
Abstract
ABCC6 deficiency promotes ectopic calcification; however, circumstantial evidence suggested that ABCC6 may also influence atherosclerosis. The present study addressed the role of ABCC6 in atherosclerosis using Ldlr−/− mice and pseudoxanthoma elasticum (PXE) patients. Mice lacking the Abcc6 and Ldlr genes were fed an atherogenic diet for 16 weeks before intimal calcification, aortic plaque formation and lipoprotein profile were evaluated. Cholesterol efflux and the expression of several inflammation, atherosclerosis and cholesterol homeostasis-related genes were also determined in murine liver and bone marrow-derived macrophages. Furthermore, we examined plasma lipoproteins, vascular calcification, carotid intima-media thickness and atherosclerosis in a cohort of PXE patients with ABCC6 mutations and compared results to dysmetabolic subjects with increased cardiovascular risk. We found that ABCC6 deficiency causes changes in lipoproteins, with decreased HDL cholesterol in both mice and humans, and induces atherosclerosis. However, we found that the absence of ABCC6 does not influence overall vascular mineralization induced with atherosclerosis. Decreased cholesterol efflux from macrophage cells and other molecular changes such as increased pro-inflammation seen in both humans and mice are likely contributors for the phenotype. However, it is likely that other cellular and/or molecular mechanisms are involved. Our study showed a novel physiological role for ABCC6, influencing plasma lipoproteins and atherosclerosis in a haploinsufficient manner, with significant penetrance.
Similar content being viewed by others


Introduction
Vascular calcification is common in aging and can also result from diabetes, end-stage renal disease, atherosclerosis, or inherited disorders. Clinical evidence demonstrated that cardiovascular calcification is a significant risk factor for cardiovascular events1. ABCC6 is a member of the ATP binding cassette superfamily2 and is a major regulator of ectopic calcification3,4. ABCC6 regulates mineralization by modulating an extracellular purinergic pathway through several key enzymes that generate inorganic pyrophosphate (PPi) and prevent its degradation5,6,7. Indeed, PPi is a potent inhibitor of ectopic calcification8. The cellular function of ABCC6 accounts for ~ 60% of plasma PPi levels in humans and mice9,10. The highest levels of ABCC6 expression are found in liver and kidneys, though other cell types also express the protein11,12. Inherited ABCC6 deficiency causes dermal, ocular and cardiovascular calcification in pseudoxanthoma elasticum (PXE) patients4, some cases of generalized arterial calcification of infancy (GACI)13 and is likely responsible for the PXE-like manifestations in β-thalassemia patients14,15.
Multiple lines of evidence indicate a pathologic correlation between ABCC6 function and dyslipidemia. Pisciotta et al. reported a PXE patient carrying a heterozygous LDLR mutation (p.R574H) with severe vascular stenosis and calcification16. The dystrophic cardiac calcification phenotype (DCC) associated with Abcc6 deficiency in mice17,18 was first identified in response to high fat diets19,20. Quantitative trait loci analysis identified Abcc6 as one of the root causes of atherosclerosis and aortic calcification induced by hyperlipidemia in ApoE−/− mice21. Furthermore, previous publications have presented suggestive evidence, but not proven, that PXE patients are susceptible to increased atherosclerosis 22,23,24.
In this study, we explored the possibility that beyond calcification, ABCC6 dysfunction could also play a significant role in dyslipidemia and atherosclerosis. For this purpose, we crossed Abcc6−/− mice into an atherogenic Ldlr−/− background and compared the results of our investigation with a cohort of human PXE patients. We found that reduced ABCC6 function indeed leads to dyslipidemia and enhanced atherosclerosis in both Abcc6-deficient mice and in PXE patients. Vascular calcification was elevated in both Abcc6+/−; Ldlr−/− and Abcc6−/−; Ldlr−/− animals, but there was no significant difference with Ldlr−/− control mice, suggesting that the Abcc6 genotype has no effect on atherosclerotic mineralization. Overall, our data imply that changes in HDL levels and/or composition as well as systemic inflammation act as contributors to the atherosclerotic phenotype seen in the Abcc6−/− mouse model and PXE patients.
