
Abstract
β-Thalassemia/HbE disease has a wide spectrum of clinical phenotypes ranging from asymptomatic to dependent on regular blood transfusions. Ability to predict disease severity is helpful for clinical management and treatment decision making. A thalassemia severity score has been developed from Mediterranean β-thalassemia patients. However, different ethnic groups may have different allele frequency and linkage disequilibrium structures. Here, Thai β0-thalassemia/HbE disease genome-wild association studies (GWAS) data of 487 patients were analyzed by SNP interaction prioritization algorithm, interacting Loci (iLoci), to find predictive SNPs for disease severity. Three SNPs from two SNP interaction pairs associated with disease severity were identifies. The three-SNP disease severity risk score composed of rs766432 in BCL11A, rs9399137 in HBS1L-MYB and rs72872548 in HBE1 showed more than 85% specificity and 75% accuracy. The three-SNP predictive score was then validated in two independent cohorts of Thai and Malaysian β0-thalassemia/HbE patients with comparable specificity and accuracy. The SNP risk score could be used for prediction of clinical severity for Southeast Asia β0-thalassemia/HbE population.
Similar content being viewed by others


Introduction
Thalassemia, an inherited red blood cell disorder, presents a significant health problem worldwide. β-Thalassemia caused by defects on β-globin gene results in reduction or absence of β-globin chain synthesis. Co-inheritance of β0-thalassemia with hemoglobin (Hb) E resulting in β0-thalassemia/HbE genotype, the most prevalent β-thalassemia diseases in Southeast Asia including Thailand and Malaysia. In contrast to β-thalassemia major patients, carrying homozygous or compound heterozygous β0-thalassemia mutations, who have a severe phenotype of β-thalassemia disease and requiring regular blood transfusions, β0-thalassemia/HbE disease has a very heterogeneous clinical symptom. Although carrying the same β-thalassemia mutation, the patients have a remarkable variability in disease severity. The average Hb levels were 7.7 g/dL with a range from 3 to 13 g/dL1. The mild and moderate symptoms usually have no need for regular blood transfusions. Whereas severe symptoms have marked anemia, hepatosplenomegaly, heavy iron overload and required regular blood transfusions similar to β-thalassemia major patients.
Genetic factors are involved in determining the β-thalassemia clinical variability. As pathophysiological changes in β-thalassemia patients are predominantly determined by the amount of excess α-globin chains, the factor that affects the imbalance between α- and β-globin chain production is an important modifier of β-thalassemia disease. This including the nature of β-thalassemia mutation, number of functional α-globin genes and genetic factor that regulate HbF production2. Our two previous genome-wide SNPs association studies (GWAS) had been performed in order to search for genetic modifying factors in β0-thalassemia/HbE disease3,4. The GWAS analysis identified a number of highly significant SNPs located in 3 regions on chromosome 2p16.1, 6q23.3 and 11p15.4. The strongest association with the disease severity is located in the region of more than 200 kb harboring several genes in the β-globin cluster and nearby olfactory receptor gene on chromosome 11p15.4. The second most significantly associated region mapped to the HBS1L-MYB intergenic 24 kb region on chromosome 6q23.3. The third significantly disease severity associated SNPs are located in the BCL11A gene on chromosome 2p16.1.
Discovery of genetic modifiers for β0-thalassemia/HbE provided a genetic basis for clinical application such as new targets for therapeutic intervention and predicting disease severity. Predictive SNPs for β0-thalassemia/HbE disease severity have a wide range of clinical applications, such as a better informed genetic counseling, clinical management and assistance in therapeutic decisions. However, as several SNPs demonstrated to be disease modifiers, single SNP might not be a good prediction of disease severity. In addition, genetic factors modify disease through a complex mechanism of multiple genes interaction of a biological network. Here the two β0-thalassemia/HbE GWAS data3,4 of 487 cases were reanalyzed to search for minimum predictive SNPs for disease severity by using a SNP interaction prioritization algorithm, interacting Loci (iLoci)5. Combination of three SNPs located on chromosome 2p16.1 (BCL11A), 6q23.3 (HBS1L-MYB) and 11p15.4 (HBE1) could predict disease severity with more than 85% specificity and 75% accuracy. The three predictive SNPs were validated in two independent cohorts of Thai and Malaysian patients with similar accuracy and specificity.
