
Abstract
Salinity is an extensive and adverse environmental stress to crop plants across the globe, and a major abiotic constraint responsible for limited crop production threatening the crop security. Soil salinization is a widespread problem across the globe, threatening the crop production and food security. Salinity impairs plant growth and development via reduction in osmotic potential, cytotoxicity due to excessive uptake of ions such as sodium (Na+) and chloride (Cl−), and nutritional imbalance. Cotton, being the most cultivated crop on saline-alkaline soils, it is of great importance to elucidate the mechanisms involved in Na2SO4 tolerance which is still lacking in upland cotton. Zhong 9835, a Na2SO4 resistant cultivar was screened for transcriptomic studies through various levels of Na2SO4 treatments, which results into identification of 3329 differentially expressed genes (DEGs) in roots, stems and leave at 300 mM Na2SO4 stress till 12 h in compared to control. According to gene functional annotation analysis, genes involved in reactive oxygen species (ROS) scavenging system including osmotic stress and ion toxicity were significantly up-regulated, especially GST (glutathione transferase). In addition, analysis for sulfur metabolism, results in to identification of two rate limiting enzymes [APR (Gh_D05G1637) and OASTL (Gh_A13G0863)] during synthesis of GSH from SO42−. Furthermore, we also observed a crosstalk of the hormones and TFs (transcription factors) enriched in hormone signal transduction pathway. Genes related to IAA exceeds the rest of hormones followed by ubiquitin related genes which are greater than TFs. The analysis of the expression profiles of diverse tissues under Na2SO4 stress and identification of relevant key hub genes in a network crosstalk will provide a strong foundation and valuable clues for genetic improvements of cotton in response to various salt stresses.
Similar content being viewed by others

Introduction
Soil salinity is a major environmental factor which limits the growth and development of plants, resulting in decrease in crop productivity and quality1. The excess accumulation of soluble salts in the soil surface is referred to as salinization, which has an adverse impact on agricultural production, biodiversity and sustainable development and may results into delay of onset, reduce the rate, and increase the dispersion of germination events, leading to reductions in plant growth and finally crop yield2. Cotton, being a semi—halophyte with economic value, is considered to be tolerant to salinity stress3,4. So, it is of great importance to study the development of cotton under salt stress and explore new salt resistant genetic material. Salt stress adversely affect the plant growth and development and ultimately decreased the yield. High concentration of Na+/ Cl− ions in soil are the basic causative agent of salt stress, which breaks the ion and water potential balance in plants, causing phytotoxicity, diminishing plant growth, and ultimately lead to death, resulting limiting crop yield5. Because of that, plant cell is subject to high osmotic pressure under high salt condition, resulting in physiological drought, ionic toxicity and malnutrition with a high ion concentration6. Saline-alkali stress performed ionic toxicity and water shortage in leaves, which inevitably affected the synthesis of chlorophyll, and then affect the photosynthesis of plants7. Therefore, the direct damage of salt stress to plants is mainly reflected in two aspects: ionic toxicity and osmotic stress8.
Presence of excessive salts in soil is hazardous for plants normal growth and development due to toxic ions like of Na+, K+ and Cl− ions. and other ions9,10. To cope with salt stress, plants have evolved mechanisms in order to coordinate the activities of various ion transporters and thereby maintain ionic homeostasis in the cell10. In Arabidopsis, SOS (Salt Overly Sensitive) system related to salt stress was first elucidated significantly in Na+ homeostasis10,11. Under salt stress, the calcium sensor SOS3 interacts and activates the serine/threonine protein kinase SOS2 with an increase in cytosolic free calcium levels, which results in the activation of the plasma membrane-localized Na+/H+ antiporter SOS1 to export the Na+ out of the cell to prevent the accumulation of toxic material12. SOS1 plays a critical role in Na+ homeostasis against oxidative stress responses under salt stress in Arabidopsis11. SOS2 also positively controls the activities of tonoplast Na+/H+ antiporter by Sequestering the Na+ ions in vacuole and exchange of H+/Ca2+ ions through CAX111.
The Ca2+ signaling pathway and ion transport showed the crosstalk with SOS system to enhance salt tolerance13. Ca2+ binding proteins mainly include CDPKs (calcium-dependent protein kinase) containing the catalytic domain of serine/threonine protein kinase, CAMs (calcium receptors calmodulin) with an EF-hand unit that binds Ca2+, and CBLs (Calcineurin B-like proteins) which consist of a single CIPKs protein14. These have been shown to be involved in salt stress responses, such as OsCPK21 in rice15, GSCBRLK in soybean16, TaCIPK25 in wheat16, and SiCBL4 in millet17. CBL9 and CIPK23 increase salt tolerance at root tips mostly by scavenging reactive oxygen18. CBL10 regulates other unknown transport processes that act as Na+ equilibriums different from those regulated by SOS119. AtGLR3.4 regulates Ca2+ flow and controls Na+ accumulation through SOS pathway during salt-stress seed germination20. In addition, the Ca2+ signaling pathway also plays a central role in response to ROS scavenging under salt stress and induced programmed cell death through MAPK cascade in mitochondria. To cope with the oxidative damage resulting from ROS, not only the enzymatic antioxidants (SOD, APX, PRX, GPX, CAT, GRX and TRX) and non-enzymatic scavengers (ASA, GSH, metabolite of proline, tocopherols, carotenoids and phenolic compounds)21, but also the hormones signaling pathway with a lot of transcription factors (TFs), such as abscisic acid (ABA) signaling pathway22,23, developed a complex regulatory network for plant growth. Previous studies have shown that the majority of plant hormones related to salt stress are melatonin, ETH (ethylene), BR (brassinolactone), ASA (ascorbic acid), IAA/auxin and ABA (abscisic acid) 24,25. These hormones mainly increases the plant salt tolerance by scavenging active oxygen26. In recent years, there have been a large number of studies on the relationship between ABA and plant salt tolerance. Among them, salt stress can induce the accumulation of endogenous ABA, and then induce the expression of E3 ubiquitin ligase or others transcription factors25,27, ion transporter22, antioxidant enzyme28 and other salt stress-related genes to enhance plant salt tolerance29.
