
Abstract
New solid solution of Na0.5Bi0.5TiO3 with BaFeO3−δ materials were fabricated by sol–gel method. Analysis of X-ray diffraction patterns indicated that BaFeO3−δ materials existed as a well solid solution and resulted in distortion the structure of host Na0.5Bi0.5TiO3 materials. The randomly incorporated Fe and Ba cations in the host Na0.5Bi0.5TiO3 crystal decreased the optical band gap from 3.11 to 2.48 eV, and induced the room-temperature ferromagnetism. Our density-functional theory calculations further suggested that both Ba for Bi/Na-site and Fe dopant, regardless of the substitutional sites, in Na0.5Bi0.5TiO3 lead to the induced magnetism, which is illustrated in terms of the exchange splitting between spin subbands through the crystal field theory and Jahn–Teller distortion effects. Our work proposes a simple method for fabricating lead-free ferroelectric materials with ferromagnetism property for multifunctional applications in smart electronic devices.
Similar content being viewed by others
Introduction
The current research trend in materials science is injecting ferromagnetism into ferroelectric materials to create next-generation smart electronic devices1,2. The development of ferromagnetism materials based on lead-based ferroelectric by doing transition metals, such as PbTiO3, has been hampered because their materials strong adverse effect on the environment and human health3. Ferroelectric lead-based PbTiO3 materials are more commonly used than lead-free ferroelectric materials, such as those based on Bi0.5(Na,K)0.5TiO3, (K,Na)NbO3, or (Ba,Ca)(Zr,Ti)O34,5. Na0.5Bi0.5TiO3 as one of common lead-free ferroelectric compounds has been considered as a promising candidate that can be replacement for lead-based substances because of its stronger polarization3. The high polarization of Na0.5Bi0.5TiO3 materials is due to the lone pair effect of Bi3+ in comparison with that of Pb2+ in perovskite structures6,7. Therefore, injecting room-temperature ferromagnetism into lead-free ferroelectric Na0.5Bi0.5TiO3 is one of significant research interests.
Scholars have developed Na0.5Bi0.5TiO3-based materials with room-temperature ferromagnetism property by using transition metals, such as Co, Fe, Mn, or Cr8,9,10,11. Moreover, the solid solutions of Na0.5Bi0.5TiO3-based materials with BiFeO3 material exhibits room-temperature ferromagnetism12. Na0.5Bi0.5TiO3 materials are composed of ferrite compounds, such as CoMn0.2Fe1.8O413. Ju et al. predicted that substituting a transition metal cation to Ti-site results in the magnetic moments because of the spin polarization of the 3d electrons in the transition metal14. However, the origin of ferromagnetism in transition metal doped lead-free ferroelectric materials were still debated. The pure Na0.5Bi0.5TiO3 materials exhibited the weak-ferromagnetism at room temperature which were possible explained by the surface-effect and/or self-defect15,16. Zhang et al. predicted that Na and Ti vacancies induced the magnetization rather than Bi or O vacancies15. Ju et al. reported that Na vacancies located at/near the surface of nanograins of nanocrystalline Na0.5Bi0.5TiO3 materials possibly displayed the ferromagnetism16. Such predictions were well consisted with recent obtained room temperature ferromagnetism in pure Na0.5Bi0.5TiO3 materials10,11. However, the magnetization of pure Na0.5Bi0.5TiO3 materials are quite small, normally less than 1 memu/g, which hinted to apply in electronics devices. Injection of transition metal into host lead-free ferroelectric Na0.5Bi0.5TiO3 materials possibly enhanced the magnetization up to ~ 9 memu/g10. Unlikely the picture of ferromagnetism at room temperature of pure Na0.5Bi0.5TiO3, a various magnetism sources were injected to lead-free ferroelectric materials which resulted in the room temperature ferromagnetism; such as the O-vacancies (in case of Cr-doped Na0.5Bi0.5TiO3), magnetic clusters (in case of Co-doped Na0.5Bi0.5TiO3), or interaction of magnetic cations through oxygen vacancies as intrinsic phenomenon (in case of Fe-, Mn-doped Na0.5Bi0.5TiO3)8,9,10,11. Therefore, the origin of room temperature ferromagnetism needs to be deep understood to control the magnetization for smart-electronic devices application.
Thank to well solid solution of Na0.5Bi0.5TiO3 material with various type of ABO3 dopant materials, the physical properties of host Na0.5Bi0.5TiO3 materials were enhanced17,18,19,20,21,22,23,24,25,26,27. Rahman et al. reported that both ferroelectric and piezoelectric properties of Na0.5Bi0.5TiO3 increased via solid solution of BaZrO3 where the both remanent polarization and piezoelectric constant increased from 22 μC/cm2 and 60 pC/N for pure Na0.5Bi0.5TiO3 to 30 μC/cm2 and 112 pC/N for 4 mol% BaZrO3 solid solution in Na0.5Bi0.5TiO3 materials17. Yang et al. reported that the (Ba0.7Ca0.3)TiO3 solid solution in host Na0.5Bi0.5TiO3 materials resulted in greatly lowered coercive field without degrading remanent polarization18. Bai et al. reported that the Bi(Me0.5Ti0.5)O3 (Me = Zn, Ni, Mg, Co)-modified Na0.5Bi0.5TiO3 materials displayed the large strain response (> 0.3%) with a high normalized strain Smax/Emax (> 550 pm/V)19. Zhou et al. reported that BaNb2O6 diffused into lattice of Na0.5Bi0.5TiO3 to form a solid solution resulted in enhancement of the dielectric properties of host Na0.5Bi0.5TiO3 materials20. Kaswan et al. reported on ferromagnetism in Bi0.5Na0.5TiO3-Bi0.8Ba0.2FeO3 composite materials21. Pattanayak et al. observed the ferromagnetic properties of a BaFe12O19-modified Bi0.5Na0.5TiO3 system22. Recently, Singh et al. reported on the ferromagnetic properties of Bi0.5Na0.5TiO3 materials induced by the addition of LaFeO3 as solid solution23. In addition, the magnetic properties of Na0.5Bi0.5TiO3 materials were found to be strong enhancement such magnetization via solid solution with various impurities materials such as ilmenite-type materials (e.g. MnTiO3, NiTiO3, FeTiO3, or CoTiO3), or perovskite-type materials (e.g. MgFeO3−δ, SrFeO3−δ, CaFeO3−δ, SrMnO3−δ, CaMnO3−δ, BaMnO3−δ, MgMnO3−δ, SrCoO3−δ, MgCoO3−δ, BaCoO3−δ, or CaCoO3−δ)24,25,26,27,28,29,30,31,32,33,34,35,36,37,38. The double perovskite-type structural materials containing the transition metals (e.g. Bi(Ti0.5Fe0.5)O3−δ, Bi(Ti0.5Mn0.5)O3−δ, Bi(Ti0.5Co0.5)O3−δ, or Bi(Ti0.5Ni0.5)O3−δ) were also reported to enhance the magnetic properties of Na0.5Bi0.5TiO3 materials when their materials were solid solution into host materials39,40,41,42. The magnetization of modified- Na0.5Bi0.5TiO3 samples via impurities of ilmenite-type materials, perovskite-type or double perovskite-type structural materials were found to have large magnetization moment which were compared with single transition metal dopants8,9,10,11,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42.
Among alkaline-earth iron perovskite AeFeO3−δ family (Ae = Ba, Ca, Sr, and Mg), BaFeO3−δ is one of interesting materials because its ferromagnetic domains could be controllable by an applied magnetic field43. BaFeO3−δ materials exhibited complex of phase such as monoclinic, rhombohedral, pseudo-cubic and cubic which depended on the valence state of Fe and transition between them44. Mori et al. reported that BaFeO3−δ compounds existed in many forms such hexagonal phase in a wide range of oxygen content BaFeO2.63–2.92 while other phase has exhibited such triclinic I, BaFeO2.50; triclinic II, BaFeO2.64–2.67; rhombohedral I and II, BaFeO2.62–2.64; and tetragonal, BaFeO2.75–2.8145. The cubic perovskite BaFeO3 with Fe4+ state has A-type spiral spin structure as ferromagnetism below 111 K46. Clemens et al. reported that BaFeO2.5 materials with Fe3+ state exhibited the G-type antiferromagnetic structure with Neel temperature of 720 K47. Delattre et al. reported that the BaFeO2.8 with orthorhombic structural exhibited the strong couple antiferromagnet48. Theoretical simulation predicted that the BaFeO2 materials has tetragonal symmetry and the G-type antiferromagnetic spin configuration49. The highest ferromagnetic ordering around 235 K in BaFeO3−δ were obtained for thin film growing on the SrTiO3 substrate50. Recently, the new system of (1-x)Na0.5Bi0.5TiO3 + xAeFeO3−δ (Ae = Sr, Ca, and Mg) materials as solid solution were successful fabricated by using the sol–gel technique28,29,30. The results provided that the impurities cation (such as Sr, Mg) and Fe random incorporated with (Bi,Na)-site and Ti-site, respectively, were exhibited the strong ferromagnetism at room temperature where the magnetization were found to great enhancement than that of single transition metal dopants which were possible resulted from co-modification at A-site via alkaline-earth and Fe cation at B-site of host Na0.5Bi0.5TiO328,29,30. In the periodic table of elements, Ba is the largest radius in alkaline earth metals, thus, we expected that the co-modification of Ba cations at A-site and Fe cations at B-site, respectively, of host Na0.5Bi0.5TiO3 materials were resulted exhibition large magnetization during solid solution of BaFeO3−δ into Na0.5Bi0.5TiO3 materials.
