
Abstract
Development of a self-enhanced solid-state ECL platform creates a straightforward experimental design for the fabrication of point-of-care applications. Herein, we develop a promising method for self-enhanced solid-state ECL platform of polyluminol gold nanocomposite on glassy carbon electrode [(PL-Au)nano/GCE] via simple one-step electrochemical deposition process without involving any additional co-reactants. The presence of gold nanoparticles (AuNPs) augments the electron transfer kinetics of PL (polyluminol) and enhances the solid-state ECL intensity and promotes label-free, excellent sensitivity, and selectivity to detect Hg2+ in physiological pH through signal-on mode. Unlike pristine PL/GCE, electrochemically co-deposited AuNPs in the (PL-Au)nano/GCE composite, enable the co-reactant accelerator by improving the catalytic activity of PL towards oxygen reduction reaction (ORR) yielding in-situ ROS (co-reactant) generation. Further, the ECL intensity of (PL-Au)nano/GCE composite, gradually increases with each addition of Hg2+ ion. This is because of the formation of an amalgamation of Au-Hg on (PL-Au)nano/GCE composite surface which further accelerates the yield of in-situ ROS and enhances the intensity of ECL. Whereas no ECL signals changes were observed for PL/GCE composite. The proposed self-enhanced solid-state ECL platform is selectively sensing the Hg2+ ion in the linear range of 0.3–200 nM with a detection limit of 0.1 nM. The demonstrated (PL-Au)nano/GCE platform might pave new avenues for further studies in the solid-state ECL platform which could be more useful in on-site monitoring of clinical bioassay and immunosensors.
Similar content being viewed by others

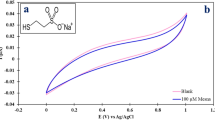

Introduction
Electrogenerated chemiluminescence (ECL) is a process of light emission which is due to an energetic interaction between electrogenerated species on electrode and electrolyte interface1. ECL has a great advantage over chemiluminescence (CL) because of low background signals, simple optical setup, good spatial–temporal control, and versatility2,3. Recently, ECL becomes a popular and widely used analytical technique in clinical diagnostics, material science, and environmental monitoring4,5. Even though Ru(bpy)32+ based ECL systems such as Ru(bpy)32+/tri-n-propylamine (TPrA)6, Ru(bpy)32+-oxalate (C2O42−)7 and Ru(bpy)32+-hydrogen peroxide (H2O2)8,9 are more acitve ECL systems in reported litrature, still luminol-H2O210 based ECL systems have more attractive glance because of its non-toxicity, cost-efficient, and high quantum yield with low oxidation potential11,12. The luminescent property of the luminol-H2O2 system mostly depends on its inherent chemical or electrochemical reaction through the intermediates of oxygen species like OH∙, OH−, and O2−13,14. The major limitation of luminol’s ECL property is hampered by its poor aqueous solubility, alkaline mediated ECL dependency. To overcome this, water-soluble luminol derivatives were chemically synthesized with enhanced ECL intensity in an aqueous electrolyte15. However, the adopted synthesis involves multiple steps, desires tedious reactions without scalability. Moreover, the co-reactant like H2O2 is also not quite stable at room temperature which also suppresses the efficiency of the ECL signal of luminol16. One can overcome this problem by generating the reactive oxygen species (ROS) by using co-reactant accelerators which can produce more ROS through the reduction of dissolved oxygen and enhances the stability of the ECL signal17. In ECL, the co-reaction or co-reactant accelerators are playing a crucial role in boosting the ECL intensity of luminophore by dissociating the co-reactant into active radicals18. For example, the ECL intensity of luminol in O2 saturated electrolyte increased by 2-fold by the co-reactant accelerator strategy19.
Because of mass transport limitation, a homogeneous phase or solution based luminol system always inhibits the efficiency of ECL emission, impeding the low molecular detection of selected analytes20,21,22. Use of heterogeneous or solid-state ECL has several advantages such as a minimum amount of luminophores is sufficient, simplifies the experimental setup, enhance more ECL intensity23,24,25. In this context, luminol is an aniline monomer derivative having an ECL inert –NH2 group facilitating the electro-polymerization in an acidic solution to form a stable PL film on the electrode surface26. However, the PL films have less electrical conductivity in neutral or alkaline solution, which needs further improvisation for highly efficient luminol system. Despite few attempts were taken in functionalizing the luminol system in solution state with metal NPs, till now no efforts were taken to hybridize PL films particularly at solid-state, which eventually have potential scope for point-of-care/on-site application.
Mercury ion (Hg2+) is a heavy metal known for the individual as well as environmental toxicity27. The presence of Hg2+ in the human body causes brain damage and other chronic diseases28,29. Enzymes like horseradish peroxidase30, glucose oxidase31, invertase32 and urease33 have a strong affinity with Hg2+ which inhibits the functions of the enzymes of the human body. Therefore, the sensitive and selective method requires in detecting Hg2+ at a lower level is highly beneficial for environmental application as well as healthcare. Though spectroscopic and chromatographic techniques are well documented as well as in practice for detection of Hg2+, nevertheless modern analytical methods demand highly sensitive yet selective portable assay platform. Even though luminol based ECL property is established for Hg2+ estimation, still it requires specific bio-receptors like DNA34 and enzymes35 for selective ECL quenching. Thus, there is a potential need for the development of a label-free solid-state ECL platform beneficial for selective detection of Hg2+ ions.