For ion transport, NHX1 regulates the export and import of Na+ in and out of vacuoles10, and HKT1 transporters have been found to reduce Na+ toxicity by regulating Na+/K+ balance in several species30. In plant cells, the proton pump, including H+-PPase (proton pump pyrophosphoric acid hydrolase), V-PPase (vacuole proton pump ATP hydrolysis enzyme) and PMH-ATPase (plasma membrane proton pump ATP hydrolysis enzyme) on the one hand provides energy for cellular metabolism, maintain normal metabolism and cell growth31. On the other hand, proton pump such as H+-PPases can improve the salt-resistance32. In Arabidopsis, 14-3-3 proteins in the SOS system are able to regulate the V-PPase (EC 3.6.1.1) gene AVP1 and mitigate the harm of Na+33. Besides, aquaporin protein, as a transporter of water molecules, responds to salt stress mainly by increasing antioxidant activity and maintaining ion balance34. The aquaporin protein related to salt resistance is mainly concentrated on the PIPs (Plasma membrane intrinsic proteins) gene. In Arabidopsis thaliana, Tip2 (Tonoplast intrinsic protein) regulates MDA (malondialdehyde) and salt-tolerance related genes (SOS1, SOS2, SOS3, DREB1A and P5CS1)35. GhSIP may be involved in endoplasmic reticulum osmotic equilibrium36.
Soil salinization has always been a major problem in global agriculture, because of inadequate irrigation and climate change, the area of saline-alkali land is likely to increase with the passage time37, so it is of great significance to study the salt tolerance in plants. The ions commonly present in saline soil are Na+, Cl− and SO42− 38. There are many studies on NaCl, however, few on Na2SO4. SO42− is different from the Na+, K+ and Cl− 39, and Na2SO4 is more toxic than NaCl40,41. Prosopis Strombulifera (Lam.) Benth, a kind of halophytic shrub with high NaCl tolerance, is found in high saline-alkali soil in Argentina, but affected with decreased Fv/Fm, ETR and Y(II) photosynthetic parameters significantly under Na2SO4 condition40. While, in Kalidium foliatum the activity of Rubisco (Ribulose-1,5-bisphosphate Carboxylase/Oxygenase) treated with NaCl was higher than that of treated with 100 mM Na2SO4, and there was no significant change under NaCl + Na2SO4 mixture treatment42. In Brassica rapa, the result of gene analysis of GSLs (Glucosinolates) biosynthetic pathway and transcription factor showed that sulfate had the strongest inhibition on growth under different treatment with NaCl, Na2SO4, KCl and K2SO4, respectively41.
So far, there is a lack of research on Na2SO4 salt stress, especially in cotton, and the mechanism of its toxicity is still unclear. In this study, through the analysis of the phenotypic, physiological and biochemical indexes and morphological analysis of Zhong 9835 in response to salt stress during 0 h, 6 h, 12 h and 24 h, we found that it is best to analysis Zhong 9835 transcriptome at 12 h under 300 mM Na2SO4 treatment. Sulfur metabolism was enriched in 3329 differentially expressed genes (DEGs) among roots, stems and leaves, especially GST, followed by Ubiquitin transcription factors. This study not only provides complement data for regulatory network at early stage under Na2SO4 stress but also provides a strong foundation and valuable clues for genetic improvements of cotton in response to various salt stresses.
Results
Phenotypic responses of Zhong 9835 to Na2SO4 stress
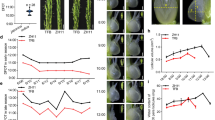
Previous studies have reported that cotton is more sensitive to abiotic stresses at three-leaf stage43. Different morphological data has been observed in G. hirsutum Zhong 9835 during its three-leaf stage under various concentrations of Na2SO4 stress. We observed the significant phenotypic difference among roots, leaves and shoots of cotton seedlings under 300 mM Na2SO4 stress after 12 h of treatment as compared to control conditions without any salt treatment (Fig. 1A). After 12 h of treatment, leaves start wilting, and roots gradually truing into black (Fig. 1A), while after 24 h, leaves and stems were seriously wilted (Fig. 1A). The result showed that under 300 mM Na2SO4 solution the pH value ranges from 6.93 at 0 h to 5.6 at 24 h, among which it becomes more acidic after 6 h (Fig. 1B). The minimum pH value was noticed at 12 h to 24 h under the treatment of Na2SO4 and control, especially in control from 7 to 6.5 (Fig. 1B). Meanwhile, SOD, POD, Proline and MDA contents in roots, stems and leaves all increased gradually from 0 to 24 h (Fig. 1 C/D/E/F). Among which, SOD up to a same content among root, stem and leaf, especially largest increase from 6 to 12 h in root and stem (Fig. 1C). POD of root increased widely from 6 to 12 h (Fig. 1D). Substantial increase of proline content of root and stem was observed at 6 h to 12 h (Fig. 1E), MDA of leaf increased greatly from 6 to 12 h, while root amplitude increased from 0 to 6 h and stem from 12 to 24 h (Fig. 1F).

Phenotypic, physiological and biochemical indexes and morphological analysis of Zhong 9835 in response to salt stress. CK: Control group; SS: 300 mM Na2SO4. (A) Phenotypic changes of Zhong 9835 under the SS and CK during 0 h, 6 h, 12 h and 24 h. (B) pH value changes of water under the SS and CK during 0 h, 6 h, 12 h and 24 h. (C) The level changes of SOD (Superoxide dismutase) under the SS and CK during 0 h, 6 h, 12 h and 24 h. (D) The level changes of Peroxidase (POD) under the SS and CK during 0 h, 6 h, 12 h and 24 h. (E) The changes of Proline (Pro) Content under the SS and CK during 0 h, 6 h, 12 h and 24 h. (F) The changes of Malondialdehyde (MDA) content under the SS and CK during 0 h, 6 h, 12 h and 24 h.
Transcriptome sequencing and alignment
Two groups (treated versus control) with three biological replications were conducted among the samples of roots, stems, and leaves, respectively. Totally 18 qualified libraries were established from the tissues of the roots, stems and leaves at 12 h with salt stress and control conditions. On average, of 42.5 million raw reads for the 18 libraries were obtained by using an Illumina Novaseq 6000 sequencing platform (Table 1). 111.34 Gb (Gigabyte) of sequence data and over 96% of the clean reads with a Q30 level were done through approximately 742 million clean valid reads. With the process of adaptor deletion, junk filtering and low copy filtering, > 95% of the sequences were confirmed as clean data, which then mapped to cotton whole genome (G. hirsutum) by using His at software44. In final, > 93.94% of the total reads were mapped to the reference genome, while the unique mapped reads were 63.46%-70.27% by the String Tie software45.