Material and methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. Because of the sensitive nature of the data from human subjects in this study, these may be subjected to approval by the authors who provided them (LM and GL).
Animals
Abcc6tm1Aabb mice were generated on a 129/Ola background25 and backcrossed into a C57BL/6J > 10 times. Ldlrtm1Her mice in the C57BL/6 background were provided by WAB who purchased them from The Jackson Laboratory (Bar Harbor, ME). These mice are hereafter designated as Abcc6−/− and Ldlr−/−, respectively. By simple crosses, we have generated Abcc6 haploinsufficient (Abcc6+/−;Ldlr−/−) and double knockout (Abcc6−/−;Ldlr−/−) animals to determine if a gene dosage affects atherosclerosis and associated calcification. Ldlr−/− mice with normal Abcc6 expression were used as controls. In this study, both male and female, age-matched mice were used, as gender had no significant impact on results. All mice were housed in pathogen-free rooms in AAALAC-accredited animal facilities at the University of Hawaii School of Medicine. Mice were kept under routine laboratory conditions with 12-h light–dark cycle with ad libitum access to water and either a standard chow diet or an atherogenic high fat diet (HFD) that contained 15.8% (wt/wt) fat, 1.25% (wt/wt) cholesterol and no cholate (diet #94059; Harlan Teklad). The University of Hawaii Institutional Animal Care and Use Committee approved these studies. Experiments were conducted according to the NIH Guide for the Care and Use of Laboratory Animals.
PXE patients and healthy controls
Fasting plasma samples and phenotypical data for PXE patients were obtained from the MAGEC PXE Reference Center of the Angers University Hospital, France. The diagnosis of PXE was based on a combination of established criteria for indisputable PXE, including clinically suggestive skin lesions, angioid streaks and histologically proven fragmented and calcified elastic fibers on skin biopsy26. For lipoprotein analyses, PXE and controls plasma samples came from subjects that were not treated with cholesterol lowering medication. Control plasma samples came from a group matched for gender and age without evidence of PXE or other pathology.
For atherosclerotic plaques assessment, results were compared to the NUMEVOX cohort. This cohort of subjects with at least one metabolic syndrome criterion with or without sleep apnea or liver steatosis is registered on clinicaltrials.gov (NCT00997165). The institution review board of the Angers University Hospital, France has approved these studies and the participants have given informed consent when required. All methods were carried out in accordance with relevant guidelines and regulations.
Calcification measurements
The level of mineralization in aortic, whisker and heart tissues was quantified using a colorimetric assay27 that directly measures the amount of excess calcium, which is normalized to the weight of the excised tissues, as described18, and expressed in mg/dL per milligram of tissue. The aorta comprised the aortic arch and the thoracic and abdominal sections, including the iliac bifurcation.
Atherosclerosis assessment
After 16 weeks of atherogenic diet, blood was collected from mice under anesthesia through cardiac puncture and the animals were euthanized using standard CO2 procedures. The animals were perfused with cold phosphate buffered saline (PBS) and a solution of 4% paraformaldehyde and 5% sucrose. After excising, aortas were cleaned of adventitial fat and opened longitudinally from the iliac bifurcation right to the common carotid artery. Aortas were pinned out onto a black wax board and stained for lipid with Oil Red O. The stained areas and overall inner surface of aortas were measured using Image J software (NIH) and the percent area stained with Oil Red O was determined.
Plasma lipoproteins and lipid analyses
Triglycerides and total cholesterol
These were either measured in fasting human samples by the clinical laboratory services at the Angers University Hospital, France, or in fasting animal samples using quantification kits from Abcam (Ab65336, Cambridge, MA) and from Wako Diagnostics (439-17501, Mountain View, CA). Apolipoprotein E (ApoE) concentrations in mouse and human plasma were assessed using ELISA kits (MBS2512274) from MyBioSources (San Diego, CA) and Abcam (Ab108813, Cambridge, MA), respectively.