Herein, a simple one-step electrochemical strategy is established for the preparation of (PL-Au)nano/GCE which displayed the self-enhanced solid-state ECL signal, thereby enabling label-free detection of Hg2+ via signal-on ECL mode. Incorporation of electrochemically co-deposited AuNPs with PL films acts as a co-reactant accelerator to enhancing the ECL intensity by producing more ROS. The optimized condition is selective for Hg2+ detection without influence from other metal ions. The obtained results were superior/specific to Hg2+ ion over the pristine PL/GCE and other noble metal composites (PL-Pt)nano/GCE and (PL-Ag)nano/GCE. Hence, the in-situ generated ROS is utilized as co-reactant, the proposed methodology does not require any addition of co-reactant into electrolyte to study the ECL of PL.
Experimental section
Chemicals
Luminol (97%), Disodium hydrogen phosphate (Na2HPO4∙7H2O), Sodium dihydrogen phosphate mono hydrate (NaH2PO4∙H2O), Tetrachloroauric (III) acid (99.9%), Hexachloroplatinic (IV) acid hydrate (99.9%), Sulphuric acid (18.3 M), Mercury chloride (HgCl2), Silver nitrate (99.99%) and all other metal salts and benzoquinone were purchased from Alfa aesar. All the chemicals were used without any further purification. O2 gas saturated electrolyte solution in electrochemical cell is achieved by continuous purging of O2 (purity of 99.99%) gas into the electrolyte solutions (0.1 M PBS) using typical design of four neck electrochemical cell with gas purging setup using capillary serves as inlet tube that inserted into the electrolytic cell which is directly connected to O2 gas cylinder. O2 gas was purged into the electrolyte solution for 45 min to complete saturation. The similar method used to saturate the electrolyte with argon gas (Ar) of 99.99% purity. Milli-Q water (18.2Ω) used as a solvent for preparing the electrolyte solution. 0.5 M H2SO4 stock solution was prepared by diluting 18.3 M H2SO4.
Electrochemistry and ECL measurements
Commercially available glassy carbon electrode (GCE) with 0.07065 cm2 surface area serves as working electrode, platinum foil as the counter electrode and Ag/AgCl is used as reference electrode respectively. Cyclic voltammetry (CV) and potential step experiments were performed with an Autolab electrochemical workstation (EcoChemie, The Netherlands). The ECL along with CV signals is measured simultaneously with a photomultiplier tube (PMT, Hamamatsu H9305-04). The PMT was held at − 500 V with a high-voltage power supply. The photo current generated at the PMT was converted to a voltage using an electrometer system (model 6517, Keithley, Cleveland, OH) and connected to the Autolab via an analog-to-digital converter (ADC).
Preparation of PL/GCE and (PL-Au)nano/GCE
PL-Au nano-composite was electrochemically deposited on GCE as follows. Initially, GCE was successively polished with Al2O3 slurry (0.3, 0.05 µm) then sonicated for 5 min in an ultrasonic bath with distilled water at room temperature. Further GCE was electrochemically cleaned in 0.5 M H2SO4 by cycling at 0.1 V/s from 0 to 1.2 V vs. Ag/ AgCl for 10 cycles. After that, polished GCE was immersed in an electrochemical cell containing 1 mM luminol in 0.5 M H2SO4 and electrochemically treated about 20 cycles in the range of 0 to 1 V at the scan rate of 0.1 V/s (Fig. 1A). After the electrochemical treatment, the electrode was washed thoroughly with milli-Q water, the modified electrode was termed to as PL/GCE. Similar way (PL-Au)nano/GCE prepared by taking 1 mM luminol + 1.5 mM HAuCl4 in 0.5 M H2SO4 (Fig. 1B). The electrode modification process is also represented in the Scheme 1A. The similar procedure was followed to deposit (PL-Pt)nano and (PL-Ag)nano on GCE using 1 mM luminol + 1.5 mM PtCl42− and 1 mM luminol + 1.5 mM AgNO3 respectively, in 0.5 M H2SO4 which can be referred as (PL-Pt)nano/GCE and (PL-Ag)nano/GCE.

Repetitive CVs of 1 mM luminol (A) and 1 mM luminol + 1.5 mM HAuCl4 (B) in 0.5 M H2SO4 at the scan rate of 0.1 V/s. FESEM images of PL/GCE (C) and (PL-Au)nano/GCE (D). The XPS spectrum (4f5/2 and 4f7/2 of Au) of (PL-Au)nano/GCE before and after etching (E,F) respectively.

Schematic illustration of PL/GCE and (PL-Au)nano/GCE preparation (A) and ECL mechanism of (PL-Au)nano/GCE before and after Hg2+ addition (B).
Characterization techniques
We used Field emission scanning electron microscope (FESEM, Supra 55 VP, Carl Zeiss), Energy dispersive X-ray (EDX, Oxford Instruments X-MAX, 20 mm2) analysis, and Atomic force microscope (AFM, Agilent technologies 5500 instruments) technique in order to know the morphological changes and elemental presence after modification of GCE. The X-ray photoelectron spectroscopic (XPS) technique used to predict the state of elements present in the outermost part of composite by using Theta Probe AR-XPS System, Thermo Fisher Scientific (UK).
ECL spectrum
ECL spectrum is recorded using the optimized PMT voltage as 950 V and slit width is 20 nm in spectrofluorimeter. Also, applied a constant potential pulse at 0.6 V vs Ag/AgCl on (PL-Au)nano/GC plate (1 × 1 cm2) in O2 saturated 0.1 M PBS at pH 7.4.