In this work, new system (1-x)Na0.5Bi0.5TiO3 + xBaFeO3−δ materials as solid solution were fabricated by sol–gel method. The BaFeO3−δ materials were well solid solution into the host Na0.5Bi0.5TiO3 materials through diffusion and random incorporation of Ba and Fe cations with host lattice of Na0.5Bi0.5TiO3 materials. The structural distortion and reduced optical band gap of host Na0.5Bi0.5TiO3 materials were obtained. The complex magnetic properties of BaFeO3−δ-modified Na0.5Bi0.5TiO3 materials was obtained as function of BaFeO3−δ amounts addition.
Results and discussion
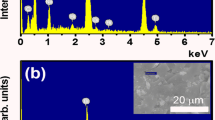
Figure 1a,b shows the EDS spectral of pure Na0.5Bi0.5TiO3 samples and BaFeO3−δ-modified Na0.5Bi0.5TiO3 sample with 5 mol.% BaFeO3−δ, respectively. The inset of each figure showed the selected area for EDS elements characterization. All expectational elements such Bi, Na, Ti and O were obtained in EDS spectral of pure Na0.5Bi0.5TiO3 samples, as shown in Fig. 1a. The addition of the Ba and Fe peaks were showed in the EDS spectral of BaFeO3−δ-modified Bi0.5Na0.5TiO3 samples, as expected, which were presented at the Fig. 1b. The results provided that the BaFeO3−δ impurities existed in our samples.

The EDS spectral of (a) pure Na0.5Bi0.5TiO3 materials, and (b) BaFeO3−δ-modified Na0.5Bi0.5TiO3 materials with 5 mol% BaFeO3−δ as solid solution. The inset of each figure shown the selected area for composition characterization.
The chemical maps of Bi0.5Na0.5TiO3 materials modified with 9 mol.% BaFeO3−δ were analyzed. The distribution of impurity elements in the Na0.5Bi0.5TiO3 materials modified with 9 mol.% BaFeO3−δ is shown in Fig. 2. The surface morphology of the area selected for chemical mapping is shown in Fig. 2a, whereas Fig. 2b presents the total contribution of all of the chemical elements in the sample. The partial chemical maps of Bi, Na, Ti, O, Ba, and Fe elements are shown in Fig. 2c–h, respectively. The results clearly demonstrated that the constituent chemical elements were homogenously dispersed in the sample.

(a) Selected area for the chemical mapping of 9 mol.% BaFeO3−δ-modified Na0.5Bi0.5TiO3 materials; (b) total chemical element distribution in samples; and (c)–(h) partial chemical element maps of Bi, Na, Ti, O, Ba, and Fe.
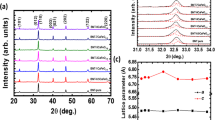
Figure 3 (a) shows the X-ray diffraction patterns of pure Na0.5Bi0.5TiO3 and BaFeO3−δ-modified Na0.5Bi0.5TiO3 with various BaFeO3−δ concentrations. On the basis of diffraction peak position and relative to intensity, all samples were indexed to a perovskite structure with the rhombohedral symmetry of the Na0.5Bi0.5TiO3 compound (JCPDS card no. 00–036-0340, space group R3c). In addition, the impurities phase or phase segregation was not founded in the X-ray diffraction patterns. All X-ray diffraction pattern of BaFeO3−δ-modified Na0.5Bi0.5TiO3 samples were indexed to follow the structural of Na0.5Bi0.5TiO3 compound. The results indicated that BaFeO3−δ materials exhibited a well solid solution in Na0.5Bi0.5TiO3 materials. In other word, the Ba and Fe cations were diffused to randomly incorporate with host lattice of Na0.5Bi0.5TiO3 compound as solid solution. In order to character the influence of Ba and Fe into host crystalline of Na0.5Bi0.5TiO3 compound, the diffraction angle of pure Na0.5Bi0.5TiO3 and BaFeO3−δ-modified Bi0.5Na0.5TiO3 samples was magnified within 31.0° to 34.0° for setline (012)/(110) peaks, as shown in Fig. 3b. The setline peaks were overloaded together which were distinguished via Lorentz fitting, as shown in dot line of Fig. 3b. The results clearly indicated that the diffraction peaks of Na0.5Bi0.5TiO3 materials trended to shift to lower diffraction angle as increasing the BaFeO3−δ amounts, which provided the evident for expansion of lattice parameter. Furthermore, the lattice parameters a and c of the pure Bi0.5Na0.5TiO3 and the BaFeO3–δ-modified Bi0.5Na0.5TiO3 as a function of BaFeO3–δ addition amounts are shown in Fig. 3c. The results show that the distorted lattice parameters of the Bi0.5Na0.5TiO3 compound are not a linear function of the concentrations of the BaFeO3–δ solid solution, which showed complex lattice parameter distortion. This result could be attributed to the different radii of Ba and Fe cations in the additives and that of Bi, Na, and Ti incorporated randomly in the lattice of the host Na0.5Bi0.5TiO3 materials. Based on the Shamon’ reported, the radius of Ba2+ and Fe2+/3+ cations were 1.61 Å and 0.645 Å/0.780 Å, respectively, while the radius of Bi3+, Na+ and Ti4+ cations were 1.17 Å, 1.39 Å and 0.605 Å, respectively51. Therefore, the Fe cations diffused to substitute for Ti-sites in perovskite structural of Na0.5Bi0.5TiO3 crystal, resulted in expansion of the lattice. The fact that the radius of Ba2+ cations are larger than that of both Bi3+ and Na+ cations were also reflected by expanding the lattice parameter of host Na0.5Bi0.5TiO3 compound. However, we noted that the oxygen vacancies were generated due to unbalance of valence states of Fe2+/3+ and Ti4+ at B-site and Ba2+ for Bi3+ at A-site. In addition, the Na vacancies were created when Ba2+ substitute Na+. The oxygen vacancies (¤) has radius of 1.31 Å which were smaller than that of oxygen anion (O2-) of 1.4 Å52. Therefore, the existence of oxygen vacancies in the structure led to reduction of the lattice parameter. The structural distortion of Na0.5Bi0.5TiO3 materials was due to co-modification at A- and B-site via alkali earth and transition metal, respectively, which was consistent with recently reported28,29,30. In other word, the X-ray diffraction characterization of BaFeO3−δ-modified Na0.5Bi0.5TiO3 samples provided that the BaFeO3−δ materials were well solid solution into host Na0.5Bi0.5TiO3 materials.

(a) X-ray diffraction pattern of BaFeO3−δ solid solution into Na0.5Bi0.5TiO3 with various concentrations within the 2θ range of 20° to 70°; (b) magnified X-ray diffraction within 2θ range of 31°–34° for comparing setline (012)/(110) peaks; and (c) dependence of the lattice parameters of pure Bi0.5Na0.5TiO3 and BaFeO3−δ-modified Bi0.5Na0.5TiO3 samples on the amounts of BaFeO3−δ solid solution.
Figure 4a show the Raman scattering of pure Na0.5Bi0.5TiO3 materials and BaFeO3−δ-modified Na0.5Bi0.5TiO3 materials with various of BaFeO3−−δ concentration as solid solutions at room temperature. The results provide that the shape of Raman scattering spectra seem to be unchanged in comparison between that of pure Na0.5Bi0.5TiO3 materials and that of BaFeO3−δ-modified Na0.5Bi0.5TiO3 s materials. In the wave number ranging from 300 cm−1 to 1000 cm−1, the Raman spectra were possible divided into three main regions and they overlapped each other. The three main band regions were in the range of 300–450 cm−1, 450–700 cm−1 and 700–1000 cm−1, respectively. The combination of experimental investigation and first principles density functional theoretical calculation for Raman vibration modes of Na0.5Bi0.5TiO3 materials exhibited that the lowest frequency modes in range of 246–401 cm−1 are dominated by TiO6 vibrations, and the higher frequency modes in the range 413–826 cm−1 are primarily associated with the oxygen atoms vibrations53. The random occupation at A-sites of Bi and Na resulted in overlap of Raman scattering peaks in range of 109–134 cm−1 and 155–187 cm−1, which were originated from Bi-O and Na–O vibration modes, respectively53. In addition, Chen et al. reported that the vibration of Ti–O bonds is related to the wave number in range of 200–400 cm−1 while the vibration of TiO6 octahedra is assigned to the wave number regions from 450 cm−1 to 700 cm−154. The overlapping of Raman scattering modes was hard to characterize the influence of Ba and Fe into vibration modes of host lattice Na0.5Bi0.5TiO3 materials. Therefore, we tried to distinguish the Raman scattering modes via the fitting with Lorentz functions (with correction of fitting over 0.99). The deconvolution Raman scattering modes (blue line) of pure Na0.5Bi0.5TiO3 samples and BaFeO3−δ-modified Na0.5Bi0.5TiO3 samples with example for 5 and 9 mol% BaFeO3−δ, as shown in Fig. 4b. The eight Raman scattering vibrational modes were obtained for both pure Na0.5Bi0.5TiO3 and BaFeO3−δ-modified Na0.5Bi0.5TiO3 samples. The results were well consistent with recently observation in vibration of Raman scattering modes of perovskite-type structural AeFeO3−δ family-modified Na0.5Bi0.5TiO3 materials28,29,30. The vibration modes at around 595 cm−1 (red dot line marked in the Fig. 4b) trended to shift to high frequency, which were suggested to be related to distorted structure of (Ti,Fe)O6 framework and/or effective mass effect because of difference between the radius and mass of impurities Fe and host Ti at B-site28,29,30. In other word, the shifted Raman scattering modes confirmed the substitution of Ba and Fe into the host lattice of Na0.5Bi0.5TiO3 materials.

