
Abstract
Hearing loss is one of the most common birth disorders in humans, with an estimated prevalence of 1–3 in every 1000 newborns. This study investigates the molecular etiology of a hearing loss cohort using a stepwise strategy to effectively diagnose patients and address the challenges posed by the genetic heterogeneity and variable mutation spectrum of hearing loss. In order to target known pathogenic variants, multiplex PCR plus next-generation sequencing was applied in the first step; patients which did not receive a diagnosis from this were further referred for exome sequencing. A total of 92 unrelated patients with nonsyndromic hearing loss were enrolled in the study. In total, 64% (59/92) of the patients were molecularly diagnosed, 44 of them in the first step by multiplex PCR plus sequencing. Exome sequencing resulted in eleven diagnoses (23%, 11/48) and four probable diagnoses (8%, 4/48) among the 48 patients who were not diagnosed in the first step. The rate of secondary findings from exome sequencing in our cohort was 3% (2/58). This research presents a molecular diagnosis spectrum of 92 non-syndromic hearing loss patients and demonstrates the benefits of using a stepwise diagnostic approach in the genetic testing of nonsyndromic hearing loss.
Similar content being viewed by others

Introduction
Hearing loss is one of the most common birth defects in humans, with an estimated prevalence of 1–3 in every 1000 newborns1. Seventy percent of hearing loss cases are nonsyndromic, and one of the primary etiologies is genetic predisposition1. To date, over 100 genes have been associated with nonsyndromic hearing loss (NSHL)2,3, and new genes are continuing to be discovered4. The timely and effective diagnosis of affected individuals is made challenging by the extreme genetic heterogeneity underlying the condition.
Interestingly, the frequency of the genes behind NSHL varies between different populations and ethnicities. The most common mutations in many populations are in the GJB2 gene, which encodes the connexin 26 protein, and cause severe-to-profound autosomal recessive NSHL1,5. Sanger sequencing of GJB2 was therefore performed in previous studies6,7. In the Saudi population, the OTOF gene, rather than GJB2, was revealed to be a major and potential contributor to hearing loss7. SLC26A4 is another common gene causing nonsyndromic hearing impairment with enlarged vestibular aqueducts in Asian and Middle Eastern populations and Ashkenazi Jews8. A single Sanger sequencing reaction is capable of covering the whole coding region of GJB2 at an affordable cost as it has only 226 amino acids. SLC26A4 has 21 exons and the coding sequencing spans 2343 bp from exon 2 to exon 21, requiring multiple Sanger sequencing reactions.
Although exome sequencing has been proposed and used as a single-step test for hearing loss patients9,10, interpreting exome sequencing data is usually laborious and time-consuming. Tiered or stepwise diagnostic approaches have been proposed multiple times in the literature4,6,11,12. Guan et al. provided a two-tier strategy which consisted of Sanger sequencing combined with targeted deletion analyses of GJB2 and STRC and two mitochondrial genes, followed by exome sequencing and targeted analysis of deafness-related genes6. Li et al. performed hotspot variant screening and subsequent exome sequencing in a family with deafness4.
This study proposes a hierarchical approach that first targets known pathogenic variants by multiplex PCR, followed by exome sequencing and a comprehensive analysis of deafness genes. We have evaluated the performance of this strategy in a cohort of 92 NSHL patients. Individuals with inconclusive or negative results in the first step were referred for exome sequencing. An analysis of the diagnostic rate in the different steps and the contribution of different genetic factors enabled us to formulate a cost-effective diagnostic paradigm which can serve as an example for other populations.
Results
Of the 92 NSHL patients, most (82%; 75/92) had no family history of hearing loss. The degree of hearing loss in the patients varied; severe-to-profound hearing loss was observed in the majority (84%, 77/92), and prelingual hearing loss was detected in 87% (80/92) patients. Of note, 17% (16/92) of the patients passed a newborn hearing screen at birth but developed hearing loss at a later age (Table 1).