Lipoprint analysis
LDL and HDL subfraction distributions were analyzed with the Quantimetrix Lipoprint system (Quantimetrix Corporation, Redondo Beach, CA) according to the manufacturer’s instructions and as described elsewhere28. The system uses 25 μl of fasting plasma samples (not previously frozen). Up to 7 LDL bands can be detected. The LDL-1 and -2 bands correspond to large particles, whereas bands LDL-3 to 7 correspond to small dense LDL particles. Similarly, 10 HDL sub-fractions are detected. HDL-1 to -3 represent large particles, sub-fractions − 4 to − 7 are reported as medium particles, and sub-fractions − 8 to − 10 as small particles. This system also calculates the cholesterol concentration for each lipoprotein fraction according to a total cholesterol value obtained for each plasma sample.
Macrophage isolation and differentiation
Bone marrow-derived macrophages were isolated and cultured as follows. Briefly, 3-month-old mice were euthanized by carbon dioxide inhalation. Hind legs were exposed and the muscle tissue removed to isolate the femurs and tibiae. The bone marrow was flushed from femurs and tibiae with culture medium (DMEM) containing 10% fetal calf serum and 1% penicillin/streptomycin/l-glutamine (Invitrogen, Carlsbad, CA) using a syringe and a 25-gauge needle. The cell suspension was drawn through a syringe with an 18-gauge needle to dissociate cell clumps and was passed through a 70-μm pore cell strainer (BD BioSciences, San Jose, CA) to remove tissue debris. The cells were plated and incubated for 5 days in the presence of 25 ng/ml macrophage colony stimulating factor (M-CSF) (PeproTech, Rocky Hill, NJ) in order to initiate macrophage differentiation. Foam cell differentiation from macrophages was achieved with 50 μg/ml acetylated LDL (Alfa Aesar, J65029) added to the cell culture media for 48 h. For macrophage polarization towards the classic M1 and alternate M2 phenotypes, macrophages were kept for 24 h in DMEM medium supplemented with 20 ng/mL TNFα for M1 differentiation and with 20 ng/mL IL-4 for M2 differentiation. Cells were harvested after 24 h for total RNA isolation.
Cholesterol efflux
Bone marrow-derived macrophages (BMDMs) were seeded in 24-well plates at a density of 5 × 105 cells per well in triplicate and allowed to adhere overnight. Cells were incubated with loading medium (DMEM/F12 Glutamax (Invitrogen), 1% penicillin/streptomycin, 0.2% fatty acid-free bovine serum albumin, 50 μg/mL acetylated LDL and 1 μCi/ml [3H]-cholesterol (NEN Life Science products, Boston, MA) for 36 h. The cells were then washed twice with PBS and equilibrated in 0.2% BSA in DMEM/F12 media for 2 h. Efflux media (DMEM, 0.2% BSA) with or without 20 μg/mL apoA1 (acceptor for ABCA1-mediated efflux) or 50 μg/mL HDL (acceptor for ABCG1-mediated efflux), was then added to each well for 7 h. Supernatants and cell lysates were then collected, and the amount of [3H]-cholesterol was determined with a scintillation counter. The percent cholesterol efflux at baseline or to ApoA1 or HDL was expressed as a percentage of the total loaded [3H]-cholesterol using the following equation: (total effluxed [3H]-cholesterol in media)/(total effluxed + total cellular [3H]-cholesterol).
Immunohistochemistry on macrophages
Bone marrow-derived macrophages were seeded in 24-well plates onto small coverslips placed inside the wells. Cells were cultured as described above with or without 50 μg/mL acetylated LDL (for foam cell differentiation). Cells were then fixed and permeabilized with cold methanol for 5 min at − 20 °C. Rabbit monoclonal anti-ABCA1 (NB400-105SS, Novus Biologicals, Littleton, CO) and anti-ABCG1 (PA5-13462, Thermo Fisher, Waltham, MA) antibodies were used to specifically detect ABCA1 and ABCG1 in BMDMs. The secondary antibody was AlexaFluor 488 (Life Technologies, CA). The coverslips with the cells from the wells were placed onto slides and immunofluorescent images were acquired using an Axioskop 2 fluorescent microscope (Zeiss, Thornwood, NY). Individual images were collected and processed with Photoshop CS6 (Adobe, San Jose, CA).