Results and discussions
Electrochemical studies of PL/GCE and (PL-Au)nano/GCE
Figure 1A,B depicts the cyclic voltammetry of PL and (PL-Au)nano growth patterns on a glassy carbon substrate. As seen in Fig. 1A, a sharp increase in peak current at 0.9 V for the 1st cycle of CV which is due to the luminol oxidation where the radical polymerization of luminol starts and decreased in the peak current (at 0.9 V) during the subsequent electrochemical cycling. Moreover, there is a reversible redox peak growth which is due to the reduction (EPc = 0.6 V) and oxidation (Epa = 0.7 V) of PL. The redox peaks current is gradually increases for initial few cycles (up to 10 cycles) and then exhibit a stable redox response without further increase in redox peak current. Interestingly, the existence of HAuCl4 with luminol causes incremental redox peak current density of PL during electrochemical cycling without change in inherent redox potential. In addition, a small reduction peak at 0.8 V observed which is due to the reduction of Au3+ to Au0 (Fig. 1B). To ensure this particular reduction reaction a control experiment with pristine HAuCl4 was performed. To this only HAuCl4 is dissolved in 0.5 M H2SO4 without luminol yield at sharp reduction peak at exactly 0.8 V and decreased upon continuous electrochemical cycling (Supplementary Fig. S1A). After electrochemical cycling, the typical redox response of modified GCE (AuNPs/GCE) in 0.5 M H2SO4 confirms the formation of AuNPs on GCE (Supplementary Fig. S1B). The CV of typical stable redox response of PL/GCE and (PL-Au)nano/GCE clearly seen in the potential window of 0–1 V in 0.5 M H2SO4 solution (Supplementary Fig. S2A,B), evidencing the deposition of PL36. However, the sweeping potential region is extended up to 1.7 V (Supplementary Fig. S3) to validate the existence of typical redox characteristics of AuNPs in line with redox peaks of PL (at 0.7 V and 0.6 V). From the second cycle onwards the redox peak of PL is started to disappear with retention of AuNPs characteristic redox behavior, suggesting an over oxidation potential resulted from either leaching or deactivating the PL in 0.5 M H2SO4. And also, the calculated AuNPs reduction peak charge in AuNPs/GCE (Supplementary Fig. S1B) is almost 2-fold less when compared with the reduction peak charge of AuNPs in the (PL-Au)nano/GCE (Supplementary Fig. S3). Moreover, the peak current and charge associated with oxidation (at 0.7 V) and reduction (at 0.6 V) peak of PL in (PL-Au)nano/GCE is also higher than that of pristine PL/GCE (Supplementary Table S1). This is because of the simultaneous growth deposition of AuNPs and PL film on GCE.
The surface morphology of electrochemically deposited PL/GCE and (PL-Au)nano/GCE was studied by using FE-SEM (Fig. 1C,D). PL/GCE shows microstructures of polymer islands (Fig. 1C), on the other hand (PL-Au)nano/GCE exhibit homogenous spherical nanostructures with an average size distribution of ~ 60 nm (Fig. 1D), evidencing the existence of nanocomposite on the GCE. To elucidate the elemental compositions and surface chemistry of the nanocomposite on GCE surface an XPS study was performed. Survey spectra of the prepared platform were presented in Supplementary Fig. S4. The high-resolution XPS spectra of Au 4f7/2 and Au 4f5/2 is denoted in Fig. 1E. The observed binding energies are comparable with the standard values of pure gold viz., 83.8 eV (CAS No.7440-57-5) and 87.43 eV (CAS No.7440-57-5) corresponded to 4f7/2 and 4f5/2, respectively. In order to understand the localization pattern of Au and PL films an in-situ etching was performed using XPS analysis. Figure 1F illustrates the XPS spectrum of etched (PL-Au)nano/GCE with an amplified signal intensity of Au 4f7/2 and 4f5/2, revealing enhanced exposure of Au via stripping of PL. This was further supported by the existence of weaker O1s and N1s peaks (Supplementary Fig. S5).
To complement the surface topography and elemental structure an AFM and EDX spectral measurements were performed and the results are presented in Supplementary Fig. S6. The EDX of PL/GCE is showing C, N, and O elements (Supplementary Fig. S6A) whereas Au along with C and O presents in (PL-Au)nano/GCE composite (Supplementary Fig. S6B). This result was evidencing that PL and AuNPs were co-deposited on the GCE surface. In addition, the AFM analysis is also displayed macrostructure kind of morphology for PL/GCE (Supplementary Fig. S6C), but nanostructured with spherical shapes presents in (PL-Au)nano/GCE composite (Supplementary Fig. S6D) as similar with FE-SEM. Electrical conductivity and charge transfer kinetics for the prepared (PL-Au)nano/GCE was measured using electrochemical impedance spectroscopy (EIS). The calculated charge transfer resistance (∆Rct) derived from the Nyquist plot of PL/GCE and (PL-Au)nano/GCE is 4076 Ω and 276 Ω, respectively (Supplementary Fig. S7). The lesser value of Rct clearly confirms that the (PL-Au)nano/GCE exhibit better electrical conductivity and charge transfer kinetic than the pristine PL/GCE.