Exploration of DEGs in roots, stems and leaves in response to Na2SO4 stress
Gene expression levels were estimated by fragments per kilo base of transcript per million fragments mapped (FPKM). Differential expression analysis of treatments and control group was performed using the DESeq among CK_R, SS_R CK_S, SS_S, CK_L, SS_L. According to root, stem, and leaf, transcriptome data between control and treatment with Na2SO4 were divided into 3 groups (CK_R vs SS_R, CK_S vs SS_S, CK_L vs SS_L) (Fig. 2A,B). The samples from Root (CK_R vs SS_R) showed 15,524 DEGs, among which 10,787 genes were up-regulated and 4,737 genes were down-regulated (Fig. 2A). A total of 20,409 genes were differentially expressed in the samples of Stem (CK_S vs SS_S), among which 6,426 genes were up-regulated and 13,983 genes were down-regulated (Fig. 2A). There were 12,146 DEGs identified from the Leaf sample (CK_L vs SS_L), among which 6,521 genes were up-regulated and 5,625 genes were down-regulated (Fig. 2A).

Expression dynamics changes and comparative analysis of differentially-expressed genes (DEGs) among CK_R, SS_R, CK_S, SS_S, CK_L, SS_L. CK: Control group; SS: 300 mM Na2SO4. (A) Number of up-regulated and down-regulated DEGs of each sample between SS and CK. (B) Number of DEGs among roots, stems and leaves under SS and CK treatments.
To further explore the DEGs between different groups, we sorted the common genes using a Venn diagram online tool (https://bioinfogp.cnb.csic.es/tools/venny/). 7,626 DEGs were identified between the samples CK_R vs SS_R and CK _S vs SS_S (Fig. 2B). among the samples of CK_R vs SS_R and CK _L vs SS_L, there were 4,716 DEGs (Fig. 2B). A total of 7,552 DEGs were identified from the samples CK_S vs SS_S and CK_L vs SS_L (Fig. 2B). finally, we found 3,329 DEGs among the samples of CK_R vs SS_R, CK_S vs SS_S and CK _L vs SS_L (Fig. 2B).
Validation of RNA-Seq data by quantitative real-time PCR
In order to validate the differential expression analysis of RNA-seq data, we performed the quantitative real-time PCR (qRT-PCR) of 20 genes to confirm the reliability of RNA-seq data by using the same RNA samples (Table S1). To corroborate the expression levels measured by RNA-Seq data, the ratio of expression levels among root, stem and leaf under Na2SO4 stress and control using RNA-Seq was compared to the ratios of expression measured by qRT-PCR. The results showed that there was a good correlation between RNA-Seq and real-time PCR results among three tissues (coefficient of determination R2 = 0.86, 0.82 and 0.91) (Fig. 3A–C). The validation experiments support the accuracy of the RNA-Seq quantification of gene expression by relative values provided by the qRT-PCR analysis.

qRT-PCR validation of transcript levels evaluated by RNA-Seq in roots, stems and leaves under 300 mM Na2SO4 stress conditions. X-axis represents log2FC (Fold Change) derived from RNA-seq; Y-axis represents log2(2−ΔΔCt) specifically from the qRT-PCR experiment. (A) Transcript level of roots. (B) Transcript level of stems. (C) Transcript level of leaves.
To study the clustering profiles of these 3329 DEGs, six samples with three biological repeats were carried out (Fig. 4). These 3329 DEGs were divided into six clusters, in which cluster had the same expression profile (Fig. 4). There were 630 genes in cluster 1, which had higher FPKM values especially in CK_L group with three biological repeats. In cluster 2, the samples SS_R possess 691 genes with three replication and harbor higher FPKM values. The FPKM values of cluster 3 were found higher than others in samples CK_S with three biological repeats, which includes 622 genes. Cluster 4 containing 320 genes was the smaller one in the clustering profile, in which the FPKM is greater in samples CK_R with three biological repeats particularly. Cluster 5 with 760 genes was the largest one in the clustering profile, which the FPKM of the samples SS_S with three biological repeats is higher. Cluster 6 of 306 genes was the smallest one, in which the FPKM of the samples SS_L with three biological repeats is greater.

Line graph for the cluster expression of 3329 DEGs. CK: Control group; SS: 300 mM Na2SO4. The X-axis shows the different treatments (from left to right: CK_L1, CK_L2, CK_L3, CK_R1, CK_R2, CK_R3, CK_S1, CK_S2, CK_S3, SS_L1, SS_L2, SS_L3, SS_R1, SS_R2, SS_R3, SS_S1, SS_S2, SS_S3), and the Y-axis shows the standardized FPKM. The dotted line shows the 0 value of FPKM. The number on the top left side of cluster panel is cluster number. The number on the bottom left side of cluster panel is genes number of each cluster. Black lines represent the average value of the relative expression level of all genes included in the cluster.
Functional enrichment of differentially-expressed genes
To further understand the molecular mechanism of 3329 DEGs, we mapped all of DEGs to the GO database and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways46. In final, the identified 3,329 DEGs were classified into 100 pathways (1877 DEGs, account for 56.90%) and 383 Gene Ontology (GO) annotations (3144 DEGs, accounts for 95.30%) which includes the biological processes, cellular components and molecular functions. Then the first 30 GO terms (Fig. 5) and a threshold of top 30 set for KEGG pathways analysis (Fig. 6) were chosen.

Gene ontology functional classification of DEGs drawn by ggplot2 (v3.3.3). X-axis represents the number of genes; Y-axis represents the GO terms names: red pillars represent biological process, green pillars represent cellular process, and blue pillars represent molecular function.

KEGG pathway enrichment analysis of DEGs drawn by ggplot2 (v3.3.3). X-axis represents the number of genes; Y-axis represents the name of KEGG pathway: the different colour represents different KEGG pathway.