Western blot
ABCA1 and ABCG1 protein expression in bone marrow-derived macrophages and foam cells were also detected using standard western blot techniques we previously described18. Proteins were extracted on ice in RIPA buffer containing 1X protease inhibitor mini complete cocktail (Roche Applied Science, Indianapolis, IN), and 5 mmol/L EDTA. Homogenates were centrifuged at 16,000 g and the protein concentration in the supernatant was determined by absorbance using a Nanodrop spectrophotometer (ThermoScientific, Wilmington, DE). 50 μg was combined with reducing Novex sample buffer (Invitrogen, Carlsbad, CA) and loaded into wells of a 4–12% polyacrylamide gel (Invitrogen, Carlsbad, CA). Proteins were transferred to nitrocellulose membrane and blotted with the anti-ABCA1 and ABCG1 antibodies (NB400-105SS, Novus Biologicals, Littleton, CO and PA5-13462, Thermo Fisher, Waltham, MA). Appropriate secondary antibodies were detected using the Odyssey infrared imaging system (Li-cor, Lincoln, NE). β-actin was used as a loading control (Abcam Ab8229, Cambridge MA) and mouse lung lysates were used as a positive control (not shown).
Reverse transcription PCR
Real-Time PCR was used to determine the level of mRNA expression of various genes in cultured macrophages and mouse tissues. Total RNA was extracted from macrophages/foam cells or approximately 20 mg of tissue samples using the RNeasy kit (Qiagen Inc., Valencia, VA). RNA was converted into first-strand cDNA using a SuperScript III First-Strand Synthesis SuperMix kit (Thermofisher, Waltham MA). The level of gene expression was detected by quantitative RT-PCR (qRT-PCR) with a StepOnePlus Real-Time System (Applied Biosystems, Foster City, CA) using commercially available TaqMan probes from Life Technologies. (Ccl2: Mm00441242_m1, TNFα: Mm00443258_m1, ApoE: Mm01307193_g1, Ccr2: Mm99999051_gH, CD36: Mm00432403_m1, Abca1: Mm00442646_m1, Abcg1: Mm00437390_m1, Hmgcr: Mm01282499_m1, SR-B1: Mm00450234_m1, Cyp7a1: Mm00484150_m1, ApoA1: Mm00437569_m1). As an endogenous control, gene Hmbs (Mm01143545_m1) was used for the macrophage/foam cell samples and Gapdh (Mm99999915_g1) was used for the liver samples.
Plasma biliary acids measurement by UPLC-MS/MS
Bile acid measurements were performed with 50 μl of mouse plasma using D4-bile acids as internal standards. Each sample was deproteinized with 200 μl of acetonitrile (ACN). After a 10.000 × g centrifugation at 15 °C for 15 min, the upper phase was collected and dried under nitrogen at 50 °C. All samples were resuspended in 100 μl of a mixture (70:30, A:B, v:v) of a solvent A (95% ultrapure H2O / 5% acetonitrile (ACN) + 10 mM of ammonium formate) and B (20% ultrapure H2O / 80% ACN + 10 mM of ammonium formate). UPLC-MS/MS measurements were performed on a Waters Acquity H-class UPLCTM associated with a Waters Xevo triple quadripole equipped with an electrospray ionization (ESI) source. The UPLC was fitted with a Cortec UPLC C18 column (1.6 μm—2.1 Å ~ 100 mm; Waters) set at 40 °C and 10 μl of sample were injected onto the column. Elution was achieved with a linear gradient of solvent B in solvent A at a flow rate of 0.3 ml/min. The elution method was as follows: 10% of solvent B up to 45% in 8 min—45% of solvent B up to 90% of solvent B in 4 min—90% of solvent B kept constant for 1 min—90% of solvent B down to 10% of solvent B in 1 min—10% of solvent B kept constant for 1 min prior to the next injection. ESI was used in negative mode with a capillary, cone and collision voltages set at 3 kV, 80 V and 12 eV, respectively. All UPLCMS/MS data were integrated and analyzed with MassLynx and TargetLynx softwares (Waters Corp. Milford, MA).
