
Abstract
Bamboos, member of the family Poaceae, represent many interesting features with respect to their fast and extended vegetative growth, unusual, yet divergent flowering time across species, and impact of sudden, large scale flowering on forest ecology. However, not many studies have been conducted at the molecular level to characterize important genes that regulate vegetative and flowering habit in bamboo. In this study, two bamboo FD genes, BtFD1 and BtFD2, which are members of the florigen activation complex (FAC) have been identified by sequence and phylogenetic analyses. Sequence comparisons identified one important amino acid, which was located in the DNA-binding basic region and was altered between BtFD1 and BtFD2 (Ala146 of BtFD1 vs. Leu100 of BtFD2). Electrophoretic mobility shift assay revealed that this alteration had resulted into ten times higher binding efficiency of BtFD1 than BtFD2 to its target ACGT motif present at the promoter of the APETALA1 gene. Expression analyses in different tissues and seasons indicated the involvement of BtFD1 in flower and vegetative development, while BtFD2 was very lowly expressed throughout all the tissues and conditions studied. Finally, a tenfold increase of the AtAP1 transcript level by p35S::BtFD1 Arabidopsis plants compared to wild type confirms a positively regulatory role of BtFD1 towards flowering. However, constitutive expression of BtFD1 had led to dwarfisms and apparent reduction in the length of flowering stalk and numbers of flowers/plant, whereas no visible phenotype was observed for BtFD2 overexpression. This signifies that timely expression of BtFD1 may be critical to perform its programmed developmental role in planta.
Similar content being viewed by others


Introduction
Bamboos belong to the subfamily Bambusoideae, family Poaceae and are widely distributed in Asia, Africa and America1,2. The plant group displays a wide range of variation across species with respect to flowering time and nature. Here flowering takes place after a prolonged vegetative phase, which may be extend up to 120 years3. When flowering occurs in a few culms of a population it is called sporadic flowering4, while in gregarious flowering a long stretch of geographical area is influenced for blooming5. Usually bamboo flowering is followed by death of each and individual culm and is known as monocarpy or semelparity.
Onset of flowering under favourable environment is decided by a complex regulatory crosstalk at molecular level and several mechanisms such as photoperiod, vernalization, autonomous, hormonal and age pathways have been characterized in plants6,7,8,9,10. In silico studies indicate that the majority of these pathways also exist in bamboo11. Non targeted transcriptome sequencing has been undertaken to identify floral tissue specifically expressed sequence tags (ESTs) of short lengths from many temperate/tropical, woody/herbaceous bamboo species such as Dendrocalamus latiflorus12,13, Phyllostachys edulis14,15,16, P. violascens17, P. aurea, Guadua inermis, Otatea acuminata, Lithachne pauciflora18 and Fargesia macclureana19. In addition, identification and expression analyses of a group of floral pathway genes or gene families have been undertaken. For example, ten genes related to floral transition and meristem identity were identified in D. latiflorus20, whereas sixteen MADS box genes were reported from Bambusa edulis21. A few studies have been conducted to functionally characterize important flowering genes such as MADS18 from D. latiflorus22, FLOWERING LOCUS T (FT) from P. meyeri23,24, TERMINAL FLOWER 1 (TFL1) like gene from B. oldhamii25, FRIGIDA (FRI) from P. violascens26, and MADS1, 2 from P. praecox27.
FD genes encoding transcription factors are members of the group A basic leucine zipper (bZIP) family28. They are ubiquitously found in angiosperms, but not in any other plant lineages29. Studies conducted on Arabidopsis (A. thaliana) and rice (O. sativa) suggest that the transition of shoot apical meristem (SAM) to inflorescence meristem (IM) is primarily governed by interaction among AtFT/OsHd3a, At14-3–3/OsGf14 and AtFD/OsFD1 proteins to form the florigen activation complex (FAC) preceding flowering30,31,32,33,34. Subsequently, FD binds to the CRE binding element (ACGT) present in the promoter of floral meristem identity gene APETALA133,35,36 (AP1). Two paralogous copies of FD genes (AtFD and AtFDP) have been identified in Arabidopsis37, whereas three copies are present in rice29. Other than these reference plants, FD homologs have been discovered from many other plants.
The loss-of-function mutation of either AtFD or AtFDP resulted in late flowering in Arabidopsis, while their overexpression demonstrated early flowering indicating their possible involvement in flowering37,38. Similarly, the RNAi lines of OsFD1 demonstrated a late flowering phenotype, while overexpression of OsFD1 resulted into early flowering29,33. In addition to flower induction, other pleiotropic roles of FD genes such as inflorescence development39,40, leaf development29 and alternative growth cessation41,42 have also been observed. This clearly indicates that FD performs diverse important roles in the vegetative and reproductive developments of plants.
Therefore, in order to understand the diverse functions of FD genes in plant growth and development, new studies need to be conducted on yet unexplored, non-reference plants demonstrating remarkable vegetative or flowering habit. Bamboos represent a particularly interesting plant group due to their semelparous life cycle and transition to flowering after decades of vegetative growth. Therefore, the main aim was to identify and characterize bamboo FD genes. This study addressed the sequence diversity and differential DNA binding properties of two FD genes isolated from Bambusa tulda in conjunction with their functional diversity based on expression patterns and impact on vegetative and flowering development in a heterologous system.
Results
Identification and sequence characterization of BtFD1 and BtFD2 genes
To study the role and diversity of FD genes (Table 1) in bamboo, B. tulda was selected, because its floral developmental stages have relatively been better characterized than any other bamboo species11,43, occurrence of sporadic flowering events in the species from time to time4,44 and its enormous economic importance in Asia. Two copies of BtFD genes have been identified by designing primers from the conserved regions of homologous FD genes, PCR and multiple sequencing (Supplementary Fig. S1, Fig. 1, BtFD1: MF983712 and BtFD2: MH142577). Homology search of the BtFD1 sequence identified Sasa veitchii (Bambusoideae) FD (SvFDL1: BAS04368.1, SvFDL2: BAS04369.1) as its closest homolog having highest similarity (77%), while the BtFD2 sequence revealed highest similarity (92%) against Panicum hallii FD homolog (XP_025812603.1). Predicted lengths of BtFD1 and BtFD2 proteins were 202 and 159 amino acids, respectively, and both of them contain the characteristic bZIP domain. However, differences between BtFD1 and BtFD2 proteins were observed with reference to other domains. BtFD1 contains motif 1 [MEDD(E/D)DMW(A/G)XTSSPSASPP], the LSL motif [T(A/V)LSLN] and the SAP motif [(S/T)LXRX(S/T)(A/T)(P/Q)F], while BtFD2 contains motif 2 [NYHHYQMAV(A/H)AA], motif 3 [(L/M/V)SGCSSLFSIS(S/T)] and a modified SAP motif (Supplementary Fig. S2). A detailed sequence comparison of the bZIP domains present in BtFD1 and BtFD2 proteins identified five amino acids changes (Supplementary Fig. S3). Out of these, only the change of Ala146 (BtFD1) > Leu100 (BtFD2) was located in the DNA binding basic region. Therefore, it was investigated whether this amino acid change may or may not impact the binding efficiency of BtFD1 and BtFD2.