Results
Characteristics of cohort study
Characteristics of the patients according to demographic information and hematological data are described in Table S1. Types of β-thalassemia mutations in this study are illustrated in Table S2. The most frequent β-thalassemia mutations in Thai β0-thalassemia/HbE patients were HBB:c.126-129delCTTT and HBB:c.52A>T in the cohort 1 (45.7% and 26.3%, respectively) and the cohort 2 (51.6% and 23.4%, respectively). While the cohort 3, Malaysian patients, HBB:c.92 + 5G>C (37.9%) and HBB:c.92 + 1G>A (22.4%) were the most frequent mutations.
Identification of predictive SNPs
As multiple SNPs demonstrated to be disease modifiers of β0-thalassemia/HbE, single SNP might not have enough power to predict disease severity. Thus, reanalysis of SNP-SNP interaction of the previously reported two GWAS data3,4 was performed by iLOCi algorithm5. The highest disease severity associated SNP pair ranked by the SNP pair interaction analysis was rs766432/rs9399137, which predicts disease severity at 62.5% accuracy (Table 1). Combination of the two highest disease severity associated SNP pairs, rs766432/rs9399137 and rs72872548/rs9399137 could increase prediction accuracy to 72.0%. While three SNP pairs, combination of the prior two pairs and rs11208724/rs1407273 have 72.3% prediction accuracy (Table 1). Although the disease severity prediction accuracy increased with the increasing number of SNP pairs, the prediction accuracy of two or more SNP pairs was not much different. Thus, only three SNPs from two SNP pairs were selected for further evaluation, which are rs766432 located in BCL11A gene on chromosome 2p16.1, rs9399137 located in HBS1L-MYB intergenic region on chromosome 6q23.3 and rs72872548, located in HBE1 gene on chromosome 11p15.4.
In addition, reanalysis for single SNP association of the disease severity was performed on the two GWAS data by FaST-LMM, which utilizes the linear mixed model to select the associated SNPs6. The three SNPs, rs766432 (BCL11A), rs9399137 (HBS1L-MYB) and rs72872548 (HBE1), showed high level of significance among the three previously reported regions that strongly associated with disease severity (chromosome 2p16.1, chromosome 6q23.3 and chromosome 11p15.4)3,4 (Table 2). The agreement between two reanalysis methods suggested that the three SNPs were good candidate predictive SNPs for constructing the disease severity predictive model. The allele frequency of the three predictive SNPs in cohort 1 Thai β0-thalassemia/HbE patients compared with another ethnic group is showed in Table S3.
Determination of risk score for β-thalassemia/HbE disease severity predictive SNPs
β0-Thalassemia/HbE patients have a wide spectrum of clinical symptoms. To determine whether the selected SNPs account for moderate symptom patients, the three predictive SNPs were genotype in 181 patients with moderate symptom as well as in the mild and severe symptoms in the discovery cohort 1. The genotype frequency of the three predictive SNPs among 668 β0-thalassemia/HbE patients in the cohort 1 with different severity (mild, moderate and severe) is showed in Table 3. Odds ratio (OR) analysis was then performed to determine risk genotypes of each SNP between two different disease severity groups; mild vs moderate, mild vs severe and moderate vs severe in the cohort 1 (Table 4). The rs766432 genotype AA has increased risk for severe symptoms (mild vs severe: OR = 0.39, 95% CI = 0.26–0.58, P = 4.98 × 10–6; moderate vs severe: OR = 0.51, 95% CI = 0.34–0.77, P = 1.45 × 10–3). The rs9399137 genotype TT has increased risk for severe symptoms (mild vs severe: OR = 0.35, 95% CI = 0.23–0.51, P = 2.16 × 10–7). While the rs72872548 genotype CC has increased risk for severe symptoms (mild vs severe: OR = 0.21, 95% CI = 0.12–0.36, P = 2.25 × 10–9; moderate vs severe: OR = 0.50, 95% CI = 0.32–0.77, P = 1.98 × 10–3).