Forty-four diagnoses by multiplex PCR
In the first step, the patients were tested via multiplex PCR. The tests yielded a positive result in 44 out of the 92 patients, while eight were inconclusive and 40 negative (Fig. 1). The genotypes of the 44 patients who tested positive are listed in Table 2. Classifications of these variants were presented in Table 3. There were 27 with a mutation in GJB2, 15 in SLC26A4, 1 with a dual molecular diagnosis of both GJB2 and SLC26A4, and 1 with a mutation in MT-RNR1. The patient who was positive with a homoplasmic m.1555A>G in the MT-RNR1 gene had aminoglycoside exposure history. Homozygous NM_004004.6:c.235delC in the GJB2 gene was the most prevalent genotype, accounting for 11% (10/92) of the study cohort. NM_004004.6:c.109G>A in GJB2 was identified in 10 of the 44 patients with positive genotypes, including three patients who were homozygous and seven patients who a compound heterozygous mutation. One patient was homozygous for both NM_000441.2:c.919-2A>G in SLC26A4 and NM_004004.6:c.109G>A in GJB2. This patient was clinically diagnosed with deafness and enlarged vestibular aqueducts, a phenotype which can be caused by deficiency of the two genes together.

Outline of the study design. Ninety-two patients with non-syndromic hearing loss were enrolled. After carrying out multiplex PCR and next generation sequencing on all the patients, the 48 undiagnosed and 10 patients diagnosed for GJB2 c.109G>A were referred for exome sequencing. NSHL non-syndromic hearing loss.
Fifteen diagnoses/probable diagnoses by exome sequencing
Two groups of patients (n = 58) were referred for exome sequencing. Group 1 was the 48 patients who received inconclusive or negative genotypes from the multiplex PCR (Fig. 1). Group 2 consisted of 10 patients who were either homozygous or compound heterozygous for NM_004004.6:c.109G>A in GJB2. Due to the variable expressivity and incomplete penetrance of NM_004004.6:c.109G>A in GJB213, these 10 patients were referred for exome sequencing in order to exclude other potential molecular etiologies.
In the first group, exome sequencing resulted in eleven diagnoses (23%, 11/48) and four probable diagnoses (8%, 4/48) (Table 4). No other causally associated variants related to hearing loss were identified in the second group. It is worth noting that patient P27 was first identified as homozygous for NM_001038603.3(MARVELD2):c.1208_1211delGACA by exome sequencing. Given that the family history did not indicate consanguinity, we performed CNV analysis of the exome data from this patient as well as all the other fifty-seven patients in step 2. The exome data revealed a heterozygous deletion of exon 3 to exon 5 in the MARVELD2 gene, which was also verified by qPCR. Variant c.1208_1211delGACA is located in exon 4, which was deleted in this CNV variant. We thus conclude that c.1208_1211delGACA is hemizygous in this case.
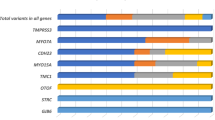
Summing up, the two-step approach identified the molecular etiology of 59/92 (64%) NSHL index patients, including 44 (48%) diagnoses by multiplex PCR in the first step and 15 (16%) diagnoses/probable diagnoses by exome sequencing (Fig. 2). Mutations in GJB2 were the most frequent (28/59), followed by mutations in SLC26A4 (18/59), MYO15A (3/59), COL11A2 (2/59), and MT-RNR1, MARVELD2, MITF, CDH23, OTOA, TRIOBP, TMPRSS3, SOX10 and BSND (1/59 each).

Genetic spectrum of enrolled non-syndromic hearing loss probands. Molecular diagnostic results were classified according to genes and detection methods. ES exome sequencing.