(a) Raman scattering spectra of BaFeO3−δ solid solution into Na0.5Bi0.5TiO3 with various concentrations from 200 cm−1 to 1000 cm−1; and (b) deconvolution Raman peaks of pure Na0.5Bi0.5TiO3 and 5 and 9 mol% BaFeO3−δ solid solution into Na0.5Bi0.5TiO3.
Figure 5a shows the optical absorption spectra of pure Na0.5Bi0.5TiO3 and BaFeO3−δ-modified Na0.5Bi0.5TiO3 with various BaFeO3−δ concentrations. Pure Na0.5Bi0.5TiO3 samples exhibited a single absorbance edge, consistent with the reported optical properties of Na0.5Bi0.5TiO3 materials9,10,11,55. However, pure Na0.5Bi0.5TiO3 materials exhibited the unsharp transition which were tailored with slightly tail. The small tail at long wavelength in absorbance spectroscopy of pure Na0.5Bi0.5TiO3 materials were suggested to be related with self-defect and/or surface effect cause of unsaturation bonding pair of atoms at the surface55,56. The addition BaFeO3−δ to Na0.5Bi0.5TiO3 material as solid solution led to a red shift of the absorbance edge. The appearance of peaks around 485 nm in the absorbance spectra of BaFeO3−δ-modified Na0.5Bi0.5TiO3 materials indicated the new local states of Fe cations in the middle electronic band structure of Na0.5Bi0.5TiO3 materials28,29,30,57. Optical band gap (Eg) values of pure Na0.5Bi0.5TiO3 and BaFeO3−δ-modified Na0.5Bi0.5TiO3 materials were calculated using the plot of (αhν)2 versus photon energy hν, as shown in Fig. 5b, where α, h and ν are the absorbance coefficient, the Planck constant and the frequency, respectively. The band gap energy of pure Na0.5Bi0.5TiO3 materials were estimated to be approximately 3.09 eV, whereas that of BaFeO3−δ-modified Na0.5Bi0.5TiO3 materials exhibited value of 2.48 eV for 9 mol. % BaFeO3−δ solid solution in host Na0.5Bi0.5TiO3 materials. The detail dependence of Eg values of BaFeO3−δ-modified Na0.5Bi0.5TiO3 compound as function of BaFeO3−δ concentration is shown in the inset of Fig. 5b. The optical band gap of Na0.5Bi0.5TiO3 material in which the Ti-sites substituted with the transition metal, as B-site modified, decreased in lead-free ferroelectric Bi-based materials; this phenomenon could be due to the presence of new local states in the electronic structure of both the highest occupied molecular orbital and the lowest unoccupied molecular orbital in the total band structure9,10,11,14,57. In addition, the reduced optical band gap in A-site modified Na0.5Bi0.5TiO3-based material was possibly a result of changes in the bonding type between hybridizations A-O58. Oxygen vacancies created because of unbalanced charges between impurities and hosts (e.g. Fe2+/3+ substitute for Ti4+, and Ba2+ replacement for Bi3+) also led to the reduction in the optical band gap because the oxygen vacancy states normally located below and near the conduction band9,10,11,59,60. Thus, we suggest that the random substitution of Ba and Fe ions into the host Na0.5Bi0.5TiO3 could alter the electronic band structure, resulting in reduction of the optical band gap.

(a) UV–Vis absorption spectra of BaFeO3−δ-modified Na0.5Bi0.5TiO3 samples as a function of BaFeO3−δ concentration; and (b) the (αhν)2 proposal with photon energy (hν) of Na0.5Bi0.5TiO3 samples as a function of the amount of BaFeO3−δ added. The inset of (b) shows the optical band gap Eg value of Na0.5Bi0.5TiO3 samples as a function of the amount of BaFeO3−δ added.
Figure 6a shows the room-temperature PL emission spectra of pure Na0.5Bi0.5TiO3 and BaFeO3−δ-modified Na0.5Bi0.5TiO3 samples with various BaFeO3−δ amount. The PL spectral of all samples exhibited a broad band emission while strong emission showed in range from 479 to 505 nm. The addition of BaFeO3−δ into host Na0.5Bi0.5TiO3 materials as solid solution suppressed the emission peak, as shown in inset of Fig. 6a. However, we noted that a slight addition of BaFeO3−δ concentration enhanced the emission intensity. In addition, the PL spectral of pure Na0.5Bi0.5TiO3 and BaFeO3−δ-modified Na0.5Bi0.5TiO3 samples was overlapped together, suggesting to multi-emission peaks with closed together. Thus, we tried to distinguish the multi-emission peaks via Lorentz fitting. The deconvoluted emission peaks of pure Na0.5Bi0.5TiO3 samples were shown in Fig. 6b. The broad band visible luminescence was also recently reported for ferroelectric titanates-based materials at room temperature such as BaTiO3, SrTiO3, PbTiO3 etc.61. The observations in broad band emission were also archived in Bi0.5K0.5TiO3 materials, which were related to the surface effect and/or self-defect effect56. Normally, the coordination status of the atoms at the surface of materials is unsaturated, resulting in unpaired states, that make them different from that in the bulk62. The unsaturated atoms that existed at the surface region of Na0.5Bi0.5TiO3 materials formed local levels in the forbidden gaps, this displayed the effect of the self-trapped excitons63. Therefore, the incident photon was absorbed by the Na0.5Bi0.5TiO3 powder as it is illuminated with the excited source. The absorption photons could create some localized levels and form small polarons. The interaction between the holes in the valence band and polarons formed by the intermediate self-trapped excitons caused blue shift of the luminescence63. In addition, the structural distortion because of coupling of TiO6-TiO6 adjacent octahedra generate the localized electronic levels above the valence band. The recombination from these levels may result in the photoluminescence of Bi0.5K0.5TiO3 materials62. The photoluminescence of ferroelectric materials is not generally governed by band-to-band transition, owning to the difficulty in recombination of electron–hole pairs and the separation of the natural polarization domain in the materials. In this kind of materials, the surface states were in charge of the luminescence, in which many unsaturated atoms that presented on the surface of the ferroelectric materials creates the localized levels in the forbidden gaps. Interestingly, the intensity of PL emission of Na0.5Bi0.5TiO3 materials was suppressed by the addition of BaFeO3−δ, as shown in the inset of Fig. 6b. The PL emission spectra of BaFeO3−δ-modified Na0.5Bi0.5TiO3 materials did not change, indicating the lack of Ba and Fe substitution at the A- and B-sites, respectively, in the new electron–hole transitions. Thus, the substitution of Fe cation with Ti at the octahedral sites created oxygen vacancies; such vacancies acted as the chapping electron generated from absorbance photon energy, thereby prevented the recombination of the electron–hole pairs to generate photons.

(a) PL spectral of pure Bi0.5Na0.5TiO3 materials and BaFeO3−δ-modified Na0.5Bi0.5TiO3 samples as a function of BaFeO3−δ concentration, and (b) deconvolution of PL spectral of pure Bi0.5Na0.5TiO3 materials. The inset of (a) shown the magnification of PL spectral of pure and BaFeO3−δ-modified Na0.5Bi0.5TiO3 samples as a function of BaFeO3−δ concentration.