In order to apply the risk genotypes for disease severity prediction, score was assigned to the SNP genotypes as 0, 1 and 2 accordingly to the risk from low to high risk of severe symptom. Therefore, SNPs predictive risk score for rs766432 were CC = 0, AC = 1 and AA = 2; for rs9399137 were CC = 0, TC = 1 and TT = 2 and for rs72872548 were AA = 0, AC = 1 and CC = 2 (Table 5). The score of each predictive SNPs was combined and used for interpretation of disease severity prediction. Three scoring models of interpretation of the combine SNPs risk score were generated (Table 5) and evaluated in the 668 β0-thalassemia/HbE cohort 1. The first scoring model, model 1, yield the highest sensitivity, 60.0%, and accuracy, 71.2%. While the model 3 resulted in the highest specificity, 81.4% (Table 6). To determine which scoring model is better in disease severity prediction, the three scoring models were further validated in two independent Thai and Malaysian β0-thalassemia/HbE cohorts.
Validation of predictive SNP scoring system
The three-SNP risk scoring models were validated in 122 cases β0-thalassemia/HbE patients in two cohorts, 64 Thai patients (cohort 2) and 58 Malaysian patients (cohort 3). The allele frequency of the three predictive SNPs in the Thai and Malaysian validation cohorts are shown in Table S3. Consistence with the results from cohort 1, the model 1 have the highest sensitivity and accuracy compared to model 2 and model 3 in every cohorts. While the model 3 have highest specificity in every cohorts (Table 6).
Discussion
β0-Thalassemia/HbE patients have a wild range of disease severity from the unrequired of regular blood transfusion to transfusion dependent. Prediction of the disease severity is helpful for clinical management and quality of life of the patients. Knowing the severity of the affected child during pregnancy could aid in genetic counseling. After birth, this could also be useful for clinical decisions of transfusion program such as frequency and age needed for blood transfusion. In addition, this will aid in deciding whether to perform hematopoietic stem cell transplantation, which is more efficient if it is performed early in life. Nevertheless, clinical phenotype can take few years to stabilize, and in such a situation the possibility of anticipating the clinical severity could be essential. Two β0-thalassemia/HbE GWAS studies have identified a number of risk SNPs. Here, FaST-LMM, iLOCi and machine learning software by WEKA was used to examine SNPs interaction and determine predictive SNP risk score.
This study explored the predictive SNPs for disease severity in 668 β0-thalassemia/HbE patients. Analysis of SNP interaction revealed that rs766432 (BCL11A), rs9399137 (HBS1L-MYB) and rs72872548 (HBE1) are highest associated with clinical symptoms. Three risk scoring models, which differ in assigning score 5 to moderate and/or severe symptoms, were evaluate for prediction of clinical severity in three independent cohorts. This due to the rs72872548 genotype AC is high frequency among mild, moderate and severe cases, 78.33, 77.35 and 63.84%, respectively. The score model 1 have the highest sensitivity and accuracy. However, it would difficult to predict whether the patients would be moderate or severe for those who have risk score 5. The score model 3 have highest specificity, 81.4–88.8%, in every cohorts. Although, assigning score 5 to moderate group might have some severe patient miss prediction as moderate, this rather better than predict moderate patients as severe patient in such cases the patients might undergo unnecessary aggressive treatments.