“Molecular function” (GO:0003674) was enriched, which mainly contain WRKY family transcription factor (46 DEGs), MYB transcription factor family protein (40 DEGs), Late embryogenesis abundant (LEA) hydroxyproline-rich glycoprotein family (16 DEGs) and HSP20-like chaperones superfamily protein (10 DEGs). In biological process, these genes were mostly Cytochrome P450 superfamily protein (53 DEGs), UDP-Glycosyltransferase superfamily protein (39 DEGs), NAD(P)-binding Rossman-fold superfamily protein (37 DEGs), RING superfamily protein (29 DEGs) and ethylene related (20 DEGs). In cellular process, the maximum number of 136 genes were found in protein kinase (including 69 DEGs of Leucine-rich protein, 43 DEGs of Protein kinase superfamily protein and 24 DEGs of serine/threonine kinases) and Protein phosphatase (32 DEGs), followed by Ca2+-binding protein (27 DEGs), heat shock protein (23 DEGs), Ubiquitin family protein (21 DEGs), and ATP binding (20 DEGs). DEGs were significantly enriched in the pathway “04075 (Plant hormone signal transduction)” (218 DEGs, account for 6.61%), “04626 (Plant-pathogen interaction) “(216 DEGs, account for 6.55%), “00500 (Starch and sucrose metabolism)” (102 DEGs, account for 3.09%), “01200 (Carbon metabolism)” (90 DEGs, account for 2.73%). JA (jasmonic acid), ETH (ethylene), BR (brassinosteroids), IAA/auxin and ABA (abscisic acid) were found among 3,329 DEGs (Fig. 7), of which Gh_A12G0212 and Gh_D12G0214 associated with ABA hormone signal transduction were up-regulated under Na2SO4. It is reported that transcription factors play an important role in hormone signaling pathways in response to salt stress22,47. 353 transcription factors (TFs) (10.70% among 3,299 DEGs) were up-regulated (Fig. 8), of which NAC (13.88%) was the maximum number of 49 genes, follow by 46 (13.03%) in WRKY, 43 (12.18%) in ERF, 40 (11.33%) in MYB. Besides, there were 108 ubiquitin, accounting for 3.30% of the 3299 DEGs (Figure S6). In addition, ion absorption, compartmentalization and the osmotic balance responded to salt stress, were detected from 3,329 DEGs (Fig. 9), which includes K+ transporter, CBL, SOS3, iron transporter and SOD, CAT, Proline transporter, P5CS, LEA48,49.

Heatmap of the standardized FPKM expression level of the DEGs related to hormones enrichment among roots, stems and leaves between SS and CK drawn by ggplot2 (v3.3.3). CK: Control group; SS: 300 mM Na2SO4. Red = high expression level of genes, and Green = low expression level of genes.

Annotation of transcription factors of specific DEGs under Na2SO4 stress created by plotrix (v3.8.1).

Heatmap of the standardized FPKM expression level of the DEGs related to ionic homeostasis enrichment among roots, stems and leaves between SS and CK performed by ggplot2 (v3.3.3). CK: Control group; SS: 300 mM Na2SO4. Red = high expression level of genes, and Green = low expression level of genes.
Metabolism analysis
So far, the research of salt stress about sulfuric acid in the cotton was still lacking. In this study, it is interesting that GSH-ASA system (Fig. 10A,B), as the roles of antioxidant genes, were significantly up-regulated under Na2SO4 conditions (Fig. 10B). GSH-ASA is an important antioxidant system, of which the performance of GSH, as three roles that one for GS (glucosinolate) as a storage material of SO42−, one for PCs (Phytochelatins) as metal-binding oligopeptides in the heavy-metal detoxify mechanism and one for antioxidant system, is most important (Fig. 10A). APR (Gh_D05G1637) and OASTL (Gh_A13G0863) identified among 3,299 DEGs are two important rate-limiting enzymes in synthesis of GSH from SO42−.

Analysis of differential genes in pathways of sulfur metabolism. CK: Control group; SS: 300 mM Na2SO4. (A) A schematic diagram of sulfur metabolism. Heatmap represents the expression level of regulatory enzyme gene for specific process performed by ggplot2 (v3.3.3). (B) Up-regulated DEGs and down-regulated DEGs under CK and SS performed by ggplot2 (v3.3.3).
Discussion
Na2SO4, being a neutral salt, is made up of Na+ and SO42−. High concentrations of Na+ and SO42− in the soil not only causes the salt toxicity to plants50, but also hindered the uptake of other minerals51. In our present study, we used Na2SO4 solution of 300 mM as a salt stress to Zhong 9835 at three leaf stage, in which the pH value ranges from 6.93 at 0 h to 6.2 at 12 h (Fig. 1B). Under the mentioned treatment plants shows significant phenotypic differences among tissues of roots, shoots and leaves. As the roots get start blacking cotyledon turn wilted and leaves lost the turgidity due to loss of water and becomes weak and thin along with stem browning. (Fig. 1A). The pH value at 24 h was 5.6 (Fig. 1B), the root blackened more significantly, the stem browned, the cotyledon shriveled, and the true leaves are wilting and turning brown (Fig. 1A). Previous studies report that the main site of Na+ toxicity for most plants is the leaf blade, where Na+ accumulates after being deposited in the transpiration stream, rather than in the roots52. And an important oxidative damage only to be induced by SO42− anion with an increase in H2O2 and MDA content40, although sulfur could be taken up by the roots and stored in the vacuoles of root and xylem parenchymal cells53. In other words, at 12 h root blackening is most likely the toxic phenomenon caused by the oxidation of SO42− in weak acidic solution.
Osmotic stress and ionic toxicity can cause oxidative damage54. In response to osmotic stress (Figure S3), we found that starch and sucrose metabolism enriched (Fig. 6) and some organic material such as LEA (Fig. 9), HSFs (Fig. 8), proline and its biosynthesis key enzymes P5CS (Fig. 9) is up-regulated consistent with previous research report49. As ion transport for a Na+ detoxification way (Figure S5), SOS system, on the one hand, can potentially ejected Na+ by Na+/H+ exchangers located in the plasma membrane: on the other hand, sequestered it into the vacuole by Na+/H+ exchangers (e.g. NHX proteins) located in the tonoplast55. As previous studies, HKT1, as a Na+/K+ transporters, regulated the equilibrium of the Na+/K+ decreasing of Na+ toxicity43. Arabidopsis K+ transporter1 (AKT1) activity is repressed by SCaBP8 (CALCINEURIN B-LIKE10 or CBL10, known as SOS3-LIKE CALCIUM-BINDING PROTEIN8, SCaBP8), which interacted with and activated by SOS3-SOS2 complex (Fig. 11) 56.

Model of the regulatory networks in response to Na+ stress and SO42−. The leftmost part of the networks is related to SO42− stress. The others part of the networks is related to ROS, which mainly include Na+ stress and ABA signal. The vacuole is the crosstalk of SO42− stress and Na+ stress.
