubated at 55 °C for 10 min, and the released pNP was quantified spectrophotometrically at 410 nm. The same conditions were applied to investigate the ideal temperature; these tests were conducted with Na2HPO4 phosphate buffer (pH 6.5, 100 mM) at a temperature range of 20 to 70 °C. Subsequently, the pH test was repeated at the identified temperature.
After performing the above tests, assays of enzymatic kinetics were performed at pH 6.5 at 55 °C for 10 min, with pNPG, pNPAp, pNPF and pNPAra as substrates. The parameters Km, Vmax, and Kcat and the Kcat/Km ratio were obtained by plotting using the Lineweaver–Burk method, where the plots were constructed by plotting the substrate concentration on the x axis and the speed of the enzymatic reaction on the y axis using the OriginPro 8.5.0 program. The experiments were performed in triplicate at an ideal temperature and pH. A unit of enzymatic activity was defined as the amount of enzyme required to release 1 µmol of p-nitrophenol per minute under the tested conditions.
Accession codes
The protein sequence in this study has been deposited at the National Center for Biotechnology Information (NCBI) under accession code MT439956.
References
Poria, V., Saini, J. K., Singh, S., Nain, L. & Kuhad, R. C. Arabinofuranosidases: characteristics, microbial production, and potential in waste valorization and industrial applications. Bioresour. Technol. 304, 123019 (2020).
Basu, P. Biomass characteristics. In Biomass Gasification Design Handbook (ed. Basu, P.) 27–63 (Elsevier Inc., 2010).
Lagaert, S., Pollet, A., Courtin, C. M. & Volckaert, G. β-Xylosidases and α-l-arabinofuranosidases: accessory enzymes for arabinoxylan degradation. Biotechnol. Adv. 32, 316–332 (2014).
Ravanal, M. C. & Eyzaguirre, J. Heterologous expression and characterization of α-l-arabinofuranosidase 4 from Penicillium purpurogenum and comparison with the other isoenzymes produced by the fungus. Fungal Biol. 119, 641–647 (2015).
Tursi, A. A review on biomass: importance, chemistry, classification, and conversion. Biofuel Res. J. 6, 962–979 (2019).
Francocci, F. & Reca, I. B. Composition of plant biomass biotech engineering of cell wall to optimize biofuel production. In Biotransformation of Agricultural Waste and By-Products (eds Francocci, F. & Reca, I. B.) 219–236 (Elsevier Inc., 2016).
Chen, H. Lignocellulose biorefinery conversion engineering. In Lignocellulose Biorefinery Engineering (ed. Chen, H.) 87–124 (Elsevier, 2015).
Zoghlami, A. & Paës, G. Lignocellulosic biomass: understanding recalcitrance and predicting hydrolysis. Front. Chem. 7, 874 (2019).
Melati, R. B. et al. Key factors affecting the recalcitrance and conversion process of biomass. Bioenergy Res. 12, 1–20 (2019).
Chandel, A. K. et al. Bioconversion of hemicellulose into ethanol and value-added products: commercialization, trends, and future opportunities. In Advances in Sugarcane Biorefinery (eds Chandel, A. K. et al.) 97–134 (Elsevier, 2018).
Adav, S. S. & Sze, S. K. Trichoderma secretome: an overview. In Biotechnology and Biology of Trichoderma (eds Adav, S. S. & Sze, S. K.) 103–114 (Elsevier, 2014).
Druzhinina, I. S. et al. Massive lateral transfer of genes encoding plant cell wall-degrading enzymes to the mycoparasitic fungus Trichoderma from its plant-associated hosts. PLoS Genet. 14, 1–33 (2018).
Horta, M. A. C. et al. Transcriptome profile of Trichoderma harzianum IOC-3844 induced by sugarcane bagasse. PLoS ONE 9, 1–17 (2014).
Dimarogona, M. & Topakas, E. Regulation and heterologous expression of lignocellulosic enzymes in Aspergillus. In New and Future Developments in Microbial Biotechnology and Bioengineering: Aspergillus System Properties and Applications (eds Dimarogona, M. & Topakas, E.) 171–190 (Elsevier B.V., 2016).