Furthermore, the role of BaFeO3−δ solid solution in Na0.5Bi0.5TiO3 materials in imparting magnetism was dependent on the applied magnetic field at room temperature, as shown in Fig. 7a–g for pure Na0.5Bi0.5TiO3 and BaFeO3−δ-modified Na0.5Bi0.5TiO3 materials with BaFeO3−δ concentration of 0.5, 1, 3, 5, 7 and 9 mol%, respectively. The pure Na0.5Bi0.5TiO3 exhibited the anti-S-shape in M-H curves, indicating the combination of diamagnetism and weak ferromagnetism, as shown in Fig. 7a. The diamagnetism in pure Na0.5Bi0.5TiO3 samples originated from the electronic configuration of Ti4+ as 3d°, whereas the weak ferromagnetism originated from self-defects9,10,11,14,16,30,64,65. The typical hysteresis loop of ferromagnetism was obtained for pure Na0.5Bi0.5TiO3 materials after subtract the diamagnetic components, as shown in inset of Fig. 7a. The saturation magnetization was estimated around 1.5 memu/g which were well consisted with recently reported by Ju et al.16. In addition, the remanent magnetization (Mr) and coercive field (HC) of pure Na0.5Bi0.5TiO3 materials were approximately 0.11 memu/g and 73 Oe, respectively, which were solid evidence for presentation of ferromagnetic state at room temperature. The estimation for their values were also performed with recently reported by Thanh et al. and Ju et al. which those were possibly originated from self-defect such as Na-, Ti- or O-vacancies10,16. The M-H curves trend to switch from anti-S-shape to S-shape in the BaFeO3−δ-modified Na0.5Bi0.5TiO3 samples as the BaFeO3−δ concentration in the solid solution increase, providing evidence regarding the strength enhancement of ferromagnetic ordering in the samples. As shown in Fig. 7c, the typical ferromagnetic hysteresis loops were obtained where the magnetization trended to saturate as the external applied magnetic field increase, and the strength of ferromagnetic increase. However, further increasing amounts of BaFeO3−δ into host Na0.5Bi0.5TiO3, the M-H curves exhibited the unsaturation with low applied external magnetic field, as shown in Fig. 7d–g. The dependence of shape in magnetic hysteresis loop of BaFeO3−δ-modified Na0.5Bi0.5TiO3 materials represented that the magnetic properties of BaFeO3−δ-modified Na0.5Bi0.5TiO3 materials were very complex, on the one hand the magnetic properties of Na0.5Bi0.5TiO3 materials were strong dependent of the concentration of BaFeO3−δ as solid solution. The Mr and HC values of BaFeO3−δ-modified Bi0.5Na0.5TiO3 materials were approximately 51–106 Oe and 0.12–0.48 memu/g, respectively. These results were consistent with the recently observed Mr and HC of transition-metal-doped lead-free and lead-based ferroelectric materials8,9,10,11,60,64,65,66,67,68. The nonzero Mr and HC values of BaFeO3−δ-modified Bi0.5Na0.5TiO3 materials provided solid evidence for the presence of the ferromagnetic state at room temperature. In additon, the maximum magnetization was estimated around 23 memu/g for 9 mol% BaFeO3−δ solid solution in host Na0.5Bi0.5TiO3 materials. That value was larger than that of self-defect induced magnetism of pure Na0.5Bi0.5TiO3 materials or single transition metals doped Na0.5Bi0.5TiO3 materials, in which around ~ 1.5 memu/g for Cr-doped Na0.5Bi0.5TiO3, ~ 3 memu/g for Co-doped Na0.5Bi0.5TiO3, ~ 9 memu/g for Mn-doped Na0.5Bi0.5TiO3, ~ 15 memu/g for Fe-doped Na0.5Bi0.5TiO3 materials, and ~ 4 memu/g for Ni-doped Na0.5Bi0.5TiO3 materials8,9,10,11,60,68. Herein, we need to remark that the origin of ferromagnetism ordering of transition metal impurities containing Na0.5Bi0.5TiO3 materials at room temperature were still debated. The weak-ferromagnetism in pure Na0.5Bi0.5TiO3 materials were possibly originated from self-defect and/or surface defect (such as Ti and Na-vacancies) while the magnetization of Na0.5Bi0.5TiO3 materials were slightly enhanced via oxygen vacancies11,14,16,30. The Mn-, Ni- and Fe-doped Na0.5Bi0.5TiO3 materials exhibited the room temperature ferromagnetism which were related to intrinsic phenomenon where the transition cations such of Mn, Ni and Fe interacted with the oxygen vacancies, like F-center interaction mechanism, e.g. Mn2+/3+–¤–Mn2+/3+ or Fe2+/3+–¤–Fe2+/3+ pairs etc., which were favored for ferromagnetic ordering8,9,10,11,60,64. Unlikely Mn-, Ni- and Fe- cations impurities in Na0.5Bi0.5TiO3 materials, the Co impurities trended to form Co-clusters embedding in host Na0.5Bi0.5TiO3 materials which displayed the room temperature ferromagnetism8. Recently, our experimental observation along with first principle calculation predicted that the interaction of Co cations into host Na0.5Bi0.5TiO3 materials possibly displayed the weak ferromagnetism at room temperature64. In addition, Hung et al. reported that MgFeO3−δ solid solution in Na0.5Bi0.5TiO3 materials exhibited strong magnetization, which were estimated to be around 39.6 memu/g, where the Mg cations played an importance role for mediating ferromagnetism28. The SrFeO3−δ- and CaFeO3−δ-modified Na0.5Bi0.5TiO3 materials also showed strong enhancement of the magnetization at room temperature29,30. Note that the Mg cations possibly substituted for both A-site (Bi3+, Na+) and B-site in Na0.5Bi0.5TiO3 crystal structure while Sr and Ca cations only replaced with A-site in host Na0.5Bi0.5TiO3 crystal structure28,29,30. Thus, we suggested that the possible room temperature ferromagnetism in BaFeO3−δ-modified Na0.5Bi0.5TiO3 materials were strongly related to the interaction of Fe cations through oxygen vacancies, like F-central interaction, which were recently suggested for Mn-, Ni-, Co- and Fe-doped Na0.5Bi0.5TiO3 materials9,10,60,64,69. A recent X-ray photoelectron spectroscopy (XPS) analysis of CaFeO3−δ-modified Na0.5Bi0.5TiO3 materials showed that Fe cations are stable in the Fe2+ and Fe3+ valence state together with O vacancies30. Therefore, we suggest that the interaction pair Fe2+/3+–¤–Fe2+/3+ favors ferromagnetic ordering69. The unsaturation in the M–H curves of magnetic hysteresis loops of Na0.5Bi0.5TiO3 materials (under 6 kOe of applied magnetic field) as the amount of the BaFeO3−δ solid solution increase suggested magnetic polaron interaction, wherein the interaction between Fe2+/3+–¤–Fe2+/3+ versus Fe2+/3+–¤–Fe2+/3+ resulted in antiferromagnetic-like ordering9,69,70,71,72,73. In addition, isolated Fe cations displayed paramagnetic properties9,69,70,71,72,73. Thus, the combination of the complex signal of ferromagnetic interaction and antiferromagnetic-like and paramagnetic properties was observed when the BaFeO3−δ solid solution was present at high concentrations in host Na0.5Bi0.5TiO3 materials. However, unlike single Fe-doped Na0.5Bi0.5TiO3 materials, the modification at A-site (Bi3+, Na+) via Ba2+ cations in host Na0.5Bi0.5TiO3 materials also possibly contributed a source to the ferromagnetism ordering, in which the substitution of Ba2+ cations for Bi3+ cations in crystal structure created the O-vacancies while Ba2+ cations incorporated for Na+ cations generated the Na-vacancies. Both O- and Na- vacancies are origin of the ferromagnetism, but they work in different ways16,30. Nevertheless, both the experimental observation and theoretical prediction have agreed that Na-vacancies induce the nonzero magnetic moment16,30. Therefore, the strength of magnetic moments can be increased by increasing the number of Na-vacancies. However, unlikely Na-vacancies, the O-vacancies were predicted to be agent of the nonmagnetic moment30. The O-vacancies were important in promoting the reduction in valence state from Ti4+ to Ti3+ (even Ti2+) because of oxygen vacancies bounding surround30,31,74,75,76. The theorey predicted that Ti4+ has no magnetic moment whereas the Ti3+ or Ti2+ have nonzero magnetic moment30. Thus, the enhancement in O-vacancies were indirectly induced by the magnetic moment along with the contribution of magnetization of Ti3+/2+ defects, resulting in increased magnetic moment that were over the self-defects compared with pure Na0.5Bi0.5TiO3 samples. Recently, XPS results have shown that the Ti4+ cations in Na0.5Bi0.5TiO3 materials were possible reduced to Ti3+ via modification of CaFeO3−δ- and SrMnO3−δ-modified as solid solution30,31. Therefore, we suggested that the increasing the O-vacancies was promoted by the magnetization of self-defect of Ti3+ cations. Recently, the interaction between Co3+/2+ cations and reduction of valence state of Ti4+-δ pair through O-vacancies were suggested to favor the ferromagnetic ordering35. Therefore, we suggested that the appearance of interaction Fe2+/3+–¤–Ti4+-δ pair may arouse increasing of the strength of ferromagnetic ordering. We recently reported that the magnetic properties of various magnetic compound, such as Mn2O3, Mn5Ge3 etc., could be tunable by a strain77,78. Therefore, due to the difference radius with ions of host lattice, the Ba cations possibly caused a chemical pressure, possibly resulting in the change of interaction between them through oxygen vacancies; finally, resulted in modification of magnetic ordering strength. Recent experimental and theoretical studies have suggested that transition metals may fill A- and B-sites in the perovskite structure of lead-free ferroelectrics, thus resulting in complex magnetic properties27,79,80,81,82,83. Liu et al. reported that K0.45Na0.49Li0.06NbO3 materials modified with Cu at the A-site show paramagnetic properties80. However, Yang et al. reported that Cu modification at the B-site of EuMnO3 materials changes magnetic properties from paramagnetic to antiferromagnetic ordering81. Deng et al. found that the paramagnetic properties of EuMnO3−δ materials were changed to antiferromagnetic via modification at the Eu site by Mn cations82. Magnetic phase transition from paramagnetic to antiferromagnetic properties has also been reported for DyMnO3 materials modified with the Mn cation at the Dy-site83. Theoretical and experimental studies have suggested that CoTiO3-modified Na0.5Bi0.5TiO3 materials have complex magnetic properties that are strongly dependent on the location of Co impurities at the A-site or B-site in the host structure27,79. Notably, the radii of Fe2+/3+ cations with VIII coordination are 0.92 Å and 0.78 Å, respectively, which are comparable with the radii of Bi3+ (1.17 Å) and Na+ (1.39 Å) cations84. Therefore, we suggested that Fe cations were random incorporated at both the A-site and B-site likely contributing to the complex magnetic properties of the host Bi0.5Na0.5TiO3 materials. The role of Fe cation substitution at the A- and B-sites in the magnetic properties of Na0.5Bi0.5TiO3 materials was further investigated by using density-functional theory (DFT) calculation.

M–H curves at room temperature of (a) undoped Na0.5Bi0.5TiO3, and BaFeO3−δ-modified Na0.5Bi0.5TiO3 samples with (b) 0.5 mol%, (c) 1 mol%, (d) 3 mol%, (e) 5 mol%, (f) 7 mol% and (g) 9 mol% BaFeO3−δ as solid solution. Inset of (a) shown the M-H curve of pure Bi0.5Na0.5TiO3 material after substrate diamagnetic components.