ROS scavenging system includes the enzymatic antioxidants (SOD, APX, PRX, GPX, CAT, GRX and TRX) and non-enzymatic scavengers (GSH-ASA system). It is well known that in cotton the enzymatic antioxidants and non-enzymatic scavengers participated in salt stress57. Among root, stem and leaf, not only the enzymatic antioxidants POD and CAT (Fig. 9), but also the non-enzymatic scavengers GPX, GST, DHAR and GST (Fig. 10B) genes were found to be up-expressed under Na2SO4 condition. And it is interesting that the number of the non-enzymatic scavengers, as a role in the reaction of peroxide detoxification by catalyzing GSH to GSSG58, especially GST is more than the enzymatic antioxidants (Fig. 10B). These results suggest that GST may be the major scavengers of ROS under Na2SO4 stress.
In response to ROS (Figure S1), both the ROS scavenging system and others signaling pathways regulated by transcription factors (Figure S4) work synergistically with efforts in avoiding PCD (Figure S2)59. Hormones including JA, ETH, BR, IAA and in particular ABA (Figure S4) although just one gene with two sub-genomes (Fig. 7), under salt stress a mass of researches had reported the crosstalk of ABA and some TFs belonging to others class such as Ubiquitin, MYB, NAC, bZIP and AP2/ERF60. Among these transcription factors, ubiquitin is most than others (Fig. 8). In apple, MdbHLH3 gene (an anthocyanin-related basic helix–loop–helix transcription factor (bHLH TF) gene) promotes ethylene production involved in ethylene biosynthesis including MdACO1, MdACS1, MdACS5A MdACS1, and MdACS5A61. U-box E3 ubiquitin ligase gene MdPUB29, highly homologous with AtPUB29, direct ubiquitination of the MdbHLH3 protein, positively regulating salt tolerance54. In addition, VTC1-CSN5B associated with the COP9 signalosome complex promotes ubiquitination-dependent VTC1 degradation through the 26S proteasome pathway, affecting the response to salt stress by regulating ASA synthesis62. In hormones, ETH and ABA induced cell senesce or cell death62,63, while auxin, BR and JA as roles related to abiotic stress can prevent the accumulation of ROS from that64. For example, IAA/auxin can reverse the hypersensitive response stimulated by purified harpin protein to extent65. AUX/IAA-mediated auxin signaling contributes to ethylene-dependent inducible aerenchyma formation in rice roots66. To Arabidopsis cell suspension cultures, auxin had an effect on the control of cell wall composition and rigidity, preventing the cell death67. In present study, the largest number of hormones is IAA/auxin, followed by JA and BR (Fig. 7). These results indicate that in Zhong 9835 the root, stem, leaf cell of treatment of 12 h under Na2SO4 stress is becoming balance by removing ROS toxicity (Fig. 11).
Materials and methods
Plant materials and salt stress treatments
The experimental materials in this study “Zhong 9835”, cotton cultivar (Gossypium hirsutum L.), was provided by Institute of Cotton Research of Chinese Academy of Agricultural Sciences. The experimental methods were as follows: Seeds were sown in sand soil pots. The sand was washed cleanly and sterilized at 121 °C for 8 h. Sterilized seeds with 1% sodium hypochlorite for 15 min and washed with sterile water for 3 times. The sterilized seeds were grown on sand with a water content of 17% under the condition of 28/25 ℃ long sunshine for 16 h/8 h with a light intensity of 150 μmol·m−2·s−1. At three true leaf stage, the samples from roots, stems and leaves with three biological repeats for each (CK_R1, CK_R2, CK_R3, CK_S1, CK_S2, CK_S3, CK_L1, CK_L2, CK_L3, SS_R1, SS_R2, SS_R3, SS_S1, SS_S2, SS_S3, SS_L1, SS_L2, SS_L3) respectively were collected under the treatment of 300 mmol L−1 Na2SO4 solution at 12 h and the control with water at 12 h. The leaves, stems and roots were used for RNA-seq and real-time fluorescence quantitative PCR (qRT-PCR). All samples were frozen in liquid nitrogen and stored at − 80 °C for further use.
Measurement of the SOD, POD, Pro and MDA content
The pH of water for plant growth during 0 h, 6 h, 12 h and 24 h was detected by Precision pH meter (S20 Mettler Toledo Instrument Co., LTD). SOD (superoxide dismutase), POD (peroxidase), Pro (proline) and MDA (malondialdehyde) contents in leaves, stems and roots were measured by SOD kits (Nanjing Jiancheng Bioengineering Research Institute), POD kits (Nanjing Jiancheng Bioengineering Research Institute), Pro kits (Nanjing Jiancheng Bioengineering Research Institute) and MDA detection kits (Nanjing Jiancheng Bioengineering Research Institute). The absorbance of SOD, POD, Pro and MDA at 450 nm, 420 nm, 520 nm and 532 nm for 0 h, 6 h, 12 h and 24 h were recorded with three replications for each sample by Ultraviolet–visible spectrophotometer (NanoDrop2000 Seymour Flight).
RNA extraction, cDNA library construction, and RNA-Seq
Total RNA from each tissue was extracted between control and treatment with 300 mM Na2SO4, based on the instruction manual of the TRlzol Reagent (Life technologies, California, USA). The integrity and concentration of total RNA was checked by Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA). The isolated mRNA by NEB Next Poly (A) mRNA Magnetic Isolation Module (NEB, E7490) were used for constructing cDNA library through the manufacturer’s instructions of NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, E7530) and NEB Next Multiplex Oligos for Illumina (NEB, E7500). And then, approximately 200nt RNA inserts were used to synthesize the first-strand cDNA and the second cDNA, according to the fragmented mRNA. In the next step, the end-repair/dA-tail and adaptor ligation were performed for double-stranded cDNA, which is to form the cDNA library by Agencourt AMPure XP beads (Beckman Coulter, Inc.) and PCR amplification. Finally, the constructed cDNA libraries of the different samples were sequenced on a flow cell using an Illumina Novaseq 6000 platform. LC-BIO Technologies (http://www.lc-bio.com/about/54.html) provides experimental procedures and commercially performed it.