Further analysis of the exome sequencing data was carried out to discover secondary findings. The cohort had two pathogenic variants from among the 59 secondary findings genes recommended by the ACMG14 (Supplementary Table S2). A heterozygous variant, GLA(NM_000169.3):c.1067G>A (p.Arg356Gln) was identified in patient P21 (female). It is related to Fabry disease, an X-linked inborn error of glycosphingolipid catabolism resulting from deficient or absent activity of the lysosomal enzyme alpha-galactosidase A15. The second pathogenic variant was RYR1(NM_000540.3):c.6502G>A (p.Val2168Met), which is associated with malignant hyperthermia. This variant was identified in P77 in a heterozygous state. The rate of secondary findings in our cohort was 3% (2/58), comparable to previously published rates of 1.8% to 4.6%16,17,18,19.
Discussion
This study applied a stepwise, genetic testing approach to explore molecular diagnoses in an NSHL cohort, achieving 64% (59/92) diagnostic yield. Although diagnostic yield varies between different patient cohorts and depends on the detection methods used, our diagnostic yield (64%) is comparable to a multi-ethnic cohort tested using exome sequencing (56%)9.
This study uncovered the etiology of 44 patients in the first step using a commercial multiplex PCR kit, providing a rapid molecular diagnosis and saving the cost of exome sequencing. The multiplex PCR contains amplicons covering the GJB2, SLC26A4 and MT-RNR1 genes. These three genes are known to have hotspot variants causing non-syndromic hearing loss in Asian populations, including c.109G>A, c.235delC, and c.299_300delAT in GJB2, c.919-2A>G, c.1229C>T, and c.2168A>G in SLC26A4, and m.1555A>G in MT-RNR120,21,22. Compared to a single test of GJB2, which is frequently used as the first-tier test to exclude hotspot variants before exome sequencing6,9, a multiplex PCR sequencing approach appears to be both efficient and cost-effective, as it is more flexible and can detect hotspot variants across multiple deafness-related genes.
It should be noted that the diagnostic rate of the multiplex PCR assay in the first step could vary dramatically in different populations depending on the prevalence of hotspot variants in targeted patients. In this study, the high diagnostic rate was attributable to the enrichment of NM_004004.6(GJB2):c.109G>A, NM_004004.6(GJB2):c.235delC, NM_004004.6(GJB2):c.299_300delAT, NM_000441.2(SLC26A4):c.919-2A>G, and NM_000441.2(SLC26A4): c.1229C>T in the Chinese population23,24. More importantly, ethnic background is relatively uniform in China. The inherent ethnic bias of the multiplex PCR assay is a drawback and might be inappropriate in a racially and ethnically diverse population25.
Allelic heterogeneity is common in hearing loss and is associated with clinical phenotype heterogeneity, with both syndromic hearing loss and NSHL being caused by mutations within the same gene26. In this study, although we only enrolled patients with nonsyndromic hearing loss, deafness-related variants were also identified in syndromic genes. P44 was heterozygous for a disease-causing nonsense variant in the MITF gene, and P111 was heterozygous for a disease-causing missense variant in the SOX10 gene. Both variants are associated with autosomal dominant inherited Waardenburg syndrome, which is considered an NSHL mimic. The variability of phenotypes makes clinical diagnosis and variant interpretation in genetic hearing loss challenging27.
The molecular diagnosis of NSHL is made yet more challenging by the variable expressivity and high prevalence of NM_004004.6:c.109G>A in the GJB2 gene13. In our cohort, one patient with enlarged vestibular aqueducts was found to be homozygous for both NM_000441.2:c.919-2A>G in SLC26A4 and NM_004004.6:c.109G>A in GJB2 in the first diagnosis step. The genotype–phenotype consistency led us to consider NM_000441.2:c.919-2A>G in SLC26A4 as the disease-causing variant. However, we cannot rule out the possibility of a blended phenotype arising from the two variants28. By contrast, 10 patients who were diagnosed with only NM_004004.6:c.109G>A in GJB2 in the first step were referred for exome sequencing, and no other potential molecular explanations were identified. These results indicate the importance of incorporating phenotype and genotype in practice and of considering dual molecular diagnoses.