To elucidate the origin of the observed ferromagnetism in BaFeO3−δ-doped Na0.5Bi0.5TiO3, the density-functional theory (DFT) calculations were performed using the Vienna ab initio Simulation Package (VASP)85,86. The generalized gradient approximation (GGA) formulated by Perdew, Burke, and Ernzerhof (PBE) was used for the electron exchange correlation potential87. Figure 8a shows the side and top views of the rhombohedral crystal structure of the 24 formula unit (f.u.) cell (120-atom) adopted for Bi0.5Na0.5TiO3 (BNT). As model systems shown in Fig. 8b,c, we have considered the substitution of one Ba atom for the Bi-site, denoted as B(Ba)NT, and Na-site [BN(Ba)T], in the 24 f.u. cell structure of the BNT. This corresponds to about 0.83 at.% doping for the A-site (Bi and Na) substitution, which is within the range of the present experimental doping concentrations (0.5–9 mol.%). To represent the presence of the Fe substitution in a sample, we replaced one Ti [BNT(Fe)], Ba [B(Fe)NT], and Na [BN(Fe)T] atom with the Fe atom in the same unit cell, as shown in Fig. 9a–c. For all systems, we used an energy cutoff of 500 eV for the plane-wave basis and a k-point mesh of 5 × 5 × 5 for the Brillouin zone integration. To obtain optimized atomic structures, the atomic positions as well as lattice parameters were fully relaxed until the largest force becomes less than 10−2 eV/Å and the change in the total energy between two ionic relaxation steps is smaller than 10−5 eV. Note that the severe distortions of octahedral TiO3 lattice were observed for all geometries after optimization.

(a) Side and top views of the optimized atomic structure of Na0.5Bi0.5TiO3 (BNT). The same with (b) Bi-site Ba [B(Ba)NT] and (c) Na-site Ba [BN(Ba)T] substitution.

(a) Side views of the optimized atomic structure of the (a) Ti-site [BNT(Fe)], (b) Bi-site [B(Fe)NT], and (c) Na-site Fe [BN(Fe)T] substitution. The atomic symbols follow the same convention used in Fig. 8. Brown spheres are the Fe substitutional atoms.
We first investigate the energetics of the Ba- and Fe-doped BNT. Here, the formation energy (Hf) is defined as \({H}_{f}=H-\sum_{i}{\mu }_{i}{n}_{i}\), where H is the total energy of the system, and \({\mu }_{i}\) and \({n}_{i}\) are the chemical potential and the number of species i in the unit cell. The calculated Hf values of BNT, B(Ba)NT, BN(Ba)T, B(Fe)NT, BN(Fe)T, and BNT(Fe) are shown in Table 1. We find that the Hf of BNT is -2.385 eV/atom, which indicates the pure BNT is quite stable. Our calculations further indicate that the Ba substitute prefers either the Bi or Na sites (A-site), as their enthalpies of formation are competitive (− 2.402 and − 2.392 eV/atom). The Fe dopant atoms may also occupy both the A- and B-site (Ti), although the absolute values of Hf for the A-site (− 2.376 eV/atom for the Bi and − 2.355 eV/atom for the Na) are higher than that (− 2.335 eV/atom) of the B (Ti)-site in magnitude. Nevertheless, in a real sample, the latter substitution (B-site) might appear more than the A-site (Bi and Na) substitution, as the A-site is mainly occupied by the Ba atoms.
Table 1 shows the calculated magnetic energy (ΔE = Esp – Enon-sp, where Esp and Enon-sp are the total energies of the spin-polarized and non-spin-polarized states, respectively), total magnetization per f.u. (M), and atom resolved magnetic moment (m) of the dopant atoms. Our calculations show that all the Fe-doped structures are magnetic, while the Ba-doped ones are nonmagnetic. The calculated magnetization ranges from 0.14 to 0.21 µB/f.u. (or approximately 3.6 to 5.5 emu/g with [emu/g] = 1.078⋅1020 (Mf.u./NA) [µB/f.u.], where the NA and Mf.u. are the Avogadro constant and the molar mass per f.u., respectively) at 0.83 at. % doping. These theoretical magnetization values are much higher than the maximum experimental value of 23 memu/g for 9 mol.% BaFeO3−δ-modified Na0.5Bi0.5TiO3. The induced magnetization mainly comes from the local atomic moment of the Fe dopant atoms, as shown in Table 1.
Figure 10 presents the spin-resolved density of states (DOS) of BNT, B(Ba)NT, BN(Ba)T, B(Fe)NT, BN(Fe)T, and BNT(Fe) compounds. We have also analyzed the orbital projected DOS (PDOS) of the Fe 3d orbital states in Fig. 11 for the selected B(Fe)NT, BN(Fe)T, and BNT(Fe) as they exhibit magnetic nature. For BNT, the valence and conduction bands are characterized by the O-2p and Ti-3d orbital states with a band gap of ~ 2.25 eV, respectively. The majority- and minority-spin states are entirely degenerate, which indicates a feature of nonmagnetic ground state. The calculated band gap of BNT is smaller than the measured value (3.08 eV), which is quite typical in DFT calculations for oxide perovskites88. The Bi-site Ba gives rise to the upward shift of the valence band states toward the Fermi level. The opposite appears for BN(Ba)T, where the Fermi level shifts upward and touches the minimum of the conduction bands. Thus, the former and latter systems are referred to as p- and n-doped semiconductors, while kept the absolute value of the band gap. On the other hand, as shown in the bottom panels in Fig. 10, for B(Fe)NT, BN(Fe)T, and BNT(Fe), there are some midgap states around the Fermi level. In particular, for BNT(Fe), a finite DOS peak state appears right at the Fermi level in the majority-spin state while the other spin channel exhibits an insulating behavior. This is a feature of the half-metallic electronic nature. Furthermore, for all the Fe-doped compounds, substantially large exchange splitting between the spin subbands (i.e., majority-spin and minority-spin) is prominent (Fig. 10). These peak states are due to the strong orbital hybridization between the Fe 3d and O 2p states. As shown in Fig. 11, the majority-spin bands of the Fe are fully occupied, and the minority-spin states are almost unoccupied for the BNT(Fe) and B(Fe)NT but partially occupied for the BN(Fe)T. Overall, one can expect the large magnetic moment at the Fe site, as addressed in Table 1. Induced moments at the neighboring sites to the Fe are rather small.

Top to bottom: The DFT results of the spin-resolved DOS for the BNT, B(Ba)NT, BN(Ba)T, BNT(Fe), B(Fe)NT, and BN(Fe)T. The Fermi level is set to zero energy.

The DFT results of the d-orbital decomposed PDOS of the Fe substitutional atom for the (a) BNT(Fe), (b) B(Fe)NT, and (c) BN(Fe)T. The black, orange, red, green, and blue lines represent the dxy, dyz, dz2, dxz, and dx2–y2 orbital states, respectively. The Fermi level is set to zero energy.
Based on the PDOS analyses for the BNT(Fe) and B(Fe)NT compounds, we infer that the six (five) d-orbitals of Fe2+ (Fe3+) ion split by high-spin state through the crystal field theory are filled by the 5 majority-spin electrons in the low-lying t2g orbital levels and 1 electrons (no electron) in the minority-spin t2g state. Thus, according to Hund's rule, the calculated magnetic moments of 4 and 5 μB of the Fe replacement for the Ti and Bi sites can be explained by the electronic configuration of the high-spin state in the crystal field theory through the unpaired electron spin count. For the BN(Fe)T, the magnetic moment of the Fe atom is reduced compared with those for the other two systems, as some minority-spin states are partially occupied (Fig. 11c). Furthermore, for all compounds, both the t2g and eg states in PDOS are slightly split, which is mainly due to the Jahn–Teller effect as the severe octahedron distortion occurs in the presence of the Fe substitution.
We now investigate the doping concentration dependent magnetization of the Fe and Ba doped BNT. Figure 12 shows the calculated M of BNT(Fe), B(Fe)NT, and BN(Fe)T as function of the concentration of the dopant atoms. For the 2 (or 1.67 at.%) and 4 (or 3.33 at.%) Fe atoms in the 24 f.u. cell, we have also considered the spin antiparallel coupling between the Fe dopant atoms. For both cases, the spin parallel coupling (ferromagnetic) is more preferable than the spin antiparallel coupling.

The DFT results of the magnetization (µB/f.u.) of BNT(Fe), B(Ba)NT, and BN(Ba)T as function of the concentration of the dopant atoms.
Clearly, the magnetization increases linearly from 0.16 µB/f.u. (4.2 emu/g) to 0.66 µB/f.u. (17.3 emu/g) as the Fe concentration increases from 0.83 at.% to 3.33 at.%, as shown in Fig. 12. Our calculated magnetization is much larger than the measured values (23 memu/g at 9 mol.%), which is presumably due to the different concentrations and stoichiometries between the theory (here only the Fe doping) and experiment (Ba and Fe co-doping). Interestingly, the Ba doping for the A-site (Bi and Na) induces magnetism (M = 0.05 µB/f.u.) at about 2.5 at.% (Fig. 12). It is further found that the calculated magnetization increases as the doping concentration increases. Our atom resolved magnetization analyses indicate that the induced magnetization mainly comes from the Ti and O atoms neighboring to the Ba dopant site. The underlying mechanism can be explained by the spin-polarized charge transfer between the dopant and neighboring atoms in the unit cell, as revealed from the Ti- and O-PDOS analyses shown in Fig. 13.

The DFT results of PDOS of the Ti and O atoms for (a) and (c) (Ba)NT and (b) and (d) BN(Ba)T at 3.33 at./% Ba doping. The Fermi level is set to zero energy.