Mapping and differential expression genes analysis
Based on the quality results of the paired-end reads, we removed the low-quality reads by per script, which included only adaptor, unknown nucleotides > 5% and Q20 < 20% (percentage of sequences with sequencing error rates < 1%). After filtering from the raw reads, the clean reads were mapped to cotton genome (G.hirsutum) by Hisat software44. According to the mapped reads from the reference cotton genome, String Tie software44 was used to estimate quantification of the gene expression levels with fragments per kilobase of transcript per million fragments mapped (FKPM)68. And an edger package, one of R packages, was applied for differential expression analysis between two groups with three tissues respectively. Fold Change ≥ 2 and FDR (false discovery rate) < 0.01 were taken as the threshold of the P-value in multiple tests for computing the significance of the differences.
Gene ontology and pathway enrichment analysis
DEGs for Gene Ontology (GO) terms enrichment analysis implemented by GO seq R packages69, were divided into 3 classes, molecular function, cellular process, and biological process. KEGG enrichment analysis of the DEGs was applied to summary the statistical enrichment of differential expression of genes in KEGG pathways (http://www.genome.jp/kegg/) by KOBAS software70. The results of the number of genes that mapped to annotated genes in GO and KEGG database was printed using ggplot2 (https://ggplot2.tidyverse.org). The heatmap analysis of DEGs was performed with R (v4.0.2) language software (https://www.r-project.org/)71,72,73,74 and the model of the regulatory networks in response to Na+ stress and SO42− was drawn by Science Slides (http://scienceslides.com/).
Validation of RNA-Seq by qRT-PCR
Each sample with 3 biological replicates was performed by Real-time RT-PCR (qRT-PCR)75,76. A set of 20 genes was chosen randomly by the FPKM. Specific primers for the chosen genes were designed through Primer 3 software. cDNA was synthesized by using an EASY spin Plus Plant RNA Kit (TIANGEN) with gDNA Eraser (TaKaRa)77. The qRT-PCR reactions were conducted using a SYBR Green I Master mixture (Bio-Rad, CFX96, Switzerland) according to the manufacturer’s protocol on a Light Cycler 480II system (Bio-Rad, CFX96, Switzerland). The results of qRT-PCR were analysed via the ΔΔCt method78, the cotton histone (His) 3 gene (GenBank accession no. AF024716) was used as a standardcontrol79. Histone (His) 3 (AF024716) (F: TCAAGACTGATTTGCGTTTCCA, R: GCGCAAAGGTTGGTGTCTTC). Each reaction was carried out in a final volume of 20 µL, 7.8 µL ddH2O and containing 10 µL of SYBR Green PCR master mix, 0.4 µL of each gene-specific primer and 1.4 µL of diluted cDNA. The PCR thermal cycling conditions were applied as follows: 95 °C for 5 min; 40 cycles of 95 °C for 5 s, 60 °C for 30 s and 72 °C for 30 s. Data were collected during the extension step: 95 °C for 15 s, 60 °C for 1 min, 95 °C for 30 s and 60 °C for 15 s. Three biological replicates were performed, and three technical replicates were designed per cDNA sample.
References
Xie, W. et al. Effects of straw application on coastal saline topsoil salinity and wheat yield trend. Soil Tillage Res. 169, 1–6 (2017).
Ashraf, M. & Foolad, M. R. Pre-sowing seed treatment—A shotgun approach to improve germination, plant growth, and crop yield under saline and non-saline conditions. Adv. Agron. 88, 223–271 (2005).
Qiu, Q.-S. et al. Regulation of vacuolar Na+/H+ exchange in Arabidopsis thaliana by the salt-overly-sensitive (SOS) pathway. J. Biol. Chem. 279, 207–215 (2004).
Katiyar-Agarwal, S. et al. The plasma membrane Na+/H+ antiporter SOS1 interacts with RCD1 and functions in oxidative stress tolerance in Arabidopsis. Proc. Natl. Acad. Sci. 103, 18816–18821 (2006).
Zhu, J.-K. Cell signaling under salt, water and cold stresses. Curr. Opin. Plant Biol. 4, 401–406 (2001).
Zhu, J.-K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 53, 247–273 (2002).
Tuteja, N. In Methods in Enzymology Vol. 428 419–438 (Elsevier, 2007).
Tester, M. & Davenport, R. Na+ tolerance and Na+ transport in higher plants. Ann. Bot. 91, 503–527 (2003).
Deinlein, U. et al. Plant salt-tolerance mechanisms. Trends Plant Sci. 19, 371–379 (2014).
Zhang, W.-D. et al. SOS1, HKT1; 5, and NHX1 synergistically modulate Na+ homeostasis in the halophytic grass Puccinellia tenuiflora. Front. Plant Sci. 8, 576 (2017).
Yang, Q. et al. Overexpression of SOS (salt overly sensitive) genes increases salt tolerance in transgenic Arabidopsis. Mol. Plant 2, 22–31 (2009).
Roosens, N. H., Thu, T. T., Iskandar, H. M. & Jacobs, M. Isolation of the ornithine-δ-aminotransferase cDNA and effect of salt stress on its expression in Arabidopsis thaliana. Plant Physiol. 117, 263–271 (1998).
Monihan, S. M., Ryu, C.-H., Magness, C. A. & Schumaker, K. S. Linking duplication of a calcium sensor to salt tolerance in Eutrema salsugineum. Plant Physiol. 179, 1176–1192 (2019).
Asano, T., Hayashi, N., Kikuchi, S. & Ohsugi, R. CDPK-mediated abiotic stress signaling. Plant Signal. Behav. 7, 817–821 (2012).
Asano, T. et al. Functional characterisation of OsCPK21, a calcium-dependent protein kinase that confers salt tolerance in rice. Plant Mol. Biol. 75, 179–191 (2011).
Sun, X. et al. A Glycine soja methionine sulfoxide reductase B5a interacts with the Ca2+/CAM-binding kinase Gs CBRLK and activates ROS signaling under carbonate alkaline stress. Plant J. 86, 514–529 (2016).
Jin, X. et al. Wheat CBL-interacting protein kinase 25 negatively regulates salt tolerance in transgenic wheat. Sci. Rep. 6, 28884 (2016).
Nath, M. et al. Salt stress triggers augmented levels of Na+, Ca2+ and ROS and alter stress-responsive gene expression in roots of CBL9 and CIPK23 knockout mutants of Arabidopsis thaliana. Environ. Exp. Bot. 161, 265–276 (2019).