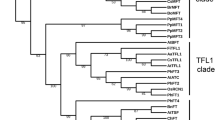
Phylogenetic analysis and predicted gene models of FD homologs identified in monocotyledonous plants. The Neighbour Joining (NJ) tree of the FD homologs was constructed with the full-length protein sequences by Mega 7.0 using default parameters and bootstrap value 2000. In the gene structures exons were marked as rectangles having conserved motifs marked in solid boxes and introns as solid lines. Annual species were marked in blue and perennials in red rhomboids. The motifs conserved are M 1 [MEDD(E/D)DMW(A/G)XTSSPSASPP], M 2 [NYHHYQMAV(A/H)AA], M 3 [(L/M/V)SGCSSLFSIS(S/T)], M 4 [(M/V)EEVWKDINLSSLHD], LSL [T(A/V)LSLN], Bzip and SAP [(S/T)LXRX(S/T)(A/T)(P/Q)F].
Phylogenetic relationship of BtFD1 and BtFD2 genes with homologs obtained from other Poaceae and non-Poaceae members
The phylogenetic analysis of BtFD1 and BtFD2 genes with homologs obtained from Poaceae and non-Poaceae members identified three major clusters. The cluster 1 was comprised of FD1 homologs obtained from all the Poaceae plants along with three bamboos (B. tulda, S. veitchii, P. heterocycla). The cluster 2 was comprised of FD1 homologs obtained from all the non-Poaceae members, while the cluster 3 was comprised of all FD2 homologs (Fig. 1). Cluster 1 specific for Poaceae FD1s was subdivided into two major sub-clusters. The sub-cluster 1 hosted FD1 sequences obtained from annual (Z. mays, S. bicolor, S. italica, S. viridis, O. sativa, O. brachyantha) and perennial (Z. japonica, D. oligosanthes, P. hallii, P. virgatum) plants, whereas sub-cluster 2 hosted only annual plants such as H. vulgare, T. aestivum, A. tauschii, B. distachyon, B. stacei. The B. tulda FD1 was placed in sub-cluster 1 along with two other bamboos P. heterocycla and S. veitchii FD1 (Fig. 1). Similarly, the FD2 specific cluster 3 was also subdivided into two sub-clusters. Here, B. tulda FD2 was clustered with P. heterocycla along with other annuals and perennial plants (Fig. 1).
Expression analyses of BtFD1 and BtFD2 genes in different tissues, diurnal conditions and seasons
Transcriptional expression patterns of BtFD1 and BtFD2 genes were investigated in diverse vegetative as well as reproductive tissues, diurnal conditions and seasons to understand the functions of these genes in bamboo vegetative as well as reproductive development. Among nine different tissues studied, expression of BtFD1 was highest in shoot apex, followed by YLF and culm-sheath in comparison to rhizome. In contrary, the expression level of BtFD2 was consistently very low in majority tissues studied (Fig. 2a). When diurnal expression patterns were analysed, expression level of BtFD1 in YLF was highest in the afternoon (4 pm), which was not the case for YLN. However, the expression level of BtFD2 was consistently very low except in a single time point i.e. afternoon (4 pm) in YLF (Fig. 2b).

Comparison of tissue specific and diurnal expression pattern of BtFD1 and BtFD2 genes. (a) Tissue specific expressions of BtFD1 and BtFD2 in nine different tissue stages of B. tulda. Each bar represents mean of three biological replicates ± SE. (b) Diurnal expressions of BtFD1 and BtFD2 in YLF and YLN in SD and LD. Each data point represents mean of three biological replicates ± SE. Transcript expression of eIF4α was used to normalize expression data. The relative fold change was calculated by 2−ΔΔCT method using the expression data in rhizome as calibrator and is plotted using Y axis. CS culm sheath, YLF young leaf from flowering culm, YLN young leaf from non-flowering culm, I inter node, SA shoot apex, IFB immature floral bud, MFB mature floral bud, R root.
Close observation of B. tulda flowering habit from 2015 to 2018 revealed that sporadic flowering events usually recurred in spring every year. Therefore, to get further insight into the functions of BtFD1 and BtFD2 genes, their expression in young leaves were studied at three time points before onset of flowering, i.e., summer (April-June), monsoon (July–August), autumn (September–October), during onset of flowering i.e., winter (November-January) and after i.e., spring (February–March, Fig. 3). The expression level of BtFD1 was notably higher in winter compared to other seasons (Fig. 3). In contrary, no such pattern was found for BtFD2 expression. It was also barely detectable and quite comparable in YLF and YLN in all the seasons except a little increase in YLN in spring.

Expression analyses of BtFD1 and BtFD2 genes in YLF and YLN of B. tulda in five different seasons. Each bar represents mean expression of three biological replicates ± SE. The eIF4α was used to normalize expression data of the targeted flowering genes. The relative fold change was calculated by 2−ΔΔCT method using the expression level observed in rhizome as the calibrator. YLF young leaf from flowering culm, YLN young leaf from non-flowering culm.
In silico and EMSA analyses to study interaction between bZIP domains of BtFD proteins and ACGT motif
The bZIP domain of FD proteins needs to interact with the conserved CRE binding element (ACGT) present in the promoter of AP1 in order to perform DNA binding activity, Therefore, the overall potential of bZIP domains present in BtFD1/BtFD2 to bind to the ACGT motif was analysed. Comparison of the bZIP domains of BtFD1, BtFD2 and their homologous sequences revealed a striking difference, i.e. Ala146 of BtFD1 was replaced by Leu100 in BtFD2 (Supplementary Fig. S3). In order to assess the impact of such an amino acid change, a two-pronged approach was adopted—(1) in silico prediction of overall DNA binding ability of BtFD1 and BtFD2, and (2) validation of the in silico prediction using EMSA analyses.
Docked structures of both BtFD1 and BtFD2 bZIP models predicted positive interactions with CRE DNA containing ACGT motif. Superimposed docked structures also revealed that the interactions of both BtFD1 and BtFD2 could take place in a similar manner (Fig. 4a). Several amino acid residues located at the basic region, spanning from His133 to Gln153 in BtFD1 and Arg87 to Arg107 in BtFD2 were found to interact with the CRE consensus sequence. In silico docking analysis suggested that Arg142, Ser144, Arg147, Ser148 and Arg149 of BtFD1 and Arg96, Leu100, Arg101, Ser102 and Arg103 of BtFD2 were particularly found to be directly interacting with TGACGTCA consensus CRE DNA. Additionally, in silico analysis predicted direct contact for Leu100 in BTFD2 with a conserved dT residue of ACGT motif, whereas the corresponding Ala146 in BtFD1 had no interfering interactions with DNA (Fig. 4b). Even though BtFD2 gained additional interaction in this way, this non-polar-polar interaction was unfavourable in nature and therefore, could interfere with its DNA binding specificity. Ala146 on the other hand, though also non-polar, might be advantageous in this position because of its smaller size. To validate this result further, EMSA studies were conducted using mimics of BtFD1 and BtFD2 bZIPs, which only differed by a single amino acid (Ala146 of BtFD1 vs. Leu100 of BtFD2, Fig. 5a). Varying concentrations of bZIP mimics of BtFD1 and BtFD2 proteins were used for EMSA analysis, which showed that reappearance of free DNA begins after 0.30 µM in case of BtFD1 and 3.12 µM in case of BtFD2 (Fig. 5b,c). Free DNA is observed at 0.28 µM in case of BtFD1, which is 2.81 µM for BtFD2 (Supplementary Figs. S5a, S5b). Therefore, the concentration ranges of ‘binding to no-binding’ for bZIP mimic of BtFD1 was 0.31–0.28 µM, whereas it was 3.12–2.81 µM for BtFD2. Taken together, the finding clearly demonstrated a tenfold enhanced DNA binding specificity for BtFD1 bZIP mimic compared to its BtFD2 analogue. This means the required threshold value for BtFD2-CRE DNA interaction is much higher than that of BtFD1 (Fig. 5b,c). This further consolidated the consequence of the single amino acid substitution (Supplementary Figs. S5a–d).