We finally explored the magnetic and electronic properties of the O-vacancy defected BNT(Fe). We have considered the presence of a single vacancy and double vacancies in the Fe-included octahedral cell to imitate different valence states of the Fe dopant atom. Our calculations show that the total magnetization (4 μB) of the BNT(Fe) compound decreases by 1 and 2 μB for the single-vacancy and double-vacancy systems, respectively. From the electronic structure analyses shown in Fig. 14a–c, the PDOS of the Fe dopant atom in BNT(Fe) shift toward the low energy region and some minority-spin states are partially occupied in the presences of the single and double oxygen vacancies. This is because of the extra electrons accumulated at the Fe site, originated from the O deficiency in the unit cell. Furthermore, the obtained magnetic moments are simply the reflections of the Fe2+ and Fe3+ ionic states in the high spin states and mixture of them in a real sample, as addressed in the previous experiment30.

The DFT results of the d-orbital decomposed PDOS of the Fe substitutional atom for the BNT(Fe) with the (a) vacancy-free, (b) single-vacancy, and (c) double-vacancy. The black, orange, red, green, and blue lines represent the dxy, dyz, dz2, dxz, and dx2–y2 orbital states, respectively. The Fermi level is set to zero energy.
Conclusion
The solid solution of BaFeO3−δ and Na0.5Bi0.5TiO3 ceramics have been successfully synthesized by a chemical route sol–gel method. The Ba and Fe ions were distributed randomly into the Na0.5Bi0.5TiO3 lattices which caused in the distortion of lattice structure and decreased the optical band gap. The complex magnetic properties were observed in this solid solution. This work shows a simple way for enhancement of room-temperature ferromagnetism in lead-free ferroelectric materials by solid solution.
Experiment
(1 − x)Na0.5Bi0.5TiO3 + xBaFeO3−δ (BNT-xBFO; x = 0.5%, 1%, 3%, 5%, 7% and 9%) samples were fabricated by sol–gel method. The raw materials were consisted of Bi(NO3)3.5H2O, NaNO3, Fe(NO3)3.9H2O, tetraisopropoxytitanium (IV) (C12H28O4Ti) and BaCO3. The solution was chosen which are acetic acid (CH3COOH) and deionized water with volume ratio of \(V_{H_{2}O}\):\(V_{CH_{3}COOH}\) = 5:2 while an acetylacetone (CH3COCH2COCH3) were selected as ligand. Fist, the BaCO3 were weighted and distinguee under mix acid acetic and deionized water. Thus, the raw materials such Bi(NO3)3.5H2O, NaNO3, Fe(NO3)3.9H2O were weighted to add the solution. To following, the solution was added with tetraisopropoxytitanium after adding to avoid hydrolysis. The solution was magnetic stirred under several hours to make homogeneous solution of sol. The sol was dried under 100 °C to prepare gels in oval. The dried gel was rout grounded and annealed under 800 °C for three hours in air then nature cooling down to room temperature. The as-prepared samples were rout ground for further samples characterization.
The chemical composition and chemical mapping of samples was carried out via energy dispersive spectroscopy (EDS, S-4800 Hitachi). The sodium is lighter element which were easy evaporated during the gelling and annealing processing that make samples nonstoichiometric composition. Therefore, the sodium nitrate was weighed to extra around 40–50 mol% to prevent the sodium evaporation9,10,11,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,60. The crystalline structural of pure Na0.5Bi0.5TiO3 and BaFeO3−δ-modified Na0.5Bi0.5TiO3 samples were characterized through X-ray diffraction (XRD, Brucker D8 Advance). The vibration modes of samples were measured by using Raman spectroscopy (with a 475 nm LASOS laser and a DU420A-Oe detector). The optical properties were studied by Ultraviolet–Visible (UV–Vis, Jasco V-670) and photoluminescence (PL, excited with 475 nm LASOS laser and a DU420A-Oe CCD detector) spectroscopy. Magnetic properties were characterized by a vibrating sample magnetometer (VSM, Lakeshore 7404). All experimental were performed at room temperature.
References
Eerenstein, W., Mathur, N. D. & Scott, J. F. Multiferroic and magnetoelectric materials. Nature 442, 759. https://doi.org/10.1038/nature05023 (2006).
Hu, J. M., Nan, T., Sun, N. X. & Chen, L. Q. Multiferroic magnetoelectric nanostructures for novel device applications. MRS Bull. 40, 728. https://doi.org/10.1557/mrs.2015.195 (2015).
N. D. Quan, L. H. Bac, D. V. Thiet, V. N. Hung, & Dung, D. D. Current development in lead-free Bi0.5(Na,K)0.5TiO3-based piezoelectric materials. Adv. Mater. Sci. Eng. 2014, 1–13, https://doi.org/10.1155/2014/365391 (2014).
Jo, W. et al. Giant electric-field-induced strains in lead-free ceramics for actuator applications – Status and perspective. J. Electroceram. 29, 71. https://doi.org/10.1007/s10832-012-9742-3 (2012).
Rodel, J. et al. Transferring lead-free piezoelectric ceramics into application. J. European Ceram. Soc. 35, 1659. https://doi.org/10.1016/j.jeurceramsoc.2014.12.013 (2015).
Baettig, P., Schelle, C. F., Lesar, R., Waghmare, U. V. & Spaldin, N. A. Theoretical prediction of new high-performance lead-free piezoelectrics. Chem. Mater. 17, 1376. https://doi.org/10.1021/cm0480418 (2005).
He, X. & Jin, K. J. Persistence of polar distortion with electron doping in lone-pair driven ferroelectrics. Phys. Rev. B 94, 224107. https://doi.org/10.1103/PhysRevB.94.224107 (2016).
Wang, Y., Xu, G., Ji, X., Ren, Z., Weng, W., Du, P., Shen, G., & Han, G. Room-temperature ferromagnetism of Co-doped Na0.5Bi0.5TiO3: Diluted magnetic ferroelectrics. J. Alloy Compound. 475, L25–L27. https://doi.org/https://doi.org/10.1016/j.jallcom.2008.07.073 (2009).
Dung, D. D. et al. Tunable magnetism of Na0.5Bi0.5TiO3 materials via Fe defects. J. Supercond. Novel Magn. 32, 3011. https://doi.org/10.1007/s10948-019-05163-z (2019).
Thanh, L. T. H. et al. Making room-temperature ferromagnetism in lead-free ferroelectric Bi0.5Na0.5TiO3 material. Mater. Lett. 186, 239–242. https://doi.org/10.1016/j.matlet.2016.09.105 (2017).
Thanh, L. T. H. et al. Origin of room temperature ferromagnetism in Cr-doped lead-free ferroelectric Bi0.5Na0.5TiO3 materials. J. Electron. Mater. 46, 3367. https://doi.org/10.1007/s11664-016-5248-0 (2017).
Zheng, G. P., Uddin, S., Zheng, X. & Yan, J. Structural and electrocaloric properties of multiferroic-BiFeO3 doped 0.94Bi0.5Na0.5TiO3–0.06BaTiO3 solid solutions. J. Alloys Compound. 663, 294. https://doi.org/10.1016/j.jallcom.2015.12.056 (2016).
Kumar, Y. & Yadav, K. L. Dielectric, magnetic and electrical properties of Bi0.5Na0.5TiO3 – CoMn0.2Fe18O4 composites. AIP Conf. Proc. 1731, 050090. https://doi.org/10.1063/1.4947744 (2016).
Ju, L., Xu, T. S., Zhang, Y. J. & Sun, L. First-principles study of magnetism in transition metal doped Na0.5Bi0.5TiO3 system. Chinese J. Chem. Phys. 29, 462. https://doi.org/10.1063/1674-0068/29/cjcp1602023 (2016).
Zhang, Y., Hu, J., Gao, F., Liu, H. & Qin, H. Ab initio calculation for vacancy-induced magnetism in ferroelectric Na0.5Bi0.5TiO3. Comput. Theor. Chem. 967, 284. https://doi.org/10.1016/j.comptc.2011.04.030 (2011).
Ju, L. et al. Room-temperature magnetoelectric coupling in nanocrystalline Na0.5Bi0.5TiO3. J. Appl. Phys. 116, 083909. https://doi.org/10.1063/1.4893720 (2014).
Rahman, J. U. et al. Dielectric, ferroelectric and field-induced strain response of lead-free BaZrO3-modified Bi0.5Na0.5TiO3 ceramics. Curr. Appl. Phys. 14, 331. https://doi.org/10.1016/j.cap.2013.12.009 (2014).
Yang, J. et al. Dielectric, ferroelectric and piezoelectric properties of Bi0.5Na0.5TiO3–(Ba0.7Ca0.3)TiO3 ceramics at morphotropic phase boundary composition. Mater. Sci. Eng. B 176, 260. https://doi.org/10.1016/j.mseb.2010.12.007 (2011).
Bai, W. et al. Phase evolution and correlation between tolerance factor and electromechanical properties in BNT-based ternary perovskite compounds with calculated end-member Bi(Me0.5Ti0.5)O3 (Me = Zn, Mg, Ni, Co). Dalton Trans. 45, 14141. https://doi.org/10.1039/C6DT01788F (2016).
Zhou, C. R. & Liu, X. Y. Dielectric properties and relaxation of Bi0·5Na0·5TiO3-BaNb2O6 lead-free ceramics. Bull. Mater. Sci. 30, 575. https://doi.org/10.1007/s12034-007-0090-x (2007).
Kaswan, K. et al. Crystal structure refinement, enhanced magnetic and dielectric properties of Na0.5Bi0.5TiO3 modified Bi0.8Ba0.2FeO3 ceramics. Ceram. Inter. 43, 4622–4629. https://doi.org/10.1016/j.ceramint.2016.12.128 (2017).
Pattanayak, R., Raut, S., Kuila, S., Chandrasekhar, M. & Panigrahi, S. Multiferroism of [Na0.5Bi0.5TiO3-BaFe12O19] lead-free novel composite systems. Mater. Lett. 209, 280–283. https://doi.org/10.1016/j.matlet.2017.08.023 (2017).