ECL experiments and ECL mechanism
Unlike the redox response of (PL-Au)nano/GCE in 0.5 M H2SO4 (Supplementary Fig. S2A,B), the simultaneously recorded CV showed an irreversible peak at − 0.4 V and 0.6 V which is due to the dissolved oxygen reduction reaction (ORR)37 and PL oxidation peak (Fig. 2A,a) in O2 gas saturated 0.1 M PBS (pH 7.4) during scanning from 0 to − 0.8 V to + 1 V. In contrast, the pristine PL/GCE showed oxidation peak at 0.6 V but the oxygen reduction peak observed at high cathode potential of − 0.65 V (Fig. 2A,b). The less cathode peak potential shift approximately − 0.25 V and high reduction current density for dissolved O2 reduction clearly indicates that the (PL-Au)nano/GCE is highly catalytic than pristine PL/GCE. To understand the role of AuNPs, the PL film also deposited on a polycrystalline gold surface (PL/pc-Au) which exhibits the redox response in O2 gas saturated 0.1 M PBS (pH 7.4) (Fig. 2A,c). Although the peak potentials of PL oxidation and ORR are similar to the (PL-Au)nano/GCE, the peak current density of PL oxidation and ORR is quite decreased. This further confirms the (PL-Au)nano/GCE shows superior electrocatalytic behavior towards ORR. The simultaneous record of ECL from these modified electrodes shown in Fig. 2B.

Simultaneously obtained CV (A) and its corresponding ECL responses (B) of (PL-Au)nano/GCE (a), PL/GCE (b), PL/pc-Au (c), in O2 saturated and (PL-Au)nano/GCE (d) in Argon saturated 0.1 M PBS (pH 7.4) at 0.1 V/s.
As expected, a high intense ECL peak was observed at 0.6 V where exactly an oxidation of PL peak exists (Fig. 2A,a) in (PL-Au)nano/GCE. The relative intensity of the observed ECL signal of (PL-Au)nano/GCE is almost three times higher than PL/GCE (Fig. 2B,b) and PL/pc-Au (Fig. 2B,c) composites. Under completely argon gas saturated 0.1 M PBS, the (PL-Au)nano/GCE shows the disappearance of ORR and ECL peak (Fig. 2A,d,B,d). All these results clearly confirm that, obtained ECL signals are totally dependent on the concentration of dissolved O2 present in the electrolyte solution. The in-situ generated reactive oxygen species (ROS) from dissolved O2 act as co-reactant which eventually react with PL anion to produce ECL signal. The ECL enhancement of (PL-Au)nano/GCE is perhaps due to the presence of AuNPs which behave as a co-reactant accelerator by producing the ROS radicals via ORR. In order to understand the significance of AuNPs on PL film, the same PL film electrodeposited in the presence of H2PtCl4 and AgNO3 with in the same experimental conditions of (PL-Au)nano/GCE (Supplementary Fig. S8A,B). Interestingly, there is no ECL observed for (PL-pt)nano/GCE and (PL-Ag)nano/GCE composites (Supplementary Fig. S8C,D). Owing to its well-known catalytic behavior of Pt in ORR, the reduction of O2 to H2O occurs via a direct 4e− pathway and it may not follow the 2e− pathway which is necessary for H2O2 and ROS generation38,39. Even though luminol and Ag have good interaction and enables excellent chemiluminescence activity in presence of H2O240. The present experimental conditions do not show any ECL for (PL-Ag)nano/GCE suggesting (PL-Ag)nano/GCE film could not act as ROS generator in solid-state ECL platform. Thus, the only AuNPs present in (PL-Au)nano/GCE has effective co-competence of a co-reactant accelerator to generate more ROS thereby promoting the ECL activity. Further, the effect of potential window on ECL intensity was studied by varying the initial potentials. Figure 3A,B shows the CV and corresponding ECL responses of (PL-Au)nano/GCE composite at various scan directions such as 0 to 1 V, − 0.3 to 1 V, − 0.6 to 1 V, and − 0.8 to 1 V respectively in O2 saturated 0.1 M PBS (pH 7.4). As illustrated in Fig. 3B, ECL intensities of (PL-Au)nano/GCE composite vary on change of potential scan direction. We observed a high intense ECL signal for − 0.8 to 1 V (Fig. 3B,d) scans direction, because at this particular direction maximum amount of ROS generated by the reduction of O2 (Fig. 3A,d). A small intense ECL signal is observed when we scan 0–1 V. Overall, the ECL intensity of (PL-Au)nano/GCE composite gradually increases by changing the potential scan towards a more negative to positive direction and concluded that ECL of (PL-Au)nano/GCE composite depends on the in-situ generated ROS. The ROS further oxidizes during anodic direction then reacts with poly luminol anion to emit light. The charge associated with ORR peak of Fig. 3A,b–d is calculated by integrating the peak area and the value is given in Supplementary Table S2. The maximum charge is obtained for the ORR peak of − 0.8 to 1 V scan direction, which is an evidence for more amounts of ROS generated at this potential scan.

Cyclic voltammograms (A) and corresponding ECL signals (B) of (PL-Au)nano/GCE in O2 saturated 0.1 M PBS (pH 7.4) at several scanned potentials, (a) 0 to 1 V, (b) − 0.3 to 1 V, (c) − 0.6 to 1 V and (d) − 0.8 to 1 V.