The three predictive SNPs identified here are located in three loci that associated with HbF levels, which affect the central mechanism underlying disease pathophysiology, the degree of the excess of α-globin chains and globin chain imbalance. Several GWAS studies showed the three loci are associated with HbF level3,7,8. The BCL11A is a transcription factor act as the specific erythroid repressor of HbF expression in the developmental silencing of the mouse and human HBG genes9,10. The HBS1L-MYB intergenic region contains distal enhancers required for MYB activation11. The c-MYB, a transcription factor, is key regulator of hematopoiesis and erythropoiesis. cMYB represses HBG genes expression via KLF1 activation of BCL11A12. In addition, several GWAS studies showed association of SNPs on BCL11A and HBS1L-MYB with red blood cells parameters such as mean corpuscular volume (MCV)13,14. The β-globin cluster, Xmn1-HBG2 (rs782144) influence on HbF level has long been discovered through candidate and genetic linkage studies and later confirmed by GWAS studies. The rs72872548 (HBE1) is located in the same haplotype block as the Xmn1-HBG2.
The use of multiple genetic factors to predict β-thalassemia was first reported in a study in Sardinian homozygous β0-thalassemia. Three genetic factors, α-thalassemia, rs11886868 in BCL11A and rs9389268 in HBS1L-MYB were account for 75% of the phenotype severity15. A study of a mix of Mediterranean (3/4) and Asian (1/4) patients from France used five genetic factors, β-thalassemia mutations, α-thalassemia, the Xmn1-HBG2, rs11886868 in BCL11A and rs9399137 in HBSB1L-MYB have 83.2% predictive accuracy16. A thalassemia severity score (TSS) was developed from 890 homozygous β-thalassemia patients of the Mediterranean basin using these different genetic modifiers including sex, β-thalassemia mutations, α-thalassemia, the Xmn1-HBG2, rs1427407 and rs1018957 in BCL11A and rs9399137 in HBS1L-MYB17. In this study, the β0-thalassemia/HbE patients who carry β+-thalassemia mutations or were found positive for the common Thai α-thalassemia mutations were excluded. Hence, the predictive score comprise of only three SNPs, which are located at the same loci as the previous studied.
The limitation of the predictive SNPs scoring is that different ethnic groups may have different allele frequency and linkage disequilibrium structures. This might render the scoring less accurate in untested populations. The TSS score was validated in a North African cohort which showed that allelic frequencies of the SNPs are different compared to the Mediterranean18. According to the International HapMap Project, the rs1018957 (BCL11A) allele A/G frequency are quite different among European (0.580/0.420), African (0.722/0.278) and this study Thai β0-Thalassemia/HbE patients (0.182/0.818). The allele frequency of the predictive SNPs in this study of both Thai and Malaysian population are comparable. This suggested that the three SNPs can be used as predictive SNPs for disease severity at least for Southeast Asia where β0-thalassemia/HbE is prevalent.
This study, to the best of our knowledge, is the first SNP risk score for prediction of clinical severity developed for a β0-thalassemia/HbE Southeast Asia population. This may assist in inform prognosis and guide therapeutics. However, the predictive SNPs of this study might not predict disease severity in β+(severe)/β0-thalassemia and β0/β0-thalassemia patients who a have higher degree of α- to non-α-globin imbalance and being the cause of the severe thalassemia phenotype. Nevertheless, the predictive SNPs validation also required in other populations.
Methods
Subjects and thalassemia diagnosis
This study was performed in accordance with the Helsinki Declaration and was approved by the Mahidol University Institutional Review Board, Nakhon Pathom, Thailand (approval number MU-IRB 2007-083, 2008/303.2001, 2009/128.2306, 2010/010.0701 and 2012/035.2802) and The Ethical Review Committee of Pathology Department, Hospital Sultanah Bahiyah, Kedah, Malaysia (approval number NMRR-12-980-13829 (IIR)). Written informed consent was obtained from all individual participants included in the study.