Singh, S., Kaur, A., Kaur, P. & Singh, L. An investigation on cubic and monoclinic phase coexistence in sol-gel derived LaFeO3-Na0.5Bi0.5TiO3 ceramics. J. Alloys Compound. 857, 158284. https://doi.org/10.1016/j.jallcom.2020.158284 (2021).
Hue, M. M. et al. Magnetic properties of (1–x)Bi0.5Na0.5TiO3 + xMnTiO3 materials. J. Magn. Magn. Mater. 471, 164. https://doi.org/10.1016/j.jmmm.2018.09.087 (2019).
Hue, M. M. et al. Tunable magnetic properties of Bi0.5Na0.5TiO3 materials via solid solution of NiTiO3. Appl. Phys. A 124, 588. https://doi.org/10.1007/s00339-018-2002-x (2018).
Dung, D. D. et al. Enhancing room-temperature ferromagnetism in Bi0.5Na0.5TiO3 via FeTiO3 solid solution. J. Electroceram. 44, 129–135. https://doi.org/10.1007/s10832-020-00203-w (2020).
Dung, D. D. et al. Experimental and theoretical studies on the room-temperature ferromagnetism in new (1–x)Bi1/2Na1/2TiO3+xCoTiO3 solid solution materials. Vacuum 179, 109551. https://doi.org/10.1016/j.vacuum.2020.109551 (2020).
Hung, N. T. et al. Room-temperature ferromagnetism in Fe-based perovskite solid solution in lead-free ferroelectric Bi0.5Na0.5TiO3 materials. J. Magn. Magn. Mater. 451, 183. https://doi.org/10.1016/j.jmmm.2017.11.015 (2018).
Hung, N. T. et al. Structural, optical, and magnetic properties of SrFeO3−δ-modified Bi0.5Na0.5TiO3 materials. Phys. B 531, 75. https://doi.org/10.1016/j.physb.2017.12.021 (2018).
Hung, N. T. et al. Intrinsic and tunable ferromagnetism in Bi0.5Na0.5TiO3 through CaFeO3−δ modification. Sci. Rep. 10, 6189. https://doi.org/10.1038/s41598-020-62889-w (2020).
Dung, D. D., Hung, N. T. & Odkhuu, D. Structure, optical and magnetic properties of new Bi0.5Na0.5TiO3-SrMnO3−δ solid solution materials. Sci. Rep. 9, 18186. https://doi.org/10.1038/s41598-019-54172-4 (2019).
Dung, D. D., Hung, N. T. & Odkhuu, D. Magnetic and optical properties of new (1–x)Bi0.5Na0.5TiO3+xCaMnO3−δ solid solution materials. Mater. Sci. Eng. B 263, 114902. https://doi.org/10.1016/j.mseb.2020.114902 (2021).
Dang, D. D., Hung, N. T. & Odkhuu, D. Magnetic and optical properties of new (1–x)Bi0.5Na0.5TiO3+xBaMnO3−δ solid solution materials. Appl. Phys. A 125, 465. https://doi.org/10.1007/s00339-019-2571-3 (2019).
Dung, D. D., Hung, N. T. & Odkhuu, D. Magnetic and optical properties of MgMnO3−δ-modified Bi0.5Na0.5TiO3 materials. J. Magn. Magn. Mater. 482, 31–37. https://doi.org/10.1016/j.jmmm.2019.03.029 (2019).
Dung, D. D. & Hung, N. T. Magnetic properties of (1–x)Bi0.5Na0.5TiO3+xSrCoO3−δ solid-solution materials. Appl. Phys. A 126, 240. https://doi.org/10.1007/s00339-020-3409-8 (2020).
Dung, D. D. & Hung, N. T. Structural, optical, and magnetic properties of the new (1–x)Bi0.5Na0.5TiO3+xMgCoO3−δ solid solution system. J. Supercond. Novel Magn. 33, 1249–1256. https://doi.org/10.1007/s10948-020-05451-z (2020).
Dung, D. D. & Hung, N. T. Structural, optical and magnetic properties of (1–x)Bi0.5Na0.5TiO3+xBaCoO3−δ solid solution systems prepared by the sol-gel method. J. Nanosci. Nanotech. 21, 2604–2612. https://doi.org/10.1166/jnn.2021.19090 (2021).
D. D. Dung, and N. T. Hung. Magnetic properties of (1-x)Bi0.5Na0.5TiO3+xCaCoO3−δ solid-solution system. J. Electron. Mater. 49, 5317–5325 (2020). https://doi.org/https://doi.org/10.1007/s11664-020-08233-4.
Dung, D. D. et al. Design and characterization of a new (1–x)Na1/2Bi1/2TiO3+xBi(Ti1/2Fe1/2)O3 solid solution. Vacuum 183, 109815. https://doi.org/10.1016/j.vacuum.2020.109815 (2021).
Phuong, L. T. K. et al. Structural, optical, and magnetic properties of a new system Bi(Mn0.5Ti0.5)O3-modified Bi0.5Na0.5TiO3 materials. Mater. Res. Exp. 6, 106112. https://doi.org/10.1088/2053-1591/ab3ce0 (2019).
Dung, D. D., Hue, M. M. & Bac, L. H. Growth and magnetic properties of new lead-free (1–x)Bi1/2Na1/2TiO3+xBi(Ti1/2Co1/2)O3 solid solution materials. Appl. Phys. A 126, 533. https://doi.org/10.1007/s00339-020-03718-9 (2020).
Dung, D. D. et al. Influenced of Bi(Ti1/2Ni1/2)O3 concentration on the structural, optical and magnetic properties of lead-free Bi1/2Na1/2TiO3 materials. Vacuum 177, 109306. https://doi.org/10.1016/j.vacuum.2020.109306 (2020).
Hong, N. H. et al. Shaping the magnetic properties of BaFeO3 perovskite-type by alkaline-earth doping. J. Phys. Chem. C 122, 2983. https://doi.org/10.1021/acs.jpcc.7b10127 (2018).
Suga, Y., Hibino, M., Kuno, T. & Mizuno, N. Electrochemical oxidation of BaFeO2.5 to BaFeO3. Electrochim. Acta 137, 359. https://doi.org/10.1016/j.electacta.2014.05.162 (2014).
Mori, S. Phase transformation in barium orthoferrate, BaFeO3−x. J. Am. Ceram. Soc. 49, 600. https://doi.org/10.1111/j.1151-2916.1966.tb13176.x (1966).
Hayashi, N. et al. BaFeO3: A ferromagnetic iron oxide. Angew. Chem. Int. Ed. 50, 12547. https://doi.org/10.1002/anie.201105276 (2011).
Clemens, O. et al. Crystallographic and magnetic structure of the perovskite-type compound BaFeO2.5: Unrivaled complexity in oxygen vacancy ordering. Inorg. Chem. 53, 5911. https://doi.org/10.1021/ic402988y (2014).
Delattre, J. L., Stacy, A. M. & Siegrist, T. Structure of ten-layer orthorhombic Ba5Fe5O14 (BaFeO2.8) determined from single crystal X-ray diffraction. J. Solid State Chem. 177, 928. https://doi.org/10.1016/j.jssc.2003.09.032 (2004).
Ju, S. & Cai, T. Y. Magnetic and optical anomalies in infinite-layer iron oxide CaFeO2 and BaFeO2: A density functional theory investigation. J. Appl. Phys. 106, 093903. https://doi.org/10.1063/1.3238271 (2009).
Callender, C., Norton, D. P., Das, R., Hebard, A. F. & Budai, J. D. Ferromagnetism in pseudocubic BaFeO3 epitaxial films. Appl. Phys. Lett. 92, 012514. https://doi.org/10.1063/1.2832768 (2008).
Shannon, R. D. & Prewitt, C. T. Effective ionic radii in oxides and fluorides. Acta Cryst. B 25, 925. https://doi.org/10.1107/S0567740869003220 (1969).
Chatzichristodoulou, C., Norby, P., Hendriksen, P. V. & Mogensen, M. B. Size of oxide vacancies in fluorite and perovskite structured oxides. J. Electroceram. 34, 100. https://doi.org/10.1007/s10832-014-9916-2 (2015).
Niranjan, M. K., Karthik, T., Asthana, S. & Pan, J. Theoretical and experimental investigation of Raman modes, ferroelectric and dielectric properties of relaxor Na0.5Bi0.5TiO3. J. Appl. Phys. 113, 194106. https://doi.org/10.1063/1.4804940 (2013).
Chen, Y. et al. Structural and electrical properties of Mn-doped Na0.5Bi0.5TiO3 lead-free single crystal. Int. Ferroelectric 141, 120. https://doi.org/10.1080/10584587.2013.780141 (2013).
Thanh, L. T. H., Tuan, N. H., Bac, L. H., Dung, D. D., Bao, P. Q.. Influence of fabrication condition on the microstructural and optical properties of lead-free ferroelectric Bi0.5Na0.5TiO3 materials. Commun. Phys. 26, 51, https://doi.org/10.15625/0868-3166/26/1/7354 (2016).
Bac, L. H. et al. Tailoring the structural, optical properties and photocatalytic behavior of ferroelectric Bi0.5K0.5TiO3 nanopowders. Mater. Lett. 164, 631. https://doi.org/10.1016/j.matlet.2015.11.086 (2016).
Dung, D. D. et al. Room-temperature ferromagnetism in Fe-doped wide band gap ferroelectric Bi0.5K0.5TiO3 nanocrystals. Mater. Lett. 156, 129. https://doi.org/10.1016/j.matlet.2015.05.010 (2015).