Yeo, J. Y., Chin, B. L. F., Tan, J. K. & Loh, Y. S. Comparative studies on the pyrolysis of cellulose, hemicellulose, and lignin based on combined kinetics. J. Energy Inst. 92, 27–37 (2019).
Biely, P., Singh, S. & Puchart, V. Towards enzymatic breakdown of complex plant xylan structures: state of the art. Biotechnol. Adv. 34, 1260–1274 (2016).
Fry, S. C. Cell walls and fibers. In Encyclopedia of Applied Plant Sciences (ed. Fry, S. C.) 75–87 (Elsevier, 2003).
Chen, D. et al. Investigation of biomass torrefaction based on three major components: hemicellulose, cellulose, and lignin. Energy Convers. Manag. 169, 228–237 (2018).
Margolles-Clark, E., Tenkanen, M., Nakari-Setälä, T. & Penttilä, M. Cloning of genes encoding alpha-l-arabinofuranosidase and beta-xylosidase from Trichoderma reesei by expression in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 62, 3840–3846 (1996).
Hazes, B. The (QxW)3 domain: a flexible lectin scaffold. Protein Sci. 5, 1490–1501 (1996).
Spinette, S. et al. Ufd2, a novel autoantigen in scleroderma, regulates sister chromatid separation. Cell Cycle 3, 1612–1618 (2004).
Hirabayashi, J., Dutta, S. K. & Kasai, K. I. Novel galactose-binding proteins in annelida. J. Biol. Chem. 273, 14450–14460 (1998).
Naumoff, D. G. Phylogenetic analysis of -galactosidases of the GH27 family. Mol. Biol. 38, 388–400 (2004).
Miyanaga, A. et al. The family 42 carbohydrate-binding module of family 54 alpha-l-arabinofuranosidase specifically binds the arabinofuranose side chain of hemicellulose. Biochem. J. 399, 503–511 (2006).
Li, S. et al. Family 13 carbohydrate-binding module of alginate lyase from Agarivorans sp. L11 enhances its catalytic efficiency and thermostability, and alters its substrate preference and product distribution. FEMS Microbiol. Lett. 362, 1–7 (2015).
Gielkens, M. et al. The abfB gene encoding the major α-l-arabinofuranosidase of Aspergillus nidulans: nucleotide sequence, regulation and construction of a disrupted strain. Microbiology 145, 735–741 (1999).
Koseki, T. et al. Role of two α-l-arabinofuranosidases in arabinoxylan degradation and characteristics of the encoding genes from shochu koji molds, Aspergillus kawachii and Aspergillus awamori. J. Biosci. Bioeng. 96, 232–241 (2003).
Santos, C. A. & Souza, A. P. Solubilization, folding, and purification of a recombinant peptidoglycan-associated lipoprotein (PAL) expressed in Escherichia coli. Curr. Protoc. Protein Sci. 92, e53 (2018).
Santos, C. A. et al. Characterization of the TolB–Pal trans-envelope complex from Xylella fastidiosa reveals a dynamic and coordinated protein expression profile during the biofilm development process. Biochim. Biophys. Acta Proteins Proteom. 1854, 1372–1381 (2015).
de Wet, B. J. M., Matthew, M. K. A., Storbeck, K. H., van Zyl, W. H. & Prior, B. A. Characterization of a family 54 α-l-arabinofuranosidase from Aureobasidium pullulans. Appl. Microbiol. Biotechnol. 77, 975–983 (2008).
Wan, C. F., Chen, C. T., Li, Y. K. & Huang, L. Expression, purification and characterization of a bifunctional α-l-arabinofuranosidase/β-d-xylosidase from Trichoderma koningii G-39. J. Chin. Chem. Soc. 54, 109–116 (2007).
Höhn, S., Virtanen, S. & Boccaccini, A. R. Protein adsorption on magnesium and its alloys: a review. Appl. Surf. Sci. 464, 212–219 (2019).