Yang, Y. et al. Calcineurin B-Like proteins CBL4 and CBL10 mediate two independent salt tolerance pathways in Arabidopsis. Int. J. Mol. Sci. 20, 2421 (2019).
Cheng, Y., Zhang, X., Sun, T., Tian, Q. & Zhang, W.-H. Glutamate receptor homolog3.4 is involved in regulation of seed germination under salt stress in Arabidopsis. Plant Cell Physiol. 59, 978–988 (2018).
Pang, C.-H. & Wang, B.-S. In Progress in Botany 231–245 (Springer, 2008).
Dai, W., Wang, M., Gong, X. & Liu, J. H. The transcription factor Fc WRKY 40 of Fortunella crassifolia functions positively in salt tolerance through modulation of ion homeostasis and proline biosynthesis by directly regulating SOS 2 and P5 CS 1 homologs. New Phytol. 219, 972–989 (2018).
Zhang, H. et al. Populus euphratica JRL mediates ABA response, ionic and ROS homeostasis in Arabidopsis under salt stress. Int. J. Mol. Sci. 20, 815 (2019).
Xu, L. et al. Melatonin enhances salt tolerance by promoting MYB108A-mediated ethylene biosynthesis in grapevines. Horticult. Res. 6, 1–14 (2019).
Denver, J. B. & Ullah, H. miR393s regulate salt stress response pathway in Arabidopsis thaliana through scaffold protein RACK1A mediated ABA signaling pathways. Plant Signal. Behav. 14, 1600394 (2019).
Zhang, K., Wang, G., Bao, M., Wang, L. & Xie, X. Exogenous application of ascorbic acid mitigates cadmium toxicity and uptake in maize (Zea mays L.). Environ. Sci. Pollut. Res. 26, 19261–19271 (2019).
Wang, J., Liu, S., Liu, H., Chen, K. & Zhang, P. PnSAG1, an E3 ubiquitin ligase of the Antarctic moss Pohlia nutans, enhanced sensitivity to salt stress and ABA. Plant Physiol. Biochem. 141, 343–352 (2019).
Li, Z. et al. GhWRKY6 acts as a negative regulator in both transgenic Arabidopsis and cotton during drought and salt stress. Front. Genet. 10, 392 (2019).
Perin, E. C. et al. ABA-dependent salt and drought stress improve strawberry fruit quality. Food Chem. 271, 516–526 (2019).
Ali, A., Maggio, A., Bressan, R. A. & Yun, D.-J. Role and functional differences of HKT1-type transporters in plants under salt stress. Int. J. Mol. Sci. 20, 1059 (2019).
Maeshima, M. Vacuolar H+-pyrophosphatase. Biochim. Biophys. Acta (BBA)-Biomembr. 1465, 37–51 (2000).
Primo, C. et al. Plant proton pumping pyrophosphatase: The potential for its pyrophosphate synthesis activity to modulate plant growth. Plant Biol. 21, 989–996 (2019).
Hsu, Y.-D. et al. Regulation of H+-pyrophosphatase by 14-3-3 proteins from Arabidopsis thaliana. J. Membr. Biol. 251, 263–276 (2018).
Kapilan, R., Vaziri, M. & Zwiazek, J. J. Regulation of aquaporins in plants under stress. Biol. Res. 51, 1–11 (2018).
Feng, Z.-J. et al. Identification of the AQP members involved in abiotic stress responses from Arabidopsis. Gene 646, 64–73 (2018).
Braz, L. C. et al. Expression of aquaporin subtypes (GhPIP1; 1, GhTIP2; 1 and GhSIP1; 3) in cotton (Gossypium hirsutum) submitted to salt stress. AoB Plants 11, plz072 (2019).
Van Zelm, E., Zhang, Y. & Testerink, C. Salt tolerance mechanisms of plants. Annu. Rev. Plant Biol. 71 (2020).
Flowers, T. Improving crop salt tolerance. J. Exp. Bot. 55, 307–319 (2004).
Richter, J. A., Behr, J. H., Erban, A., Kopka, J. & Zörb, C. Ion‐dependent metabolic responses of Vicia faba L. to salt stress. Plant Cell Environ. 42, 295–309 (2019).
Reginato, M. A., Turcios, A. E., Luna, V. & Papenbrock, J. Differential effects of NaCl and Na2SO4 on the halophyte Prosopis strombulifera are explained by different responses of photosynthesis and metabolism. Plant Physiol. Biochem. 141, 306–314 (2019).
Aghajanzadeh, T., Reich, M., Kopriva, S. & De Kok, L. Impact of chloride (NaCl, KCl) and sulphate (Na2SO4, K2SO4) salinity on glucosinolate metabolism in Brassica rapa. J. Agron. Crop Sci. 204, 137–146 (2018).
Gong, D. et al. Effects of salt stress on photosynthetic pigments and activity of ribulose-1, 5-bisphosphate carboxylase/oxygenase in Kalidium foliatum. Russ. J. Plant Physiol. 65, 98–103 (2018).
Guo-Wei, Z., Hai-Ling, L., Lei, Z., Bing-Lin, C. & Zhi-Guo, Z. Salt tolerance evaluation of cotton (Gossypium hirsutum) at its germinating and seedling stages and selection of related indices. Yingyong Shengtai Xuebao 22 (2011).
Kim, D. et al. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Paterson, A. H. et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492, 423–427 (2012).
Tatusov, R. L., Galperin, M. Y., Natale, D. A. & Koonin, E. V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36 (2000).
Hubbard, K. E., Nishimura, N., Hitomi, K., Getzoff, E. D. & Schroeder, J. I. Early abscisic acid signal transduction mechanisms: Newly discovered components and newly emerging questions. Genes Dev. 24, 1695–1708 (2010).
de Freitas, P. A. F., de Souza Miranda, R., Marques, E. C., Prisco, J. T. & Gomes-Filho, E. Salt tolerance induced by exogenous proline in maize is related to low oxidative damage and favorable ionic homeostasis. J. Plant Growth Regulat. 37, 911–924 (2018).
Huang, Z. et al. Salt stress encourages proline accumulation by regulating proline biosynthesis and degradation in Jerusalem artichoke plantlets. PLoS ONE 8, e62085 (2013).
Collins, N. C., Tardieu, F. & Tuberosa, R. Quantitative trait loci and crop performance under abiotic stress: Where do we stand?. Plant Physiol. 147, 469–486 (2008).