In silico interactions of bZIP domains of BtFD1 and BtFD2 with ACGT motif. (a) Superimposed structures of bZIP domains of BtFD1 (magenta) and BtFD2 (green) interacting with ACGT motif in a similar manner. (b) Arg149/Arg103 of BtFD1/BtFD2 interact with cognate DNA sequence containing ACGT motif and Leu100 of BtFD2 making additional contact with DNA (dT residue).

EMSA study to compare the DNA binding efficiency of bZIP domains of BtFD1 and BtFD2 proteins. (a) Sequence alignment of bZIP domains of TobZL (experimental template) vs. BtFD1 and BtFD2 reveals changes at two amino acids residues (His to Ser and Lys to Ala/Leu). (b) EMSA studies of BtFD1 and (c) BtFD2 mimics show overall ability to bind with CRE DNA. Lanes 1–7 in both the gels contain twofold serially diluted proteins (0.080 to 5.000 μM for BtFD1 and 0.800 to 50.00 μM for BtFD2 bZIP mimics), lane 8 contains free DNA.
Constitutive expression of BtFD1 and BtFD2 genes in Arabidopsis
In order to study roles of BtFD1 and BtFD2 genes on the vegetative as well as reproductive development of plants, these homologs were constitutively expressed in Arabidopsis (Columbia) plants. The phenotypes of transgenic p35S::BtFD1 Arabidopsis plants revealed drastic suppression of vegetative and floral growth in short day (SD) and long day (LD) conditions (Fig. 6a). Leaf numbers observed in three independent p35S::BtFD1 transgenic lines after four weeks of growth were 8 to 9 in LD and 6 to 7 in SD, which were 10 and 14 in wild-type plants, respectively (Fig. 6b). The reduction in leaf number in p35S::BtFD1 plants in comparison to WT was statistically significant in SD (p.adj = 0.000), but not in LD (p.adj = 0.193), when one-way ANOVA was conducted. In contrary, change in leaf numbers of p35S::BtFD2 transgenic plants in comparison to WT were statistically insignificant in SD (p.adj = 0.007) as well as LD (p.adj = 0.040). Apart from the numbers, size of leaves were also significantly reduced in p35S::BtFD1 plants in SD (p.adj = 0.000) and LD (p.adj = 0.000) compared to WT (Fig. 6c). In contrary, the difference in leaf size between p35S::BtFD2 and WT plants were statistically significant in SD (p.adj = 0.000), but not in LD (p.adj = 0.725) (Fig. 6c). In order to simultaneously consider the effects of genetic background (WT, p35S::BtFD1, p35S::BtFD2) as well as duration of light (SD, LD), two-way ANOVA was also conducted. The genetic background had significant effect on leaf numbers (p = 0.000), whereas the light duration did not (p = 0.356). Number of leaves were significantly reduced in p35S::BtFD1 plants compared to the WT (p.adj = 0.000). In contrary, no significant difference was obtained for leaf numbers of p35S::BtFD2 plants compared to WT (p.adj = 0.952). However, both the genetic background (p = 0.000) as well as the light duration have significant effects (p = 0.000) on leaf perimeter. Also, change in perimeter was significant in both the cases for p35S::BtFD1 plants compared to the WT (p.adj = 0.000), which was not the case for the p35S::BtFD2 plants compared to the WT (p.adj = 0.022).

Phenotypic comparisons of wild type (WT) and transgenic p35S::BtFD1 and p35S::BtFD2 A. thaliana plants. (a) Plants were grown in LD (16-h light and 8-h dark) and SD (10-h light and 14-h dark) for four weeks. Arrow indicates emerged inflorescence axis. The scale bar represents 1 cm. (b) Comparisons of rosette leaf numbers of four-week-old transgenic plants in (c) Comparisons of perimeters of rosette leaves of transgenic vs. WT plants. Each bar represents mean perimeter of eight individual mature leaves ± SE. comparison to WT in SD and LD. Each bar represents mean leaf numbers obtained from four individual plants ± SE. One-way ANOVA analyses were performed to test statistical significance at p.adj ≤ 0.0001. LD long day, SD short day.
In order to obtain kinetic differences in leaf growth, perimeter of first true leaves were measured in four-day intervals in SD (Fig. 7a). Consistently, the leaf perimeter of p35S::BtFD1 trasgenic plants were significantly lower than WT and p35S::BtFD2 (Fig. 7b). Further, histological observation on leaf epidermal cells of first true leaves of these plants revealed that the perimeter were significantly lower (0.234 ± 0.005 cm to 0.299 ± 0.007 cm) in p35S::BtFD1 (p.adj = 0.000) plants compared to WT, but not in case of p35S::BtFD2 (p.adj = 0.066, Fig. 7c,d). Like vegetative growth, the flowering time was extremely delayed in p35S::BtFD1 Arabidopsis plants compared to p35S::BtFD2 and WT (Supplementary Figs. S4a, S4b). This was apparent by the significant increase of leaf number in p35S::BtFD1 plants compared to WT (p.adj = 0.000), but not in case of p35S::BtFD2 (p.adj = 0.558). Additionally, the length of the flowering stalk and the numbers of flowers/plant were strongly reduced in p35S::BtFD1 transgenic plants, while no obvious difference was noticed for p35S::BtFD2 and WT plants in LD (Supplementary Fig. S4a). In order to promote flowering, FD binds to AP1 to induce it at the transcriptional level. Therefore, the expression of AtAP1 was measured in the wild type, p35S::BtFD1, and p35S::BtFD2 Arabidopsis plants. Indeed, the expression of the AtAP1 gene in the four-week-old leaves of p35S::BtFD1 Arabidopsis plants grown under LD was tenfold higher compared to WT, which was only twofold in case of p35S::BtFD2 plants (Supplementary Fig. S4c).

Comparison of leaf sizes of wild type A. thaliana (WT) with that of transgenic p35S::BtFD1 and p35S::BtFD2 plants. (a) Representative first true leaves of WT, p35S::BtFD1 and p35S::BtFD2 plants in 14, 18, 22, 26, 30, 34, 38 days after germination (DAG). (b) Perimeter of first true leaves measured in every four DAG in SD (10-h light and 14-h dark). Each data represents mean of ten individual data ± SE. The scale bar represents 5 mm. (c) Leaf epidermal cells obtained from the first true leaves of 22, 26 and 30 DAGs in SD. In each picture, the cell wall of a representative cell is marked by red outline. The scale bar represents 0.1 mm. (d) Perimeter of leaf epidermal cells measured in 22, 26 and 30 DAGs in. SD. Each data represents mean of thirty (fifteen apical and fifteen basal) individual cells ± SE. Mixed three-way ANOVA were performed to test statistical significance at p.adj ≤ 0.0001.
Discussion
Bamboo FD genes are similar to other Poaceae FD homologs in terms of sequence similarity and phylogenetic relationships
FD is a bZIP family protein and plays important roles in controlling the timing of reproductive phase transition in angiosperms29,33,45. In addition, its role in vegetative development has also been observed29. In this study two bamboo FD genes (BtFD1 and BtFD2) were identified and their sequences were characterized to study phylogenetic relationships of these genes to other homologous monocot genes. Characterization of the amino acid sequences inferred that like other Poaceae FD1s, bamboo BtFD1 possessed motifs 1, LSL, bZIP and SAP, but not motif 4, which is usually characteristic of non-Poaceae FD1s29 (Supplementary Fig. S2). Among these motifs, bZIP and SAP were found absolutely necessary for the interaction with AP136 and 14–3-3, respectively. However, the functional significance of other motifs in flowering needs further investigation33. In contrary, the LSL motif is absent in all FD2 homologs including bamboo that have been identified so far suggesting their less likely involvement in flowering (Supplementary Fig. S2).