Madeira, J. V. et al. Agro-industrial residues and microbial enzymes: an overview on the eco-friendly bioconversion into high value-added products. In Biotechnology of Microbial Enzymes: Production, Biocatalysis and Industrial Applications (eds Brahmachari, G. et al.) 475–511 (Elsevier Inc., 2017).
Liu, X. & Kokare, C. Microbial Enzymes of Use in Industry. Biotechnology of Microbial Enzymes: Production, Biocatalysis and Industrial Applications (Elsevier Inc., 2017).
Ebringerová, A., Hromádková, Z. & Heinze, T. Hemicellulose. In Polysaccharides I (ed. Heinze, T.) 1–67 (Springer, 2005).
Pearson, W. R. An introduction to sequence similarity ("homology") searching. In Current Protocol in Bioinformatics, Chapter 3, Unit 3.1 3.1.1-7 (2013).
Horta, M. A. C. et al. Network of proteins, enzymes and genes linked to biomass degradation shared by Trichoderma species. Sci. Rep. 8, 1–11 (2018).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Jones, D. T., Taylor, W. R. & Thornton, J. M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282 (1992).
Felsenstein, J. Confidence limits on phylogenies: an approach using the bootstrap. Soc. Study Evol. 39, 1–15 (1985).
Ferreira Filho, J. A., Horta, M. A. C., Beloti, L. L., Dos Santos, C. A. & de Souza, A. P. Carbohydrate-active enzymes in Trichoderma harzianum: a bioinformatic analysis bioprospecting for key enzymes for the biofuels industry. BMC Genom. 18, 1–12 (2017).
Dong, L., Lv, L. B. & Lai, R. Molecular Cloning. Dong wu xue yan jiu = Zoological research/“Dong wu xue yan jiu” bian ji wei yuan hui bian ji (Yunnan Ren Min Chu Ban She, 2012).
Acknowledgements
This work was supported by grants from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2015/09202-0 and 2018/19660-4), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) Computational Biology Program (CBP: Process number 88882.160095/2013-01) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq: Process number 312777/2018-3; 430350/2018-0). MLLM received an MS fellowship from CAPES (CBP—88887.200427/2018-00); JAFF received a PhD fellowship from CNPq (170565/2017-3) and a PD fellowship from CAPES (CBP—88887.334235/2019-00); RRM received a PD fellowship from FAPESP (17/14253-9); LMZ is a recipient of a research fellowship from FAPESP (19/08855-1); CAS received a PD fellowship from FAPESP (2016/19775–0) and an SWE PD fellowship from CAPES (CBP); and APS is the recipient of a research fellowship from CNPq (312777/2018–3). We thank the National Biorenewables Laboratory (LNBR).
Author information
Authors and Affiliations
Contributions
M.L.L.M. carried out all the experiments and wrote the manuscript. J.A.F.F. helped with the in silico analyses and wrote the manuscript. R.R.M. and L.M.Z. helped with the biochemical characterization. C.A.S. designed the experiments and wrote the manuscript. A.P.S. directed the general study and wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Table 1
. ThABF Blastp against the CAZy database.
Supplementary Table 2
. GH54 sequences used in the phylogenetic tree.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Motta, M.L.L., Filho, J.A.F., de Melo, R.R. et al. A novel fungal metal-dependent α-l-arabinofuranosidase of family 54 glycoside hydrolase shows expanded substrate specificity. Sci Rep 11, 10961 (2021). https://doi.org/10.1038/s41598-021-90490-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90490-2
This article is cited by
-
Microbial α-L-arabinofuranosidases: diversity, properties, and biotechnological applications
World Journal of Microbiology and Biotechnology (2024)
-
Two α-Arabinofuranosidases from Chrysoporthe cubensis and Their Effects on Sugarcane Bagasse Saccharification
BioEnergy Research (2024)
-
Functional characteristics of an α-L-arabinofuranosidase secreted by the basidiomycete Coriolopsis byrsina
Biologia (2023)
-
Whole-genome sequencing and comparative genomic analysis of potential biotechnological strains of Trichoderma harzianum, Trichoderma atroviride, and Trichoderma reesei
Molecular Genetics and Genomics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