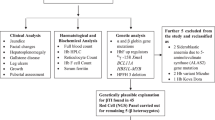
Three cohorts of β0-thalassemia/HbE patients were enrolled, Thai discovery cohort (cohort 1), Thai validation cohort (cohort 2) and Malaysian validation cohort (cohort 3). In this study, the β0-thalassemia/HbE patients who carry β+-thalassemia mutations or were found positive for the common Thai α-thalassemia mutations were excluded. The cohort 1 comprised 668 cases (180 mild, 181 moderate and 307 severe) of Thai β0-thalassemia/HbE patients. A total of 487 cases (180 mild and 307 severe) were randomly selected from the two GWAS analysis of mild and severe cases3,4. In addition 181 moderate patients were newly combining in the cohort 1. Two independent validation cohorts of Thai and Malaysian patients comprised 64 Thai cases and 58 Malaysian cases, respectively.
After informed consent was obtained, EDTA blood was collected and hematological data analysis was performed using an automated cell counter (ADVIA 120, Bayer, Tarrytown, NY). Hemoglobin analysis and quantification were determined by the automated hemoglobin cation exchange high-performance liquid chromatography (Bio-Rad variant II, Bio-Rad Hercules, CA). The β-thalassemia mutations were characterized using reverse dot blot hybridization19. The α-thalassemia deletional mutations were genotyped by multiplex GAP-PCR20. The α-thalassemia point mutations, Hb Constant Spring and Hb Pakse, were determined by dot blot hybridization21. The disease severity of β0-thalassemis/HbE presenting for mild, moderate and severe symptoms was classified by scoring system based on 6 parameters; hemoglobin at steady state, age at receiving first blood transfusion, requirement for blood transfusion, size of spleen, age at thalassemia presentation and the growth and development22.
Predictive SNP discovery
The disease severity association analysis was performed on the two separated GWAS analysis datasets3,4 of 180 mild and 307 severe of cohort 1 by two techniques i.e. iLOCi5 for SNP interaction and FaST-LMM6 for single SNP association. The predictive model was later constructed using a standard machine learning software, Waikato Environment for Knowledge Analysis or WEKA (http://www.cs.waikato.ac.nz/ml/weka/). The classification accuracy of disease severity was measured from the Hidden Naive Bayes model included in WEKA.
Genotyping of predictive SNPs
Genomic DNA was extracted from peripheral blood using the Gentra Puregene Blood Kit (Qiagen, Hilden, Germany). The three predictive SNPs; rs766432, rs9399137 and rs72872548, located on chromosome 2p16.1 (BCL11A), 6q23.3 (HBS1L-MYB) and 11p15.4 (HBE1), respectively, were genotyped by high-resolution melting (HRM) analysis using primers indicated in Table S4 and SsoFast EvaGreen Supermix (Bio-Rad, Hercules, CA) on CFX96 real-time PCR system (Bio-Rad).
Statistical analysis
All statistical procedures were performed using SPSS v.18.0 software package (SPSS Inc., Chicago, IL). Odds ratio (OR) analysis was used to identify risk genotypes of the predictive SNPs associated with disease severity.
References
Fucharoen, S., Winichagoon, P., Pootrakul, P., Piankijagum, A. & Wasi, P. Variable severity of Southeast Asian β0-thalassemia/Hb E disease. Birth Defects Orig. Artic. Ser. 23, 241–248 (1987).
Mettananda, S. & Higgs, D. R. Molecular basis and genetic modifiers of thalassemia. Hematol. Oncol. Clin. N. Am. 32, 177–191 (2018).
Nuinoon, M. et al. A genome-wide association identified the common genetic variants influence disease severity in β0-thalassemia/hemoglobin E. Hum. Genet. 127, 303–314 (2010).
Sherva, R. et al. Genetic modifiers of Hb E/β0 thalassemia identified by a two-stage genome-wide association study. BMC Med. Genet. 11, 51 (2010).