Quan, N. D., Hung, V. N., Quyet, N. V., Chung, H. V. & Dung, D. D. Band gap modification and ferroelectric properties of Bi0.5(Na, K)0.5TiO3-based by Li substitution. AIP Adv. 4, 017122. https://doi.org/10.1063/1.4863092 (2014).
Tuan, N. H. et al. Defect induced room temperature ferromagnetism in lead-free ferroelectric Bi0.5K0.5TiO3 materials. Phys. B 532, 108. https://doi.org/10.1016/j.physb.2017.04.02560 (2018).
Dung, D. D. et al. Defect-mediated room temperature ferromagnetism in lead-free ferroelectric Na0.5Bi0.5TiO3 materials. J. Supercond. Novel Magn. 33, 911–920. https://doi.org/10.1007/s10948-019-05399-9 (2020).
Meng, J., Rai, B. K., Katiyar, R. S. & Zou, G. T. Study of visible emission and phase transition in nanocrystalline A1−xA′xTiO3 systems. Phys. Lett. A 229, 254. https://doi.org/10.1016/S0375-9601(97)00152-7 (1997).
Leite, E. R. et al. Amorphous lead titanate: a new wide-band gap semiconductor with photoluminescence at room temperature. Adv. Funct. Mater. 10, 235. https://doi.org/10.1002/1099-0712(200011/12)10:6%3c235::AID-AMO409%3e3.0.CO;2-6 (2000).
Lin, Y. et al. Photoluminescence of nanosized Na0.5Bi0.5TiO3 synthesized by a sol–gel process. Mater. Lett. 58, 829. https://doi.org/10.1016/j.matlet.2003.07.025 (2004).
Dung, D. D. et al. Role of Co dopants on the structural, optical and magnetic properties of lead-free ferroelectric Na0.5Bi0.5TiO3 materials. J. Sci. Adv. Mater. Dev. 4, 584–590. https://doi.org/10.1016/j.jsamd.2019.08.007 (2019).
Shah, J. & Kotnala, R. K. Induced magnetism and magnetoelectric coupling in ferroelectric BaTiO3 by Cr-doping synthesized by a facile chemical route. J. Mater. Chem. A 1, 8601–8608. https://doi.org/10.1039/C3TA11845B (2013).
Cuong, L. V. et al. Observation of room-temperature ferromagnetism in Co-doped Bi0.5K0.5TiO3 materials. Appl. Phys. A 123, 563. https://doi.org/10.1007/s00339-017-1173-1 (2017).
Ren, Z. et al. Room-temperature ferromagnetism in Fe-doped PbTiO3 nanocrystals. Appl. Phys. Lett. 91, 063106. https://doi.org/10.1063/1.2766839 (2007).
Oanh, L. M., Do, D. B., Phu, N. D., Mai, N. T. P. & Minh, N. V. Influence of Mn doping on the structure, optical, and magnetic properties of PbTiO3 material. IEEE Trans. Magn. 50, 2502004. https://doi.org/10.1109/TMAG.2013.2297516 (2014).
Coey, J. M. D., Douvalis, A. P., Fitzgerald, C. B. & Venkatesan, M. Ferromagnetism in Fe-doped SnO2 thin films. Appl. Phys. Lett. 84, 1332. https://doi.org/10.1063/1.1650041 (2004).
Tuan, N. H., Linh, N. H., Odkhuu, D., Trung, N. N. & Dung, D. D. Microstructural, optical, and magnetic properties of BiCoO3-modified Bi0.5K0.5TiO3. J. Electron. Mater. 47, 3414–3420. https://doi.org/10.1007/s11664-018-6166-0 (2018).
Tuan, N. H. et al. Structural, optical, and magnetic properties of lead-free ferroelectric Bi0.5K0.5TiO3 solid solution with BiFeO3 materials. J. Electron. Mater. 46, 3472–3478. https://doi.org/10.1007/s11664-017-5328-9 (2017).
Durst, A. C., Bhatt, R. N. & Wolff, P. A. Bound magnetic polaron interactions in insulating doped diluted magnetic semiconductors. Phys. Rev. B 65, 235205. https://doi.org/10.1103/PhysRevB.65.235205 (2002).
Coey, J. M. D., Venkatesan, M. & Fitzgerald, C. B. Donor impurity band exchange in dilute ferromagnetic oxides. Nat. Mater. 4, 173–179. https://doi.org/10.1038/nmat1310 (2005).
Qiao, Y. et al. Local order and oxygen ion conduction induced high-temperature colossal permittivity in lead-free Bi0.5Na0.5TiO3-based systems. ACS Appl. Energy Mater. 1, 956–962. https://doi.org/10.1021/acsaem.7b00347 (2018).
Liu, X., Fan, H., Shi, J., Wang, L. & Du, H. Enhanced ionic conductivity of Ag addition in acceptor-doped Bi0.5Na0.5TiO3 ferroelectric. RSC Adv. 6, 30623–30627. https://doi.org/10.1039/C6RA00682E (2016).
Hejazi, M. M., Taghaddos, E. & Safari, A. Reduced leakage current and enhanced ferroelectric properties in Mn-doped Bi0.5Na0.5TiO3-based thin films. J. Mater. Sci. 48, 3511–3516. https://doi.org/10.1007/s10853-013-7144-9 (2013).
Dung, D. D., Thiet, D. V., Tuan, D. A. & Cho, S. Strain effects in epitaxial Mn2O3 thin film grown on MgO(100). J. Appl. Phys. 113, 17A314. https://doi.org/10.1063/1.4794720 (2013).
Dung, D. D. et al. Strain modified/enhanced ferromagnetism in Mn3Ge2 thin films on GaAs(001) and GaSb(001). J. Appl. Phys. 113, 153908. https://doi.org/10.1063/1.4802591 (2013).
Lan, Y. et al. Semiconducting tailoring and electrical properties of A-site Co substituted Bi0.5Na0.5TiO3−δ ferroelectric ceramics. Mater. Chem. Phys. 260, 124100. https://doi.org/10.1016/j.matchemphys.2020.124100 (2021).
Liu, L. et al. Ferroic properties of Fe-doped and Cu-doped K0.45Na0.49Li0.06NbO3 ceramics. J. Mater. Sci. Mater. Electron. 26, 6592–6598. https://doi.org/10.1007/s10854-015-3257-z (2015).
Yang, A. M. et al. Copper doped EuMnO3: synthesis, structure and magnetic properties. RSC Adv. 6, 13982–13933. https://doi.org/10.1039/C5RA27426E (2016).
Deng, J. et al. Synthesis, structure and magnetic properties of (Eu1-xMnx)MnO3−δ. RSC. Adv. 7, 2019–2024. https://doi.org/10.1039/C6RA25951K (2017).
Deng, J. et al. The origin of multiple magnetic and dielectric anomalies of Mn-doped DyMnO3 in low temperature region. J. Alloys Comp. 725, 976–983. https://doi.org/10.1016/j.jallcom.2017.07.223 (2017).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A 32, 751–765. https://doi.org/10.1107/S0567739476001551 (1976).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558. https://doi.org/10.1103/PhysRevB.47.558 (1993).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169. https://doi.org/10.1103/PhysRevB.54.11169 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865. https://doi.org/10.1103/PhysRevLett.77.3865 (1996).
Padilla, J. & Vanderbilt, D. Ab initio study of BaTiO3 surfaces. Phys. Rev. B 56, 1625. https://doi.org/10.1103/PhysRevB.56.1625 (1997).
Acknowledgements
This research is funded by The Ministry of Science and Technology, Viet Nam, under project number ĐTĐLCN.29/18. Prof. Yong Soo Kim thanks for the supporting from Priority Research Centers Program (2019R1A6A1A11053838), and the Basic Science Research Program (2021R1A2C1004209, 2020R1F1A1048657) through the National Research Foundation of Korea (NRF), funded by the Korean government. The research work at the Incheon National University was supported by Future Materials Discover Program (Grant No. 2016M3D1A1027831) and the Basic Research Program (Grant No. 2020R1F1A1067589) through the NRF grant funded by the Ministry of Science and ICT.
Author information
Authors and Affiliations
Contributions
D.D.D. and N.H.L. conceived the idea and designed the experiments. N.H.L., N.A.D., N.N.T., N.V.D., and N.N.T. performed the experiments and measurements. D.O. performed the theoretical calculations and wrote the corresponding paragraphs. D.D.D. and N.T.H. wrote the paper. Y.S.K. and A.D.N. reviewed and commented on the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dung, D.D., Lam, N.H., Nguyen, A.D. et al. Experimental and theoretical studies on induced ferromagnetism of new (1 − x)Na0.5Bi0.5TiO3 + xBaFeO3−δ solid solution. Sci Rep 11, 8908 (2021). https://doi.org/10.1038/s41598-021-88377-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-88377-3
This article is cited by
-
Enhanced room temperature ferromagnetism in YMnO3-modified lead-free ferroelectric Bi0.5Na0.5TiO3 materials
Applied Physics A (2023)
-
Fabrication and Optical Properties of (1−x)Bi½Na½TiO3−xEr½Na½TiO3 Solid Solution System
Journal of Electronic Materials (2023)
-
Room temperature magneto-dielectric coupling in the CaMnO3 modified NBT lead-free ceramics
Applied Physics A (2023)
-
Optical properties of a new binary lead-free ferroelectric semiconductor Bi1/2Na1/2TiO3–Nd1/2Na1/2TiO3 solid solution system
Applied Physics A (2022)
-
Synthesis and Characterization of (1−x)Bi1/2Na1/2TiO3+xSrNiO3-δ Solid Solution System
Journal of Electronic Materials (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