Phylogenetic analyses of the FD homologs obtained from monocotyledonous plants revealed the presence of three major clusters. Cluster 1 and 2 were comprised of FD1 homologs of Poaceae and non-Poaceae members respectively, while all FD2s from the Poaceae species were placed in cluster 3 (Fig. 1). All the bamboo FD1 and FD2 homologs (P. heterocycla, B. tulda and S. veteichii) were found in the Poaceae specific clades of FD1 and FD2 respectively (Fig. 1). It had previously been found that the FD gene clade could be broadly classified into four subgroups, which were Poaceae specific FD1, Poaceae specific FD2, Poaceae specific FD3 and FDs obtained from eudicots as well as non-Poaceae members of monocots29.
Bamboo FD1 and FD2 genes are divergent in expression patterns with respect to tissues, diurnal conditions and seasons
The detailed expression analyses of BtFD1 and BtFD2 genes in diverse tissues, diurnal conditions and seasons may provide clues about their possible functionality. It is well established that in SAM, FT interacts with FD to form FAC and consequently floral meristem identity genes are activated to induce flowering45. Therefore, FD expression has primarily been observed in SAM tissues of Arabidopsis, rice, P. sativum, P. tremula x P. alba and P. aphrodite plants29,30,33,40,42,46. In addition, expression of FD1 was also detected in leaves of O. sativa29, T. aestivum36 and A. chinensis41 plants. In bamboo, the expression level of BtFD1 gene was highest in shoot apex. However, the expression of BtFD2 was very low in all the tissues. This is unlike rice, where FD2 was primarily expressed in leaves29. Like many other flowering genes, FD also was found diurnally regulated in rice34 and P. tremula x P. alba42. In bamboo, expression of BtFD1 in YLF was highest in the afternoon (4 pm), but in YLN it was in the morning (8am). In poplar, similar diurnal regulation of FD was observed in SD, where it attained its maximum expression at mid night, whereas no such pattern was observed in LD42. In contrary, the diurnal expression of BtFD2 remained consistently low throughout the day. Expression analyses across seasons also point towards a role of BtFD1in flower induction. Transcript accumulation of BtFD1 in the floral inductive tissue YLF began in autumn and reached the maximum level in winter, i.e. just before sprouting (Fig. 3). This observation was comparable to perennial dicots poplar and kiwifruit, where FD was transiently expressed just before flowering every year41,42. In contrary, expression of BtFD2 was almost negligible throughout the year. Taken together, the analysed expression data suggest that BtFD1 may perform important roles in flower and vegetative development of bamboo, whereas the function of BtFD2 is yet to be discovered.
A single amino acid change resulting into differential binding efficiency between bZIP domains of BtFD1/BtFD2 and CRE DNA
Sequence analyses and in silico characterization of the two BtFD proteins confirmed that they belong to bZIP transcription factor family. Among several different subfamilies of bZIPs, BtFDs were found to be homologous to CREB. Co-crystal structure of CRE DNA—CREB bZIP of Mus musculus35 (PDB ID IDH3) was chosen for homology modelling purpose. Generally, the CREB family bZIP members are capable to interact with A box (TACGTA), G box (CACGTG) or C box (GACGTC) elements present in the promoter region of their target genes causing transcriptional upregulation47,48. In plants, the FD1 members of CREB family are involved in the establishment of floral meristem identity30,36,39. Overall, the bZIP regions of BtFD1 and BtFD2 proteins differ in five different amino acid positions. Particularly one position (Ala146 is BtFD1 vs. Leu100 in BtFD2) at the crucial DNA binding site (NXXAAXXSR) was interesting. Therefore, it was asked whether any of these amino acid changes, in particular this single amino acid substitution, could have any impact on their DNA binding activity. Our in silico analyses revealed that the bZIP domains identified in BtFD1 and BtFD2 were capable to dimerise and form a bZIP structure. They also demonstrated specific interaction with the TGACGTCA sequence. In particular, Asn141, Arg149 of BtFD1 and Asn95, Arg103 of BtFD2 can directly interact with cognate DNA substrate. Similar interaction has also been found in maize39 and wheat36. EMSA analysis highlighted that the change of Ala146 in BtFD1 vs. Leu100 in BTFD2 resulted into ten times enhanced binding of BtFD1 than BtFD2. This may be the result of an additional, yet unfavourable contact between Leu100 of BtFD2 and dT residue of ACGT motif, apparent in the docked structure. It might be possible that Leu100 interfered with the interaction of target DNA by making a polar vs. non-polar interaction. In contrary, the shorter Ala146 residue, which was present in BtFD1 could not interfere and thus enabling higher DNA binding efficiency of BtFD1 (Fig. 5b,c).
Ectopic expression of BtFD1 severely suppressed vegetative growth and flowering in Arabidopsis, but BtFD2 did not
In order to study the functions of BtFD1 and BtFD2 genes in planta, transgenic alterations of these genes needed to be carried out. Altering activities of these genes in bamboo itself were difficult due to many reasons such as long-life cycle, difficulty with in vitro regeneration and unavailability of efficient transformation methods49,50. Therefore, BtFD1 and BtFD2 genes were ectopically expressed in Arabidopsis plants and their phenotypes were compared.
The vegetative growth of transgenic Arabidopsis plants overexpressing BtFD1 gene was severely suppressed with respect to the number and size of the rosette leaves (Fig. 6a–c). Similar phenotypes had been noticed when AtFD and AtFDP together were overexpressed in rice45 and also when poplar FDL1 was overexpressed in Populus tremula × tremuloides42,51. The involvement of FD in controlling vegetative growth has been observed in a pea loss-of-function mutant, demonstrating severe branching even after flower induction40. A few molecular players in connection to the growth retardation due to FD1 overexpression have been identified. For instance, in poplar, BRANCHED1 and 2 genes, which promote shoot growth by maintaining proper auxin and cytokinin levels were downregulated52,53. Overexpression of Arabidopsis FD and FDP in rice resulted in the down-regulation of many cell wall growth responsive genes such as EXTENSIN, EXPANSIN and XTH145. Similar to all these previous observations, in this study the p35S::BtFD1 Arabidopsis plants revealed reduced leaf and leaf epidermal cell sizes compared to p35S::BtFD2 and wild-type plants.
The role of FD1 in flower induction has already been established by a large body of literature and a variety of mutant phenotypes have been observed: (a) Delay in flowering was observed in the loss-of-function mutants of Arabidopsis30,37,38, pea40 and maize39, while early flowering was observed in the FD1 overexpressing lines of rice29,33 and Phalaenopsis46. (b) However, exceptions to this line of observation have also been noticed42,45,51. When AtFD and AtFDP together were overexpressed in rice, flowering and vegetative growth has been retarded45. Similarly, overexpression of poplar FDL1 in Populus tremula × tremuloides resulted into delayed flowering in SD42,51. Our results demonstrated that transgenic Arabidopsis plants overexpressing BtFD1 exhibited a delay in flowering time and numbers of flowers/plant compared to p35S::BtFD2 and WT plants (Supplementary Fig. S4a). However, expression of AtAP1 was remarkably higher in p35S::BtFD1 Arabidopsis than p35S::BtFD2 and WT plants. Similar observations were also reported in AtFD and AtFDP overexpressing lines of rice45 and FDL overexpressing lines of poplar51, which, nevertheless, led to late flowering phenotypes. Together, it can be concluded that timely expression of BtFD1 may be critical to perform its programmed flower specific role in planta, which was altered in the transgenic Arabidopsis plants constitutively overexpressing BtFD1 in a spatially and timely improper manner. Therefore, the apparent delay in flowering time could be an indirect effect of extensive suppression of vegetative growth, while in contrast, the flowering program is still enhanced based on the marker gene AtAP1 induction. It is already well accepted that flowering can only be induced after plants attain sufficient vegetative growth54.