Ahmadizadeh, M. et al. Reproductive stage salinity tolerance in rice: A complex trait to phenotype. Indian J. Plant Physiol. 21, 528–536 (2016).
Munns, R. Comparative physiology of salt and water stress. Plant Cell Environ. 25, 239–250 (2002).
De Kok, L. J. et al. Pathways of plant sulfur uptake and metabolism-an overview. Landbauforschung Völkenrode (Special Issue) 283, 5–13 (2005).
Han, P.-L., Dong, Y.-H., Jiang, H., Hu, D.-G. & Hao, Y.-J. Molecular cloning and functional characterization of apple U-box E3 ubiquitin ligase gene MdPUB29 reveals its involvement in salt tolerance. J. Integr. Agric. 18, 1604–1612 (2019).
Qiu, Q.-S., Guo, Y., Dietrich, M. A., Schumaker, K. S. & Zhu, J.-K. Regulation of SOS1, a plasma membrane Na+/H+ exchanger in Arabidopsis thaliana, by SOS2 and SOS3. Proc. Natl. Acad. Sci. 99, 8436–8441 (2002).
Quan, R. et al. SCABP8/CBL10, a putative calcium sensor, interacts with the protein kinase SOS2 to protect Arabidopsis shoots from salt stress. Plant Cell 19, 1415–1431 (2007).
Keunen, E., Peshev, D., Vangronsveld, J., Van Den Ende, W. & Cuypers, A. Plant sugars are crucial players in the oxidative challenge during abiotic stress: Extending the traditional concept. Plant Cell Environ. 36, 1242–1255 (2013).
Mari, M., Morales, A., Colell, A., García-Ruiz, C. & Fernández-Checa, J. C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 11, 2685–2700 (2009).
Deng, X. et al. A preliminary study on PCD aspects and roles of reactive oxygen species during aerenchyma formation in wheat roots under waterlogging. J. Triticeae Crops 5, 832–838 (2009).
Yang, O. et al. The Arabidopsis basic leucine zipper transcription factor AtbZIP24 regulates complex transcriptional networks involved in abiotic stress resistance. Gene 436, 45–55 (2009).
Hu, D. G. et al. The regulatory module Md PUB 29-Mdb HLH 3 connects ethylene biosynthesis with fruit quality in apple. New Phytol. 221, 1966–1982 (2019).
Yu, Y. et al. Ascorbic acid integrates the antagonistic modulation of ethylene and abscisic acid in the accumulation of reactive oxygen species. Plant Physiol. 179, 1861–1875 (2019).
Xu, W. & Huang, W. Calcium-dependent protein kinases in phytohormone signaling pathways. Int. J. Mol. Sci. 18, 2436 (2017).
Ma, F., Yang, X., Shi, Z. & Miao, X. Novel crosstalk between ethylene-and jasmonic acid-pathway responses to a piercing–sucking insect in rice. New Phytol. 225, 474–487 (2020).
Song, J. et al. Indole-3-acetic acid reverses the harpin-induced hypersensitive response and alters the expression of hypersensitive-response-related genes in tobacco. Biotech. Lett. 36, 1043–1048 (2014).
Yamauchi, T., Tanaka, A., Tsutsumi, N., Inukai, Y. & Nakazono, M. A role for auxin in ethylene-dependent inducible aerenchyma formation in rice roots. Plants 9, 610 (2020).
Zhang, T. et al. Hormone crosstalk in wound stress response: Wound-inducible amidohydrolases can simultaneously regulate jasmonate and auxin homeostasis in Arabidopsis thaliana. J. Exp. Bot. 67, 2107–2120 (2016).
Florea, L., Song, L. & Salzberg, S. L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research 2 (2013).
Young, M. D., Wakefield, M. J., Smyth, G. K. & Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 11, R14 (2010).
Mao, X., Cai, T., Olyarchuk, J. G. & Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793 (2005).
Futschik, M. E. & Carlisle, B. Noise-robust soft clustering of gene expression time-course data. J. Bioinform. Comput. Biol. 3, 965–988 (2005).
Wickham, H. et al. (Springer, 2016).
Lemon, J. Plotrix: A package in the red light district of R. R-News 6, 8–12 (2006).
Wickham, H. The split-apply-combine strategy for data analysis. J. Stat. Softw. 40, 1–29 (2011).
Wu, A. R. et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods 11, 41 (2014).
Pombo, M. A., Zheng, Y., Fei, Z., Martin, G. B. & Rosli, H. G. Use of RNA-seq data to identify and validate RT-qPCR reference genes for studying the tomato-Pseudomonas pathosystem. Sci. Rep. 7, 1–11 (2017).
Wan, C.-Y. & Wilkins, T. A. A modified hot borate method significantly enhances the yield of high-quality RNA from cotton (Gossypium hirsutum L.). Anal. Biochem. 223, 7–12 (1994).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408 (2001).
Zhang, B. et al. Transcriptome analysis of Gossypium hirsutum L. reveals different mechanisms among NaCl, NaOH and Na2CO3 stress tolerance. Sci. Rep. 8, 1–14 (2018).
Acknowledgements
This work was supported by grants from National key research and development program (2016ZX08005004-005).
Author information
Authors and Affiliations
Contributions
W.Y. conceived and designed the experiments. Q.W., X.L. and X.C. performed the experiments, prepared the materials and conducted the manuscript. W.A.M., reviewed the manuscript, edited and added the conformational changes. D.W., L.Z., R.C., M.H., C.R., Y.Z. and Y.F. participated in data analyses. Thanks, S.W., J.W., L.G. and C.C. for providing some significant advices. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Q., Lu, X., Chen, X. et al. Transcriptome analysis of upland cotton revealed novel pathways to scavenge reactive oxygen species (ROS) responding to Na2SO4 tolerance. Sci Rep 11, 8670 (2021). https://doi.org/10.1038/s41598-021-87999-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-87999-x
This article is cited by
-
Transcriptome Analysis Reveals Key Molecular Pathways in Response to Alkaline Salt Stress in Canola (Brassica napus L.) Roots
Journal of Plant Growth Regulation (2023)
-
Overexpression of cotton GhNAC072 gene enhances drought and salt stress tolerance in transgenic Arabidopsis
BMC Genomics (2022)
-
Reactive oxygen species signalling in plant stress responses
Nature Reviews Molecular Cell Biology (2022)
-
Genome-wide identification and function analysis of HMAD gene family in cotton (Gossypium spp.)
BMC Plant Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