Copy number variations are common causes of nonsyndromic hearing loss29. Exome sequencing data can be analyzed for CNVs, although such analyses suffer from low sensitivity and uncertain specificity30. In this study, one patient in our cohort was diagnosed to have a SNV compounded with a CNV in MARVELD2. The homozygous c.1208_1211delGACA(p.Arg403Lysfs*11) in the MARVELD2 gene was initially thought to be the causal etiology. The lack of consanguineous history led us to reanalyze the coverage depth of the exons in the MARVELD2 gene, resulting in the identification of an EX3_EX5 Del. This finding highlights the importance of considering CNV deletions in a non-consanguineous family when a pathogenic variant is identified in a homozygous state.
It is worth noting that 17% (16/92) of the patients passed a newborn hearing screening at birth but developed hearing loss at a later stage. Seven of these patients received a positive molecular diagnosis, with variants in the GJB2 and SLC26A4 genes (Supplementary Table S3). These results are consistent with recent findings showing that newborns with positive genotypes can be missed by physiologic newborn hearing screens but identified by genetic screens, highlighting the necessity of concurrent hearing and genetic screening in newborns21,31.
This study has several limitations which should be noted. First, the stepped approach may miss a dual molecular diagnosis if a patient is diagnosed in the first step. A dual molecular diagnosis was reported in 4.9% of patients with multiple phenotypic traits28. For single phenotype traits such as NSHL, dual molecular diagnosis might be rare, but this is worth considering when deciding on a diagnostic approach. Second, CNV analysis of the STRC gene is absent in our exome data pipelines due to the presence of a pseudogene. This might result in an underestimate of the contribution of disease-causing CNVs in this study.
In conclusion, this work demonstrates the benefits of a stepwise approach to diagnose non-syndromic hearing loss patients. Instead of starting with exome sequencing, multiplex PCR targeted hotspot variants across multiple genes can provide a molecular etiology for 48% of Eastern Asian patients in a prompt and efficient manner. It seems likely that this will result in significant savings, but a future cost-effectiveness analysis will address that question.
Methods
Participants
A total of 92 patients with NSHL in Eastern Asian ethnicity were recruited. No other visible phenotype was reported. We obtained informed consent from the patients. This study was approved by the Institutional Review Board of BGI. All methods were performed in accordance with the relevant guidelines and regulations.
Study design
The enrolled patients were first assayed with a commercial multiplex PCR to analyze common variants in the Asian population. Patients undiagnosed by the multiplex PCR assay were then referred for exome sequencing. Because of the variable expressivity and penetrance of NM_004004.6:c.109G>A in GJB2, patients diagnosed with this variant were also referred for exome sequencing to explore other potential molecular etiologies.
Multiplex PCR
Genetic variant detection was carried out in all patients by applying multiplex PCR combined with next-generation sequencing. The commercial multiplex PCR kit (BGI Biotech, Wuhan, China) was designed to cover certain pathogenic variants of 22 genes, including the complete coding region of GJB2 and most of the coding regions of SLC26A4. Genomic DNA was extracted from 2 ml of peripheral blood using a DNA Extraction Kit (BGI Biotech, Wuhan, China). Targeted variants were amplified by multiplex PCR using 2 × KAPA 2G Fast Multiple PCR Mix (KAPA BIOSYSTEMS, Wilmington, MA, USA). The PCR program consisted of one round of 95 °C for 3 min, then 30 cycles of 95 °C for 15 s, 62 °C for 30 s, and 72 °C for 90 s.
Library preparation, sequencing and bioinformatics
PCR products were pooled to prepare a library. Briefly, ~ 3.5 μg purified products were sheared by ultrasonoscope and quality-controlled using an Agilent Bioanalyzer DNA 2100 kit (Agilent, Santa Clara, CA, USA). Subsequently, end-repair and A-tailing were performed before adapters were ligated to both ends of the fragments. Finally, the adapter-ligated products were amplified by 8-cycle PCR and purified using Agencourt AMPure XP beads (Beckman Coulter, Fullerton, CA, USA). The prepared libraries were subjected to single-strand circularized DNA and DNA nanoball preparation before being sequenced on a BGISEQ-500 sequencer (BGI, Shenzhen, China) with PE5032. Raw sequence reads were mapped to the human reference genome (hg19) using Bowtie 2.3.3 with SAMtools 1.6 used to create BAM and index files. For variant calling, Genome Analysis Tool Kit (GATK 3.7)33 was used to analyze the alignment data.