To validate the ECL experimental conditions and its associated effect of ECL intensity of (PL-Au)nano/GCE, the different concentrations of luminol and HAuCl4.3H2O were taken for electro-deposition (see in Supplementary Sect. 2). From the experimental observation it is found that 1.5 mM of HAuCl4·3H2O and 1 mM luminol is sufficient for deposition of (PL-Au)nano/GCE composite, which apparently enabled a high intense ECL signal (Supplementary Figs. S9, S10). The reproducibility test was carried out by repeating the ECL experiments of (PL-Au)nano/GCE composite by four different time intervals in O2 saturated 0.1 M PBS (pH 7.4) at 0.05 V/s. The ECL intensity at different repetitions is shown as bar chart diagram (Supplementary Fig. S11), the ECL intensity was almost const at each repeated experiments.
Since the ECL signal of (PL-Au)nano/GCE composite depends on the in-situ generated ROS, we performed a time based-ECL transient experiments by changing the initial stepping potentials to study the ECL stability of ROS in O2 saturated 0.1 M PBS (pH 7.4). Figure 4A shows the ECL intensity vs. time transients of (PL-Au)nano/GCE composite at various initial potentials such as 0 (about 6 s) to 0.6 V (2.5 s), − 0.3 (6 s) to 0.6 V (2.5 s), − 0.6 (6 s) to 0.6 V (2.5 s) and − 0.8 (6 s) to 0.6 V (2.5 s) respectively in O2 saturated 0.1 M PBS (pH 7.4). The obtained ECL signals are quite stable even up to 18 consecutive cycles at various initial step potentials. Among different initial potential pulse, the potential pulse between − 0.8 to 0.6 V, displays more intense and stable ECL signals which may be ascribed to the generation of more number of ROS at the interface of (PL-Au)nano/GCE (Fig. 4A).

ECL intensity vs. time curves of (PL-Au)nano/GCE (A) and ECL spectrum (B) in O2 saturated 0.1 M PBS (pH 7.4) at several pulse potentials such as 0 to 0.6 V (a), − 0.3 to 0.6 V (b), − 0.6 to 0.6 V (c) and − 0.8 to 0.6 V (d).
The ECL spectrum of (PL-Au)nano/GCE (Fig. 4B) also performed to elucidate the wavelength of PL at various initial potentials in O2 saturated 0.1 M PBS (pH 7.4). As seen from Fig. 4B, ECL signals were observed only at 430 nm which is consistent with the photoluminescence spectrum. Further, the highest intensity of ECL spectrum is obtained for the pulse potential of − 0.8 to 0.6 V, indicating the highest ROS generation in the specified potentials. This again complements the consistent of ECL transient experiment results (vide supra). Further to prove the concept that ROS playing a crucial role in enhancing the ECL intensity of (PL-Au)nano/GCE, we studied ECL experiments in the presence of ROS scavengers such as superoxide dismutase (SOD) and benzoquinone (BQ)17,41. It is worthy to note that OH∙ and O2∙− selectively quenched by SOD and BQ respectively (Supplementary Fig. S12A,B) which confirm the ECL emission is due to formation of ROS radicals (OH∙ and O2−). Based on all the above results, the ECL reaction mechanism of (PL-Au)nano/GCE in the presence of dissolved O2 is as follows.
Determination of Hg2+ ion
It has been identified in the literature that, the AuNPs and Hg2+ ion has strong specific metallophilic interaction and form spontaneous Au-Hg amalgam which tunes the catalytic properties of AuNPs and accelerate the rate of decomposition of H2O242. Such study can be escalated for various applications particularly in environmental pollutant monitoring, and detection of Hg2+ as preservatives in vaccines. Inspired from that herein, the effect of Hg2+ ion with (PL-Au)nano/GCE by exploring the various Hg2+ ion concentrations from 10 to 150 nM in O2 saturated 0.1 M PBS (pH 7.4) at 0.1 V/s (Supplementary Fig. S13A). Interestingly, the ECL intensity of (PL-Au)nano/GCE linearly increases with each addition of Hg2+ ion (Supplementary Fig. S13B) and reaches the maximum limit up to 150 nM, after that there is no much enhancement was observed. The observed enhancement of ECL intensity (signal on mechanism) during the addition of Hg2+ ion is due to the affinity between AuNPs and Hg2+ ion, leading to the formation of Au–Hg amalgam via aurophilic interactions42. As expected, the surface properties of AuNPs has been further changed upon interaction with Hg2+, and accelerate the in-situ generation of highly reactive ROS through catalytic reduction of dissolved oxygen43. The high ORR activity is a great sign in enhancing the ECL intensity of (PL-Au)nano/GCE and lead to detect the Hg2+ ion at trace level. The calibration curve of ECL intensity versus Hg2+ ion concentrations was represented in Supplementary Fig. S13B. A perfect linear relationship between ECL efficiency and Hg2+ concentrations were obtained from 10 to 150 nM. The detailed ECL mechanism of (PL-Au)nano/GCE before and after Hg2+ addition is shown in Scheme 1B.
As seen in Scheme 1B, dissolved O2 get reduced on the electrode surface to produce highly reactive ROS. When the Hg2+ added into the electrolyte it was reduced and forms Au–Hg amalgam. Freshly formed Au–Hg amalgam further reduces the O2 to produces more number ROS which enhances the ECL intensity of (PL-Au)nano/GCE. Further, we performed a time vs. ECL intensity transient experiment to gain better sensitivity. Figure 5A depicts the obtained ECL transient curves at potentials pulse of holding the potential of − 0.8 V about 10 s and then 0.6 V held about 0.5 s in O2 saturated 0.1 M PBS (pH 7.4). The ECL intensity of (PL-Au)nano/GCE enhanced by each addition of Hg2+ ion in the linear range of 0.3–200 nM (Fig. 5B). The detection limit or limit of detection (LOD) is obtained by using 3× standard deviation/slope and the value is observed to be 0.1 nM and LOD is comparable with previous methods (Table 1). It is worthy to note that similar experiments were also performed by using PL/GCE in the presence of Hg2+ ion at different concentrations and there is no change in ECL intensity of PL/GCE during the Hg2+ addition (Supplementary Fig. S14). These results undoubtedly confirm that (PL-Au)nano/GCE composite only capable of producing more ECL in the presence of Hg2+ ion.