The evolution of gene function within the FD family revealed the existence of functional redundancy in Arabidopsis45. In contrary, a clear functional diversification was noticed between the two rice FD genes OsFD1 vs. OsFD229. Our study revealed that the two bamboo FD genes imposed contrasting effects on shoot growth and flowering time, which may be mediated by two ways: (a) by acquiring expression divergence where BtFD1 maintained a flower associated expression pattern whereas expression level of BtFD2 was consistently low and (b) by adapting a single amino acid change (Ala146 vs. Leu 100) located in their DNA binding region which may cause a differential binding to their target protein AP1. Future studies are required to investigate the impact of residue alterations in the other four positions. Such single residue swapping was found sufficient to convert the flowering repressor TFL1 to an activator FT and vice versa by altered interaction with their interactor proteins55. Taken together, it can be concluded that regions involved in protein–protein or DNA–protein interactions can be potential targets to study the functional evolution of closely related homologous genes. Further studies are required to uncover whether BtFD1 is anyhow involved in long perennialism of bamboo and whereas its homolog BtFD2 evolved any additional function or required other interacting partner to be functional.
Materials and methods
Collection of Bambusa tulda tissues for gene expression analyses
Flowering tissues were obtained from three populations of B. tulda located in Shyamnagar, W.B. (SHYM7, SHYM16, 22.38° N. 88.40° E) and Bandel11 (BNDL22, 22.93° N. 88.38° E). Recurrent incidence of sporadic flowering was noticed every year in spring from 2015 to 2018. Corresponding voucher specimen were submitted to the Botanical Survey of India (B.S.I), Shibpur (deposition nos. 56A, 56B, 57A, 57B, 58A. 58B, 59A, 59B, 59C on 05.06.2015). To perform tissue specific gene expression analysis, six vegetative tissues such as young leaf from flowering (YLF) and nonflowering culm (YLN), culm sheath (CS), root (R), internode (I), shoot apex (SA) and two flowering tissues such as immature and mature floral buds (IFB, MFB) were collected. In order to perform diurnal expression analyses, YLF and YLN were collected at four different time points of a day- morning (8 am), noon (12 pm), afternoon (4 pm) and night (8 pm) for both long day (LD, 14 h light exposure, sunrise at 4:30 am and sunset at 6:30 pm) and short-day (SD, 11 h light exposure, sunrise at 6 am and sunset at 5 pm). Tissues were also collected in five different seasons: summer (April–June, 2017), monsoon (July–August, 2017) autumn (September–October, 2017), winter (November–January, 2017) and spring (February–March, 2018). At least three, independent biological replicates were used for each tissue stage/diurnal condition/season.
Isolation of nucleic acids and preparation of cDNA libraries
Isolation of genomic DNA was carried out from young, healthy leaves by using DNeasy Plant Mini Kit (QIAGEN, Germany). Total RNA was isolated by a combination of Trizol (INVITROGEN, USA) and RNAeasy Plant Mini Kit (QIAGEN, Germany) as per the procedure described before43,56. Samples were treated with DNase I enzyme (THERMO SCIENTIFIC, USA) to avoid genomic DNA contamination, if any. Quality and quantity of the samples were checked in a BioSpectrometer (EPPENDORF, Germany) and agarose-formamide gel elctrophoresis. Approximately 1 μg of total RNA was used for cDNA synthesis using verso cDNA synthesis kit (THERMO SCIENTIFIC, USA) following manufacturer’s protocol. For real time RT-qPCR analyses, 2 μl of tenfolds diluted stock solution of cDNA samples was used.
Analysing FD gene and amino acid sequences obtained from various genome databases
Rice gene sequences (OsFD1: OS09G36910 and OsFD2: OS06G50830) were used as queries to retrieve genomic as well as amino acid sequences of FD1 and FD2 genes available in various genome databases. BLASTP analyses were performed in Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html), Plaza_monocot_v4 (https://bioinformatics.psb.ugent.be/plaza/versions/plaza_v4_monocots/) and NCBI (https://www.ncbi.nlm.nih.gov) databases. All BLASTP hits were obtained using the set criteria of an E-value threshold ≤ e−10, identity ≥ 40% and length coverage with respect to the query sequence ≥ 40%. However, when multiple hits were obtained, only the best BLASTP hit was selected for further analyses. If no homologous genes were found using the set criteria, it is mentioned as ‘no hit found’ (NHF, Table 1).
Primer designing, PCR amplification and sequencing of B. tulda BtFD1 and BtFD2 genes
In order to obtain B. tulda genes, homologous sequences obtained from closely related monocot species were aligned and degenerate primers were designed from the conserved regions by using Primer3 program (http://bioinfo.ut.ee/primer3-0.4.0/, Supplementary Table S1). PCR amplification was conducted using high fidelity Phusion Taq DNA polymerase (THERMO SCIENTIFIC, USA). Amplified PCR products of desired molecular weight were gel purified by using GeneJET gel elution kit (THERMO SCIENTIFIC, USA) and cloned into TA vector (pGEM-T Easy Vector Systems, PROMEGA, USA) or blunt end vector (pJET PCR cloning kit, THERMO SCIENTIFIC, USA). Selection of bacterial colonies were done based on the blue-white screening and/or ampicillin sensitivity (100 µg/ml). Plasmids were isolated by GeneJET plasmid miniprep kit (THERMO SCIENTIFIC, USA). Sanger’s sequencing was undertaken and contigs were assembled by CAP3 (www. http://doua.prabi.fr/software/cap3) prior to submission to NCBI (MF983712, MH142577). The full length genomic and coding sequences (CDS) were analysed in the Gene Structure Display Server (GSDS, http://gsds.cbi.pku.edu.cn/index.php) to predict the gene models.
Sequence data and phylogenetic analyses
The FD gene sequences identified from B. tulda were used as query and BLASTP analyses were performed in NCBI (https://www.ncbi.nlm.nih.gov) database to identify its homologous sequences in related species. Amino acid sequences of BtFD1 and 2 genes were aligned with other homologous sequences using the Clustal Omega program (https: //www.ebi.ac.uk/Tools/msa/clustalo/). A phylogenetic tree was constructed by using the Neighbour Joining (NJ) method in MEGA7 tool57. Bootstrap analysis with values for 2000 replicates was conducted to estimate nodal support.
In silico modelling and docking studies
In silico analysis was performed to predict the possibility of binding between of bZIP domains of BtFD1 or BtFD2 to the ACGT motif. Due to unavailability of BtFD1 and BtFD2 crystal structures, amino acid sequences corresponding to their bZIP domains were first subjected to homology modelling by SwissModel (https://swissmodel.expasy.org). Both of them demonstrated significant sequence homology (36% identity) with their nearest structural homologue CRE binding protein known from Mus musculus35 (PDB ID IDH3). Therefore, it was chosen as template for modelling bZIPs of BtFD1 and BtFD2. Ramachandran analysis using Molprobity option from Swissmodel revealed 98.96% residues to be in favourable region for BtFD1 and 95% for BtFD2 and both were modelled in their dimeric state. The two bZIP models were then subjected to energy minimization using Chimera (https://www.cgl.ucsf.edu/chimera/). Ramachandran analysis through Procheck (https://servicesn.mbi.ucla.edu/PROCHECK/) and post energy minimization revealed complete inclusion of residues in favourable region for both the models. The energy minimized structures were then docked with 21 bp CRE DNA sequence (obtained from 1DH3 crystal structure) using NPdock (http://genesilico.pl/NPDock).