Exome sequencing and data analysis
Exome sequencing was performed following standard manufacturer protocols on a BGISEQ-500 platform. An in-house bioinformatics pipeline was employed to process the variant call format (VCF) files and to maintain variants of potential clinical usefulness, including (i) variants with minor allele frequency (MAF) < 1%, (ii) variants in genes with an OMIM disease entry. We interpreted variants in 130 genes curated by ClinGen Expert as having a limited-to-definitive relationship to hearing loss34. This interpretation was based on ClinGen Expert Specification of the ACMG/AMP Variant Interpretation Guidelines for Genetic Hearing Loss27.
Definition of molecular diagnosis
Patients were categorized as “positive” or “diagnosed” if they were homozygous or double heterozygous for a pathogenic/likely pathogenic variant(s) in a recessive inherited gene or heterozygous for a pathogenic/likely pathogenic variant in a dominant inherited gene. In addition, patients with a pathogenic/likely pathogenic variant plus a rare VUS in a recessive inherited gene were considered “probably diagnosed.” Patients with a pathogenic/likely pathogenic variant in a recessive inherited gene were considered “inconclusive”. Patients with a variant in a gene that was not inherited recessively or dominantly, for example in mitochondrial genes, were categorized as “diagnosed” if the phenotype associated with the genotype.
Sanger validation and qPCR
Sanger sequencing was carried out to validate SNPs/Indels detected by either multiplex PCR or exome sequencing. All PCR products were sequenced on an ABI 3730XL DNA Analyzer. Mutations were confirmed by comparing our sequencing data with the UCSC human reference sequences35. Verification of exon-level deletions or duplications called by exome sequencing was carried out by qPCR. The qPCR methodology has been previously described36. The primer pair sequences are shown in Supplementary Table S1.
References
Morton, C. C. N. & Walter, E. Newborn hearing screening—A silent revolution. N. Engl. J. Med. 354(20), 2151–2164 (2006).
Vona, B., Nanda, I., Hofrichter, M. A., Shehata-Dieler, W. & Haaf, T. Non-syndromic hearing loss gene identification: A brief history and glimpse into the future. Mol. Cell Probes 29(5), 260–270 (2015).
Van Camp, G. & Smith. R. Hereditary hearing loss homepage. https://hereditaryhearingloss.org. Accessed: August 2019.
Li, M. et al. Extrusion pump ABCC1 was first linked with nonsyndromic hearing loss in humans by stepwise genetic analysis. Genet. Med. 21, 2744–2754 (2019).
Korver, A. M. H. et al. Congenital hearing loss. Nat. Rev. Dis. Primers 3(1), 16094 (2017).
Guan, Q. et al. AUDIOME: a tiered exome sequencing-based comprehensive gene panel for the diagnosis of heterogeneous nonsyndromic sensorineural hearing loss. Genet. Med. 20(12), 1600–1608 (2018).
Almontashiri, N. A. M. et al. Recurrent variants in OTOF are significant contributors to prelingual nonsydromic hearing loss in Saudi patients. Genet. Med. 20(5), 536–544 (2018).
Sloan-Heggen, C. M. et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 135(4), 441–450 (2016).
Bademci, G. et al. Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. Genet. Med. 18(4), 364–371 (2016).
Zazo Seco, C. et al. The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in The Netherlands. Eur. J. Hum. Genet. 25(3), 308–314 (2017).
Baux, D. et al. Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci. Rep. 7(1), 16783 (2017).
Budde, B. S. et al. Comprehensive molecular analysis of 61 Egyptian families with hereditary nonsyndromic hearing loss. Clin. Genet. 98, 32–42 (2020).