ECL intensity vs. time signals of ECL sensor (A) of (PL-Au)nano/GCE at various Hg2+ concentrations (a) 0, (b) 0.3, (c) 1, (d) 10, (e) 30, (f) 50, (g) 100, (h) 150 and (i) 200 nM by holding the potential of − 0.8 V for 10 s and 0.6 V holds about 0.5 s, its calibration curve (B), selectivity of ECL enhancement of (PL-Au)nano/GCE against some metal ions (C) in O2 saturated 0.1 M PBS (pH 7.4).
Interference study
In order to check the selectivity towards accelerator as well as detection of Hg2+ ion on (PL-Au)nano/GCE using ECL method, other metal ions such as Fe2+, Co2+, Pb2+, As2+, Cu2+, Cd2+, Zn2+ and Mg2+ were also added during the ECL measurements. The interference study has performed by using i–t transient experiment by holding the potential of − 0.8 V for 10 s and 0.6 V about 0.5 s. Figure 5C shows the bar diagram of ECL intensity with respect to 100 µM of all metal ions using (PL-Au)nano/GCE composite in O2 saturated 0.1 M PBS (pH 7.4). It can be notified that the high intense ECL signal is only in the case of Hg2+ addition, on the other hand, ECL signal from (PL-Au)nano/GCE against other studied metal ions were extremely low and comparable to the bare system. The obtained results further evidencing the selectivity of the prepared (PL-Au)nano/GCE system against the Hg2+, suggesting an ideal platform for real sample analysis.
Real sample analysis
The Hg2+ ion in clinical samples like serum and environmental samples like tap water was tested by spiking the known concentrations. Initially, the tap water was boiled for few minutes to remove the contaminants, and then cooled at an open atmosphere until to reach room temperature. Afterward, the known concentrations of Hg2+ were spiked into the water and performed the ECL experiments. To detect the Hg2+ ion in serum samples a known amount of Hg2+ spiked and recorded the ECL experiments. The recovery of sample was also calculated in both tap water and serum indicated in Table 2.
From the observed results we can say that the proposed strategy to detect Hg2+ in real sample analysis could be possible. The percentage recoveries of the analyte were in the range of 93–104 suitable for practical application (Table 2). Further, we compared the Hg2+ recovery in tap water and serum samples by using Atomic absorption spectroscope (AAS) technique. The standard Hg2+ ion solutions of 25, 50, 100, 150, 250, 300, and 500 nM were calibrated, the linear plot shown in Supplementary Fig. S15 and the unknown concentrations of real samples were analyzed and shown in Supplementary Table S3. The obtained recovery (%) by the AAS is consistent with adopted ECL method.
Conclusions
In conclusion, we developed a co-reactant-free solid-state ECL strategy by electrodeposition of polyluminol-gold on GCE surface and observed a stable ECL signal in O2 saturated neutral buffer solutions. The observed ECL intensity effectively increases with the addition of Hg2+ ion even at picomolar range with a good linear relationship. Even though the adopted methodology is simple and one-pot synthesis procedure involved to prepare the self-enhanced solid-state ECL platform. This is the first time we utilized a luminol-gold probe to detect Hg2+ with signal-on ECL platform rather than quenching. Moreover, this co-reactant-free novel solid-state self-enhanced ECL offers good recovery in the real sample analysis of Hg2+ ion sensing and obtained results were comparable with standard spectroscopic technique of atomic absorption spectroscope.
References
White, H. S. & Bard, A. J. Electrogenerated chemiluminescence and chemiluminescence of the Ru(bpy)32+-peroxydisulfate system in acetonitrile-water solutions. J. Am. Chem. Soc. 104, 6891–6895 (1982).
Venkateswara Raju, C. & Senthil Kumar, S. Highly sensitive novel cathodic electrochemiluminescence of tris(2,2′-bipyridine)ruthenium(II) using glutathione as a co-reactant. Chem. Commun. 53, 6593–6596 (2017).
Senthil Kumar, S. & Bard, A. J. Background emission of electrogenerated chemiluminescence during oxidation of tri-n-propylamine from the dimeric 1Δg state of O2. Anal. Chem. 85, 292–295 (2013).
Zhang, Z., Cui, H., Lai, C. & Liu, L. Gold nanoparticle-catalyzed luminol chemiluminescence and its analytical applications. Anal. Chem. 77, 3324–3329 (2005).
Shen, J., Zhou, T. & Huang, R. Recent advances in electrochemiluminescence sensors for pathogenic bacteria detection. Micromachines 10, 532 (2019).
Zu, Y. & Bard, A. J. Electrogenerated chemiluminescence. 66. The role of direct coreactant oxidation in the ruthenium Tris(2,2‘)bipyridyl/tripropylamine system and the effect of halide ions on the emission intensity. Anal. Chem. 72, 3223–3232 (2000).
White, H. S. & Bard, A. J. Electrogenerated chemiluminescence. 41. Electrogenerated chemiluminescence and chemiluminescence of the Ru(2,21-bpy)32+-S2O82− system in acetonitrile-water solutions. J. Am. Chem. Soc. 104, 6891–6895 (1982).