Site-directed mutagenesis for electrophoretic mobility shift assay (EMSA)
In order to validate the prediction of binding between bZIP domains of BtFD1 and BtFD2 with the ACGT motif, electrophoretic mobility shift assay was performed. The bZIP sequence obtained from Thalassiosira oceanica LOV photoreceptor (To_bZIP + LOV − TobZL) protein, was used for site directed mutagenesis to obtain bZIP mimics of BtFD1 and BtFD2 proteins. Pairwise sequence alignment between among bZIP regions of BtFD1, BtFD2 and TobZL revealed two amino acid differences at the DNA binding basic region (Fig. 5a). Double mutations leading to conversion of His > Ser144 and Lys > Ala146 were introduced in TobZL to mimic BtFD1 bZIP and His310 > Ser98 and Lys312 > Leu100 to mimic BtFD2 bZIP. Mutations were done using standard procedures to induce site directed mutagenesis and were verified by DNA sequencing (EUROFINS GENOMICS INDIA PVT. LTD).
Over-expression and purification of BtFD1 and BtFD2 bZIP mimics
The BtFD1 and BtFD2 bZIP mimics were cloned in pET28a expression vector, transformed in E. coli [BL21(DE3)C43] and grown at 37 °C. After isopropyl β-D-1-thiogalactopyranoside induction, bacterial cells were grown at 20 °C for overnight. Cells were then centrifuged and pellets were re-suspended in buffer containing 20 mM Tris (pH 8.0), 10 mM NaCl, 10% glycerol in presence of the protease inhibitor. Following sonication on ice and centrifugation, the supernatant was incubated with Ni–NTA agarose (QIAGEN, Germany) for 2 h. After washing in 10 mM imidazole containing re-suspension buffer, proteins were finally eluted with 250 mM imidazole. The eluted fractions were next pooled and excess imidazole was removed using PD10 desalting column (SIGMA). Proteins were concentrated and stored at − 20 °C in aliquots for future use.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay was carried out to study DNA-binding activity of BtFD1 and BtFD2 proteins. A 24 bp DNA fragment [5′ d(TGTAGCGTCTGACGTGGTTCCCAC) 3′ and complementary sequence] containing the consensus CREB binding site, TGACGT, were synthesized (INTEGRATED DNA TECHNOLOGIES). Lyophilized DNA strands (labeled and un-labelled) were suspended in nuclease free water and 10 μM of it was annealed by rapid heating at 95 °C followed by gradual cooling in annealing buffer (12 mM Tris; pH 8.0, 30 mM NaCl). A final concentration of 0.5 μM double stranded DNA was used in the protein DNA binding assay buffer (50 mM Tris–HCl pH 8.0, 20 mM NaCl, 0.5 mM DNA, 1.25 mM MgCl2, 20% glycerol). Serially diluted protein was added to the solution followed by an incubation at 22 °C for 1 h. The protein DNA complex along with the control set (only DNA) was resolved in 10% polyacrylamide gels at 180 V for 35 min. The gel was then stained with SyBr Gold (THERMO SCIENTIFIC, USA) in 0.5X TBE buffer for 40 min and imaged using a gel documentation system (BIORAD, USA).
Gene expression analyses by real time RT-qPCR
To perform real time RT-qPCR analyses, gene specific primers were designed from the coding sequences of the BtFD1 and BtFD2 genes using Primer3 program (http://bioinfo.ut.ee/primer3-0.4.0/, Supplementary Table S1). The real time RT-qPCR analyses were performed by using SsoAdvanced Universal SYBR Green Supermix (BIO-RAD, USA) and CFX connect real-time PCR detection system (BIO-RAD, USA). To confirm the absence of any primer dimers in the amplified products, a standard melt curve analysis was conducted. The BteIF4α and AtACT2 genes were previously identified as ideal reference gene for normalizing expression data obtained from Bambusa58 and Arabidopsis56, respectively. The relative fold change in gene expression level was calculated by the 2−ΔΔCt method59. The PCR amplification efficiency were measured for the five pairs of primers used in RT-qPCR. Two fold serial dilutions of the pooled cDNA templates were used to obtain standard curves for each primer pair. The amplification efficiency was analyzed using the formula60 10(−1/slope) − 1 × 100. The obtained percentage of efficiency was 95%-98%.
Gateway cloning of BtFD1 and BtFD2 genes
Gateway recombination sequences were tagged to the 5′ end of the primers to PCR amplify BtFD1 and 2 genes using Phusion Taq DNA polymerase enzyme (THERMO SCIENTIFIC, USA, Supplemental Table S1). Approximately 100 ng of gel-purified PCR fragments were recombined with 100 ng of pDONR221 donor vector using BP Clonase enzyme (INVITROGEN, USA). Reactions were transformed into E. coli (DH5α) and isolated plasmids were verified by DNA sequencing before recombination into the binary pAlligator2 vector providing The CaMV 35S promoter for expression61. Finally, the expression clones were mobilized to competent Agrobacterium tumefaciens (pGV3101/pMP90) by electroporation using a BIO-RAD Gene Pulser.
In planta transformation, selection, phenotypic characterization and statistical analysis
Approximately six-week-old A. thaliana (Col-0) plants were transformed by the floral dipping method62. Transformed T1 seeds were selected on the basis of green fluorescence of the GFP reporter gene61. The number and perimeter of the rosette leaves were measured from three independent T3 plants having single insertions in order to perform phenotypic comparisons with wild-type A. thaliana Col-0 plants grown in both long day (LD, 16-h light and 8-h dark) and short day (SD, 10-h light and 14-h dark) conditions for four weeks. One-way ANOVA was carried out in R software (version 3.4.4) to find the degree of significance with respect to the difference in leaf numbers and sizes among p35S::BtFD1, p35S::BtFD2 and WT plants. For the analyses of leaf number, twelve replicates were considered for each of p35S::BtFD1 and p35S::BtFD2 transgenic plants (3 transgenic lines and 4 individual plants), whereas for WT it was 4. For the analyses of leaf perimeter, 24 replicates were considered for each of the p35S::BtFD1 and p35S::BtFD2 transgenic plants (3 transgenic lines and 8 individual leaves), whereas for WT it was 8. Since comparisons among three genetic backgrounds of plants (WT, p35S::BtFD1 and p35S::BtFD2) were performed in pairs (3 pairs), adjusted p-values (Tukey’s HSD) were considered and expressed as p.adj. In case of two-way ANOVA, one factor was considered as the genetic background (WT, p35S::BtFD1 and p35S::BtFD2), whereas the other factor was the duration of light (LD vs. SD). Here also, adjusted p-values (Tukey’s HSD) were used for conducting pairwise comparisons among three genetic backgrounds of plants (WT, p35S::BtFD1 and p35S::BtFD2). In order to study if there is any significant change in flowering time among WT, p35S::BtFD1 and p35S::BtFD2 plants, total number of rosette leaves were counted during the time of flowering from 6 independent plants/genetic background and one-way ANOVA was carried out to test significance in difference. The adjusted p values were obtained via Tukey’s HSD.