Piriyapongsa, J. et al. iLOCi: A SNP interaction prioritization technique for detecting epistasis in genome-wide association studies. BMC Genomics 13(Suppl 7), S2 (2012).
Lippert, C. et al. FaST linear mixed models for genome-wide association studies. Nat. Methods 8, 833–835 (2011).
Menzel, S. et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat. Genet. 39, 1197–1199 (2007).
Uda, M. et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc. Natl. Acad. Sci. USA 105, 1620–1625 (2008).
Sankaran, V. G. et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322, 1839–1842 (2008).
Sankaran, V. G. et al. Developmental and species-divergent globin switching are driven by BCL11A. Nature 460, 1093–1097 (2009).
Stadhouders, R. et al. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J. Clin. Investig. 124, 1699–1710 (2014).
Bianchi, E. et al. c-myb supports erythropoiesis through the transactivation of KLF1 and LMO2 expression. Blood 116, e99-110 (2010).
Soranzo, N. et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat. Genet. 41, 1182–1190 (2009).
van der Harst, P. et al. Seventy-five genetic loci influencing the human red blood cell. Nature 492, 369–375 (2012).
Galanello, R. et al. Amelioration of Sardinian β0 thalassemia by genetic modifiers. Blood 114, 3935–3937 (2009).
Badens, C. et al. Variants in genetic modifiers of β-thalassemia can help to predict the major or intermedia type of the disease. Haematologica 96, 1712–1714 (2011).
Danjou, F. et al. A genetic score for the prediction of β-thalassemia severity. Haematologica 100, 452–457 (2015).
Abdaoui, W. et al. Genetic background of β-thalassemia in Northeast Algeria with assessment of the thalassemia severity score and description of a new β0-thalassemia frameshift mutation (HBB: c.374dup; p.Pro126Thrfs*15). Hemoglobin 43, 223–228 (2019).
Winichagoon, P. et al. Prenatal diagnosis of β-thalassaemia by reverse dot-blot hybridization. Prenat. Diagn. 19, 428–435 (1999).
Chong, S. S., Boehm, C. D., Cutting, G. R. & Higgs, D. R. Simplified multiplex-PCR diagnosis of common southeast asian deletional determinants of α-thalassemia. Clin. Chem. 46, 1692–1695 (2000).
Pichanun, D. et al. Molecular screening of the Hbs Constant Spring (codon 142, TAA>CAA, α2) and Pakse (codon 142, TAA>TAT, α2) mutations in Thailand. Hemoglobin 34, 582–586 (2010).
Sripichai, O. et al. A scoring system for the classification of β-thalassemia/Hb E disease severity. Am. J. Hematol. 83(6), 482–484 (2008).
Acknowledgements
This study was supported by Research Chair Grant, National Science and Technology Development Agency (NSTDA), Thailand; Office of the Higher Education Commission and Mahidol University under the National Research University Initiative; the Program Management Unit for Human Resources & Institutional Development, Research and Innovation (B05F630062); Mahidol University (MRC-MGR 01/2563). T.M. was supported by the Thailand Research Fund through the Royal Golden Jubilee Ph.D. Program (Grant No. PHD/0277/2551).
Author information
Authors and Affiliations
Contributions
Investigation, T.M. and H.H.; Software, S.T. and C.N.; Formal Analysis, T.M., C.P. and P.W.; Writing—original draft, T.M., C.P. and S.S.; Writing—review and editing, T.M. and S.S.; Conceptualization, S.S. and S.F.; Funding acquisition, S.S. and S.F.; Supervision, S.S. S.S. was the principal investigator and takes primary responsibility for the concept and design of the project, the analysis of the data and drafting and editing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Munkongdee, T., Tongsima, S., Ngamphiw, C. et al. Predictive SNPs for β0-thalassemia/HbE disease severity. Sci Rep 11, 10352 (2021). https://doi.org/10.1038/s41598-021-89641-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-89641-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