Choi, J.-P. & Bard, A. J. Electrogenerated chemiluminescence (ECL) 79. Anal. Chim. Acta. 541, 141–148 (2005).
Irkham, et al. Electrogenerated chemiluminescence by in situ production of coreactant hydrogen peroxide in carbonate aqueous solution at a boron-doped diamond electrode. J. Am. Chem. Soc. 142, 1518–1525 (2020).
Mayer, M. et al. Shedding light on the diversity of surfactant interactions with luminol electrochemiluminescence for bioanalysis. Anal. Chem. 91, 13080–13087 (2019).
Xu, S., Liu, Y., Wang, T. & Li, J. Highly sensitive electrogenerated chemiluminescence biosensor in profiling protein kinase activity and inhibition using gold nanoparticle as signal transduction probes. Anal. Chem. 82, 9566–9572 (2010).
Gu, W. et al. Stabilized, superparamagnetic functionalized graphene/Fe3O4@Au nanocomposites for a magnetically-controlled solid-state electrochemiluminescence biosensing application. Anal. Chem 87, 1876–1881 (2015).
Vitt, J. E., Johnson, D. C. & Engstrom, R. C. The effect of electrode material on the electrogenerated chemiluminescence of luminol electrode potential. J. Electrochem. Soc. 138, 1637–1643 (1991).
Zhang, Z. et al. Utilization and prospects of electrochemiluminescence for characterization, sensing, imaging and devices. Mater. Chem. Front 3, 2246–2257 (2019).
Mayer, M. et al. Electrochemiluminescence bioassays with a water-soluble luminol derivative can outperform fluorescence assays. Angew. Chem. Int. Ed. 57, 408–411 (2018).
Jiang, X. et al. A novel metal–organic framework loaded with abundant N-(aminobutyl)-N-(ethylisoluminol) as a high-efficiency electrochemiluminescence indicator for sensitive detection of mucin1 on cancer cells. Chem. Commun. 53, 9705–9708 (2017).
Gu, W. et al. Single-atom iron boosts electrochemiluminescence. Angew. Chem. Int. Ed. 59, 3534–3538 (2020).
Hu, L., Wu, Y., Xu, M., Gu, W. & Zhu, C. Recent advances in co-reaction accelerators for sensitive electrochemiluminescence analysis. Chem. Commun. 56, 10989–10999 (2020).
Yang, L. et al. Synthesis and application of CeO2/SnS2 heterostructures as a highly efficient coreaction accelerator in the luminol-dissolved O2 system for ultrasensitive biomarkers immunoassay. Anal. Chem. 91, 14066–14073 (2019).
Gao, W., Xia, X. H., Xu, J. J. & Chen, H. Y. Three-dimensionally ordered macroporous gold structure as an efficient matrix for solid-state electrochemiluminescence of Ru(bpy)32+/TPA system with high sensitivity. J. Phys. Chem. C. 111, 12213–12219 (2007).
Haghighi, B. & Bozorgzadeh, S. Enhanced electrochemiluminescence from luminol at multi-walled carbon nanotubes decorated with palladium nanoparticles: A novel route for the fabrication of an oxygen sensor and a glucose biosensor. Anal. Chim. Acta. 697, 90–97 (2011).
Maness, K. M., Terrill, R. H., Meyer, T. J., Murray, R. W. & Wightman, R. M. Solid-state diode-like chemiluminescence based on serial, immobilized concentration gradients in mixed-valent. J. Am. Chem. Soc. 118, 10609–10616 (1996).
Collinson, M. M., Taussig, J. & Martin, S. A. Solid-state electrogenerated chemiluminescence from gel-entrapped ruthenium (II) tris (bipyridine) and tripropylamine. Chem. Mater. 11, 2594–2599 (1999).
Xue, J. et al. Efficient solid-state electrochemiluminescence from high-quality perovskite quantum dot films. Anal. Chem. 89, 8212–8216 (2017).
Maness, K. M. et al. Solid state electrochemically generated luminescence based on serial frozen concentration gradients of RuIII/II and RuII/I Couples in a molten ruthenium 2,2′-bipyridine complex. J. Am. Chem. Soc. 119, 3987–3993 (1997).
Li, G., Zheng, X. & Song, L. Electrochemiluminescence characterization of poly(luminol-benzidine) composite films and their analytical application. Electroanalysis 21, 845–852 (2009).
Bhattacharjee, Y. & Chakraborty, A. Label-free cysteamine-capped silver nanoparticle-based colorimetric assay for Hg(II) detection in water with subnanomolar exactitude. ACS Sustain. Chem. Eng. 2, 2149–2154 (2014).
Kraepiel, A. M. L. & Amyot, M. The chemical cycle and bioaccumulation. Annu. Rev. Ecol. Syst. 29, 543–566 (1998).
Onyido, I., Norris, A. R. & Buncel, E. Biomolecule-mercury interactions: Modalities of DNA base-mercury binding mechanisms. Remediation strategies. Chem. Rev. 104, 5911–5929 (2004).
Han, S., Zhu, M., Yuan, Z. & Li, X. A methylene blue-mediated enzyme electrode for the determination of trace mercury (II), mercury (I), methylmercury, and mercury–glutathione complex. Biosens. Bioelectron. 16, 9–16 (2001).