In order to obtain kinetic pattern of the differences in leaf growth, the perimeter of the first true leaves were measured in four day intervals in SD by using photographs and ImageJ software63. In addition, histological observation was performed on leaf epidermal cells since it had previously been observed that a positive correlation exists between expansion of leaf lamina and size of epidermal cells64. It was observed in the light microscope using NIS elements software and DS-Qi2 NIKON camera and the perimeter of epidermal cells were obtained from the apical and basal parts of the first true leaves of 22-, 26-, and 30-day-old plants grown in SD. Ten epidermal cells obtained from leaves of three independent plants of WT, p35S::BtFD1 and p35S::BtFD2 were subjected to mixed three-way ANOVA to test significance in difference of epidermal cell sizes. The adjusted p values were obtained via Bonferroni correction. In order to observe epidermal cells in the light microscope, first true leaves were preserved in 10% formaldehyde: 50% ethanol: 5% acetic acid solution. Leaves were dipped in absolute ethanol and boiled for 30–45 s to remove chlorophylls and were subsequently stained with 0.01% toluidine blue.
References
Clark L. G., Londoño, X., Ruiz-Sanchez, E. Bamboo taxonomy and habitat. Trop. For. 10, 1–3 (2015).
Kellogg, E. A. Flowering Plants · Monocotyledons (Springer XIII, 1998).
Janzen, D. Why Bamboos Wait So Long to Flower. Ann. Rev. Ecol. Syst. 7, 347–391 (1976).
Bhattacharya, S., Das, M. & Pal, A. Morphological and molecular characterization of Bambusa tulda with a note on flowering. Ann Bot. 98, 529–535 (2006).
Bhattacharya, S., Ghosh, J., Das, M. & Pal, A. Morphological and molecular characterization of Thamnocalamus spathiflorus subsp. spathiflorus at population level. Plant Syst Evol. 282, 13–20 (2009).
Putterill, J., Laurie, R. & Macknight, R. It’s time to flower: the genetic control of flowering time. Bioassays 26, 363–373 (2004).
Song, Y. H., Shim, J. S., Kinmonth-Schultz, H. A. & Imaizumi, T. Photoperiodic flowering: Time measurement mechanisms in leaves. Annu. Rev. Plant Biol. 66, 441–464 (2015).
Hung, C., Qiu, J., Sun, Y., Chen, J. & Kittur, F. S. Gibberellin deficiency is responsible for shy-flowering nature of Epipremnum aureum. Sci Rep. 6, 1–11 (2016).
Bouché, F., Woods, D. P., Amasino, R. M. & Wisconsin, F. B. Winter memory throughout the plant kingdom: Different paths to flowering. Plant Physiol. 173, 27–35 (2017).
Zhou, J. C. Y. & Xie, T. L. C. Research progress on the autonomous flowering time pathway in Arabidopsis. Physiol. Mol. Biol. Plants 23, 477–485 (2017).
Biswas, P., Chakraborty, S., Dutta, S., Pal, A. & Das, M. Bamboo flowering from the perspective of comparative genomics and transcriptomics. Front. Plant Sci. 7, 1–10 (2016).
Liu, M. et al. Transcriptome Sequencing and De Novo Analysis for Ma Bamboo (Dendrocalamus latiflorus Munro) Using the Illumina Platform. PLoS ONE 7, 1–11 (2012).
Zhang, X., Zhao, L., Larson-rabin, Z., Li, D. & Guo, Z. D. Novo sequencing and characterization of the floral transcriptome of Dendrocalamus latiflorus (Poaceae: Bambusoideae). PLoS ONE 7, e42082 (2012).
Gao, J. et al. Characterization of the floral transcriptome of Moso bamboo (Phyllostachys edulis) at different flowering developmental stages by transcriptome sequencing and RNA-seq analysis. PLoS ONE 9, e98910 (2014).
Ge, W. et al. Main regulatory pathways, key genes and microRNAs involved in flower formation and development of moso bamboo (Phyllostachys edulis). Plant Biotechnol J. 15, 82–96 (2017).
Zhao, H. et al. Comprehensive analysis of multi-tissue transcriptome data and the genome-wide investigation of GRAS family in Phyllostachys edulis. Sci. Rep. 6, 1–15 (2016).
Jiao, Y. et al. Comparative transcriptomic analysis of the flower induction and development of the Lei bamboo (Phyllostachys violascens). BMC Bioinform. 20, 687 (2019).
Wysocki, W. P., Ruiz-Sanchez, E., Yin, Y. & Duvall, M. R. The floral transcriptomes of four bamboo species (Bambusoideae; Poaceae): Support for common ancestry among woody bamboos. BMC Genom. 17, 1–14 (2016).
Li, Y. et al. De novo sequencing of the transcriptome reveals regulators of the floral transition in Fargesia macclureana (Poaceae). BMC Genom. 20, 1035 (2019).
Wang, X., Zhang, X., Zhao, L. & Guo, Z. Morphology and quantitative monitoring of gene expression patterns during floral induction and early flower development in Dendrocalamus latiflorus. Int. J. Mol. Sci. 15, 12074–12093 (2014).
Shih, M. et al. BeMADS1 is a key to delivery MADSs into nucleus in reproductive tissues: De novo characterization of Bambusa edulis transcriptome and study of MADS genes in bamboo floral development. BMC Plant Biol. 14, 179 (2014).
Tian, B., Chen, Y., Yan, Y. & Li, D. Isolation and ectopic expression of a bamboo MADS-box gene. Chin. Sci. Bull. 50, 217–224 (2005).
Hisamoto, Y., Kashiwagi, H. & Kobayashi, M. Use of flowering gene FLOWERING LOCUS T (FT) homologs in the phylogenetic analysis of bambusoid and early diverging grasses. J. Plant Res. 121, 451–461 (2008).
Yang, Z. et al. Identifcation and Characterization of the PEBP Family Genes in Moso Bamboo (Phyllostachys heterocycla). Sci. Rep. 9, 14998 (2019).
Zeng, H. Y., Lu, Y. T., Yang, X. M., Xu, Y. H. & Lin, X. C. Ectopic expression of the BoTFL1-like gene of Bambusa oldhamii delays blossoming in Arabidopsis thaliana and rescues the tfl1 mutant phenotype. Genet. Mol. Res. 14, 9306–9317 (2015).
Liu, S. N., Zhu, L. F., Lin, X. C. & Ma, L. Y. Overexpression of the repressor gene PvFRI-L from Phyllostachys violascens delays flowering time in transgenic Arabidopsis thaliana. Biol. Plant. 60, 401–409 (2016).
Lin, E. P. et al. Identification and characterization of two Bamboo (Phyllostachys praecox) AP1/SQUA-like MADS-box genes during floral transition. Planta 231, 109–120 (2009).
Jakoby, M. et al. bZIP transcription factors in Arabidopsis. Trend. Plant Sci. 7, 106–111 (2002).
Tsuji, H., Nakamura, H., Taoka, K. I. & Shimamoto, K. Functional diversification of FD transcription factors in rice, components of florigen activation complexes. Plant Cell Physiol. 54, 385–397 (2013).
Wigge, P. A. et al. Integration of spatial and temporal information during floral induction in Arabidopsis. Science 309, 1056–1059 (2005).
Tamaki, S., Matsuo, S., Wong, H. L., Yokoi, S. & Shimamoto, K. Hd3a protein is a mobile flowering signal in rice. Science 316, 1033–1036 (2014).
Komiya, R., Yokoi, S. & Shimamoto, K. A gene network for long-day flowering activates RFT1 encoding a mobile flowering signal in rice. Development 3450, 3443–3450 (2009).
Taoka, K. I. et al. 14-3-3 proteins act as intracellular receptors for rice Hd3a florigen. Nature 476, 332–335 (2011).