Shen, J. et al. Consensus interpretation of the p.Met34Thr and p.Val37Ile variants in GJB2 by the ClinGen Hearing Loss Expert Panel. Genet. Med. 21(11), 2442–2452 (2019).
Green, R. C. et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 15(7), 565–574 (2013).
Schiffmann, R. Fabry disease. Neurocutaneous Syndr. 2015:231–248.
Yang, Y. et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312(18), 1870–1879 (2014).
Schwartz, M. L. B. et al. A model for genome-first care: Returning secondary genomic findings to participants and their healthcare providers in a large research cohort. Am. J. Hum. Genet. 103(3), 328–337 (2018).
Dewey, F. E. et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 354(6319), aaf6814 (2016).
Haer-Wigman, L. et al. 1 in 38 individuals at risk of a dominant medically actionable disease. Eur. J. Hum. Genet. 27(2), 325–330 (2019).
Tsukada, K., Nishio, S. Y., Hattori, M. & Usami, S. Ethnic-specific spectrum of GJB2 and SLC26A4 mutations: Their origin and a literature review. Ann. Otol. Rhinol. Laryngol. 124(Suppl 1), 61S-76S (2015).
Wang, Q. et al. Nationwide population genetic screening improves outcomes of newborn screening for hearing loss in China. Genet. Med. 21(10), 2231–2238 (2019).
Qing, J. et al. Prevalence of mutations in GJB2, SLC26A4, and mtDNA in children with severe or profound sensorineural hearing loss in southwestern China. Genet. Test Mol. Biomark. 19(1), 52–58 (2015).
Dai, P. et al. GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J. Transl. Med. 7, 26 (2009).
Wang, Q. J. et al. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clin. Genet. 72(3), 245–254 (2007).
Shearer, A. E., Shen, J., Amr, S., Morton, C. C. & Smith, R. J. Newborn Hearing Screening Working Group of the National Coordinating Center for the Regional Genetics Network. A proposal for comprehensive newborn hearing screening to improve identification of deaf and hard-of-hearing children. Genet. Med. 21, 2614–2630 (2019).
Keats, B. J. & Berlin, C. I. Genomics and hearing impairment. Genome Res. 9(1), 7–16 (1999).
Oza, A. M. et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 39(11), 1593–1613 (2018).
Posey, J. E. et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 376(1), 21–31 (2017).
Shearer, A. E. et al. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 6(5), 37 (2014).
Yao, R. et al. Evaluation of three read-depth based CNV detection tools using whole-exome sequencing data. Mol. Cytogenet. 10(1), 30 (2017).
Guo, L. et al. Concurrent hearing and genetic screening in a general newborn population. Hum. Genet. 139(4), 521–530 (2020).
Huang, J. et al. A reference human genome dataset of the BGISEQ-500 sequencer. Gigascience 6(5), 1–9 (2017).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43(5), 491–498 (2011).
DiStefano, M. T. et al. ClinGen expert clinical validity curation of 164 hearing loss gene–disease pairs. Genet. Med. 21, 2409 (2019).
Rosenbloom, K. R. et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 43(Database issue), D670–D681 (2015).
Ceulemans, S., van der Ven, K. & Del-Favero, J. Targeted screening and validation of copy number variations. Methods Mol. Biol. 838, 311–328 (2012).
Author information
Authors and Affiliations
Contributions
Z.P. conceived and designed the study. L.C., J.W. and J.X. contributed to patient recruitment and phenotypic information collection. J.P. performed bioinformatics analysis. H.L., X.X., N.L., and C.C. performed the validation experiments. X.X., N.L., C.C., L.C. and N.S. performed data analysis and interpretation. J.W., J.X. and L.C. drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
J.X., L.C., H.L., J.P., N.S., and Z.P. were employed at BGI Genomics at the time of submission. No other conflicts relevant to this study should be reported.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Xiang, J., Chen, L. et al. Molecular diagnosis of non-syndromic hearing loss patients using a stepwise approach. Sci Rep 11, 4036 (2021). https://doi.org/10.1038/s41598-021-83493-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-83493-6
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