Alexander, P. W. & Rechnitz, G. A. Enzyme inhibition assays with an amperometric glucose biosensor based on a thiolate self-assembled monolayer. Electroanalysis 5, 343–350 (2000).
Soldatkin, O. O. et al. Novel conductometric biosensor based on three-enzyme system for selective determination of heavy metal ions. Bioelectrochemistry 83, 25–30 (2012).
Pirvutoiu, S. et al. Flow injection analysis of mercury(II) based on enzyme inhibition and thermometric detection. Analyst 126, 1612–1616 (2001).
Pavlov, V. et al. Amplified chemiluminescence surface detection of DNA and telomerase activity using catalytic nucleic acid labels. Anal. Chem. 76, 2152–2156 (2004).
Tao, X. et al. Development of a highly sensitive chemiluminescence enzyme immunoassay using enhanced luminol as substrate. Luminescence 29, 301–306 (2014).
Ashok Kumar, S., Cheng, H.-W. & Chen, S.-M. Electroanalysis of ascorbic acid (vitamin C) using nano-ZnO/poly(luminol) hybrid film modified electrode. React. Funct. Polym. 69, 364–370 (2009).
El-Deab, M. S., Okajima, T. & Ohsaka, T. Electrochemical reduction of oxygen on gold nanoparticle-electrodeposited glassy carbon electrodes. J. Electrochem. Soc. 150, A851 (2003).
Sanchez-Sanchez, C. & Bard, A. Hydrogen peroxide production in the oxygen reduction reaction at different electrocatalysts as quantified by scanning electrochemical microscopy. Anal. Chem. 81, 8094–8100 (2009).
Jiang, Z., Jiang, Z., Tian, X. & Chen, W. Amine-functionalized holey graphene as a highly active metal-free catalyst for the oxygen reduction reaction. J. Mater. Chem. A 2, 441–450 (2014).
He, Y., Liu, D., He, X. & Cui, H. One-pot synthesis of luminol functionalized silver nanoparticles with chemiluminescence activity for ultrasensitive DNA sensing. Chem. Commun. 47, 10692–10694 (2011).
Chu, H., Guo, W., Di, J., Wu, Y. & Tu, Y. Study on sensitization from reactive oxygen species for electrochemiluminescence of luminol in neutral medium. Electroanalysis 21, 1630–1635 (2009).
Han, K. N., Choi, J. & Kwon, J. Gold nanozyme-based paper chip for colorimetric detection of mercury ions. Sci. Rep. 7, 2806 (2017).
Zhang, S. et al. High-throughput and ultratrace naked-eye colorimetric detection of Au3+ based on the gold amalgam-stimulated peroxidase mimetic activity in aqueous solutions. Chem. Commun. 53, 5056–5058 (2017).
Yaping, L. Electrogenerated chemiluminescence detection of mercury (II) ions based on DNA probe labeled with ruthenium complex. Anal. Sci. 27, 193–196 (2011).
Tseng, C.-W., Chang, H.-Y., Chang, J.-Y. & Huang, C.-C. Detection of mercury ions based on mercury-induced switching of enzyme-like activity of platinum/gold nanoparticles. Nanoscale 4, 6823–6830 (2012).
Li, Q., Zhou, X. & Xing, D. Rapid and highly sensitive detection of mercury ion (Hg2+) by magnetic beads-based electrochemiluminescence assay. Biosens. Bioelectron. 26, 859–862 (2010).
Gao, Y., Wang, T. & Liu, F. Determination of Hg2+ in tap water based on the electrochemiluminescence of Ru(phen)32+ and thymine at bare and graphene oxide-modified glassy carbon electrodes. Chin. J. Chem 34, 1297–1303 (2016).
Zhu, X., Chen, L., Lin, Z., Qiu, B. & Chen, G. A highly sensitive and selective ‘signal-on’ electrochemiluminescent biosensor for mercury. Chem. Commun. 46, 3149–3151 (2010).
Wang, H., Wang, Y., Jin, J. & Yang, R. Gold nanoparticle-based colorimetric and ‘turn-on’ fluorescent probe for mercury(II) ions in aqueous solution. Anal. Chem. 80, 9021–9028 (2008).
Yang, K., Zeng, M., Hu, X. J., Guo, B. S. & Zhou, J. B. Layered MnO2 nanosheet as a label-free nanoplatform for rapid detection of mercury(II). Analyst 139, 4445–4448 (2014).
Acknowledgements
C.V. Raju thanks the CSIR for the award of Senior Research Fellowship (SRF) and S.S.K. thanks Department of Science and Technology (EMR/2017/004449) for their financial support. CSIR-CECRI Manuscript Communication Number: CECRI/PESVC/Pubs/2020-031.
Author information
Authors and Affiliations
Contributions
The author "C.V.R." has done the work and collected all the data and written the manuscript. The corresponding author "S.S.K." designed the overall work and corrected the entire manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Raju, C.V., Kumar, S.S. Co-reactant-free self-enhanced solid-state electrochemiluminescence platform based on polyluminol-gold nanocomposite for signal-on detection of mercury ion. Sci Rep 11, 6932 (2021). https://doi.org/10.1038/s41598-021-86195-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-86195-1
This article is cited by
-
An electrochemiluminescence aptasensor based on poly(aniline-luminol)/graphene oxide/chitosan for ultra-sensitive detection of Hg2+
Polymer Bulletin (2023)
-
Dual-target electrochemical DNA sensor for detection of Pb2+ and Hg2+ simultaneously by exonuclease I–assisted recycling signal amplification
Microchimica Acta (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