Brambilla, V. et al. Antagonistic transcription factor complexes modulate the floral transition in rice. Plant Cell 29, 2801–2816 (2017).
Schumacher, M. A., Goodman, R. H. & Brennan, R. G. The structure of a CREB bZIP somatostatin CRE complex reveals the basis for selective dimerization and divalent cation-enhanced DNA binding. J. Biol. Chem. 275, 35242–35247 (2000).
Li, C. & Dubcovsky, J. Wheat FT protein regulates VRN1 transcription through interactions with FDL2. Plant J. 55, 543–554 (2008).
Jaeger, K. E., Pullen, N., Lamzin, S., Morris, R. J. & Wigge, P. A. Interlocking feedback loops govern the dynamic behavior of the floral transition in Arabidopsis. Plant Cell 25, 820–833 (2013).
Koornneef, M., Hanhart, C. J. & van der Veen, J. H. A genetic and physiological analysis of late flowering mutants in Arabidopsis thaliana. Mol. Gen. Genet. 229, 57–66 (1991).
Muszynski, M. G. et al. Delayed Flowering1 encodes a basic leucine zipper protein that mediates floral inductive signals at the shoot apex in maize. Plant Physiol. 142, 1523–1536 (2006).
Sussmilch, F. C. et al. Pea VEGETATIVE2 is an FD homolog that is essential for flowering and compound inflorescence development. Plant Cell 27, 1046–1060 (2015).
Varkonyi-Gasic, E. et al. Homologs of FT, CEN and FD respond to developmental and environmental signals affecting growth and flowering in the perennial vine kiwifruit. New Phytol. 198, 732–746 (2013).
Parmentier-Line, C. M. & Coleman, G. D. Constitutive expression of the Poplar FD-like basic leucine zipper transcription factor alters growth and bud development. Plant Biotechnol. J. 14, 260–270 (2016).
Dutta, S. et al. Identification, characterization and gene expression analyses of important flowering genes related to photoperiodic pathway in bamboo. BMC Genom. 19, 1–19 (2018).
Mohan Ram, H. Y. & Hari, G. B. Some observations on the flowering of bamboos in Mizoram. Curr. Sci. 50, 708–710 (1981).
Jang, S., Li, H. Y. & Kuo, M. L. Ectopic expression of Arabidopsis FD and FD PARALOGUE in rice results in dwarfism with size reduction of spikelets. Sci. Rep. 7, 1–15 (2017).
Jang, S., Choi, S. C., Li, H. Y., An, G. & Schmelzer, E. Functional characterization of Phalaenopsis aphrodite flowering genes PaFT1 and PaFD. PLoS ONE 10, 1–29 (2015).
Foster, R., Izawa, T. & Chua, N. H. Plant bZIP proteins gather at ACGT elements. FASEB J. 8, 192–200 (1994).
Izawa, T., Foster, R. & Chua, N. H. Plant bZIP protein DNA binding specificity. J. Mol. Biol. 230, 1131–1144 (1993).
Das, M. & Pal, A. In vitro regeneration of Bambusa balcooa Roxb.: Factors affecting changes of morphogenetic competence in the axillary buds. Plant Cell. Tissue Organ Cult. 81, 109–112 (2005).
Das, M., Bhattacharya, S., Singh, P., Filgueiras, T. S. & Pal, A. Bamboo taxonomy and diversity in the era of molecular markers. Adv. Bot. Res. 47, 225–268 (2008).
Tylewicz, S. et al. Dual role of tree florigen activation complex component FD in photoperiodic growth control and adaptive response pathways. Proc. Natl. Acad. Sci. USA 112, 3140–3145 (2015).
Dun, E. A., Germain de Saint, A., Rameau, C. & Beveridge, C. A. Antagonistic action of strigolactone and cytokinin in bud outgrowth control. Plant Physiol. 158, 487–498 (2012).
Ferguson, B. J. & Beveridge, C. A. Roles for auxin, cytokinin, and strigolactone in regulating shoot branching. Plant Physiol. 149, 1929–1944 (2009).
Jung, J. H., Seo, P. J., Kang, S. K. & Park, C. M. miR172 signals are incorporated into the miR156 signaling pathway at the SPL3/4/5 genes in Arabidopsis developmental transitions. Plant Mol. Biol. 76(1–2), 35–45 (2011).
Hanzawa, Y., Money, T. & Bradley, D. A. single amino acid converts a repressor to an activator of flowering. Proc. Natl. Acad. Sci. USA 102, 7748–7753 (2005).
Das, M. et al. Expression pattern similarities support the prediction of orthologs retaining common functions after gene duplication events. Plant Physiol. 171, 2343–2357 (2016).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33(7), 1870–1874 (2016).
Chakraborty, S., Dutta, S., Biswas, P. & Das, M. Identification of candidate reference genes in tropical bamboos stable across species, tissues, and developmental stages. Biol. Plant. 63, 253–261 (2019).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Ginzinger, D. G. Gene quantification using real-time quantitative PCR: An emerging technology hits the mainstream. Exp. Hematol. 30, 503–512 (2002).
Bensmihen, S. et al. Analysis of an activated ABI5 allele using a new selection method for transgenic Arabidopsis seeds. FEBS Lett. 561, 127–131 (2004).
Clough, S. J. & Bent, A. F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 (1998).
Schneider, C. A., Rasband, W. S., Eliceiri, K. W. & Instrumentation, C. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2017).
Coockson, S. J., Radziejwoski, A. & Granier, C. Cell and leaf size plasticity in Arabidopsis: What is the role of endoreduplication?. Plant Cell Environ. 29, 1273–1283 (2006).
Acknowledgements
The research results reported in this paper are funded by Council of Scientific and Industrial Research, India [38(1386)/14/EMR-II], [38(1493)/19/EMR-II], Department of Biotechnology, Govt. of India (BT/PR10778/PBD/16/1070/2014) and FRPDF grant of Presidency University. A part of the study was sponsored by the Alexander von Humboldt Foundation, Germany. SD acknowledges a SRF fellowship from CSIR, India [08/l55(0055)/ 2019 EMR-I]. AD thanks DBT, Govt. of India for her PhD fellowship. DM acknowledges research funding from DBT, Govt. of India [BT/PR26435/BRB/10/1627/2017]. We thank Prof. Amita Pal for her thoughtful suggestions to improve the quality of the manuscript.
Author information
Authors and Affiliations
Contributions
M.D., A.R.S. and S.D. designed the experiments; P.B. amplified FD genes; S.D. and S.C. performed gene expression analysis; D.M. performed in silico DNA protein interaction; A.D. carried out mutagenesis, expression and purification of FD1-FD2 mimics and conducted EMSA; M.D., A.R.S. and B.G. performed in planta transformation, A.R.S. and S.D. performed phenotyping and experimental validation, S.G. performs statistical analyses related to characterization of transgenic plants, M.D. wrote the manuscript with help from all the co-authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dutta, S., Deb, A., Biswas, P. et al. Identification and functional characterization of two bamboo FD gene homologs having contrasting effects on shoot growth and flowering. Sci Rep 11, 7849 (2021). https://doi.org/10.1038/s41598-021-87491-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-87491-6
This article is cited by
-
Ectopic expression of a bamboo SVP-like gene alters flowering time and floral organs in Arabidopsis thaliana
Plant Cell, Tissue and Organ Culture (PCTOC) (2022)
-
Evidence of stress induced flowering in bamboo and comments on probable biochemical and molecular factors
Journal of Plant Biochemistry and Biotechnology (2021)
-
Genomic insights into growth and development of bamboos: what have we learnt and what more to discover?
Trees (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