Cytokine and Chemokines
TNFα, IFNγ, IL-1 β, IL-6, IL-10, IL-12p70, IL-17A, CCL2, CCL4, CCL5, CXCL8 and CXCL10 concentrations were measured in human plasma samples by multiplex fluorescent-bead-based technology (Luminex 200, Austin, TX) using a commercially available Luminex screening assay (R&D Systems, Minneapolis, MN). Cytokines/chemokines were measured in mouse plasma samples using the Th1 / Th2 / Th17 Cytokines Multi-Analyte ELISArray Kits from QIAGEN Inc. (Valencia, VA).
Data analysis
Results are presented as the mean +/− standard error of the mean (SEM). Animal numbers used for individual sets of data varied between experiments and are shown on the figures. For multigroup comparisons, one-way ANOVA (F) followed by the Tukey HSD-test was used for detecting the statistical differences. The ANOVA F values shown in the results are the ratio of two mean square values. If the null hypothesis is true, F is expected to have a value close to 1.0. A large F ratio signifies that the variation among group means is more than you'd expect to see by chance. The Student’s t-test was used for detecting differences between two groups by comparing the effect of one variable such as genotype, cell differentiation or diet. Relative gene expression was calculated using the delta-delta threshold method29. Difference between relative gene expressions greater than 1.5-fold were considered statistically significant. Results from the comparison between PXE patients and the NUMEVOX cohort (described in the results section and Table 2) were analyzed using Kruskal Wallis and performed with Stata 12.0 software (StataCorp, Texas, USA). The level of statistical significance was set at P < 0.05. Statistical comparison into the groups were performed using Wilcoxon’s test. A p value < 0.05 was considered statistically significant. Analyses were performed using GraphPad’s PRISM software v.8.4.2 (San Diego, CA).
Results
ABCC6 deficiency induces dyslipidemia in mice and humans
The premise of this study was that ABCC6 may influence the levels of plasma lipoproteins and susceptibility to atherosclerosis. To explore this possibility, we generated 3 mouse genotypes with normal, reduced and no Abcc6 expression in an atherogenic background (Ldlr−/−) to determine if gene dosage affects dyslipidemia, atherosclerosis and possibly vascular calcification.
Mice
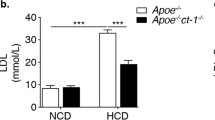
We first determined the effect of ABCC6 deficiency on lipoprotein profiles using a Lipoprint analyzer in several mouse models fed either normal chow or a high fat diet for 16 weeks. We used Abcc6+/−;Ldlr−/− and Abcc6−/−;Ldlr−/− mice in comparison with Ldlr−/− controls as well as Abcc6−/− and wild type (WT) animals. The results, summarized in Table 1, showed that under non-atherogenic conditions (i.e. normal diet and Ldlr+/+ status), ABCC6 deficiency leads to substantially reduced HDL and increased LDL levels (p = 0.03 and p = 0.0035, n = 8 versus n = 3, respectively). Lipoprint analysis allowed the quantitative evaluation of the main HDL and LDL sub-fractions and notably revealed that the large HDL subclass, which shows an inverse relationship to cardiovascular risk, was significantly reduced in Abcc6−/− mice (Supplemental Table 1). Interestingly, the highly atherogenic sub-fractions LDL 3 through 7 (labelled as “Large LDL” on Supplemental Table 1) were elevated in Abcc6−/− mice but only in animals fed normal chow. When compared to control mice, total cholesterol and HDL levels were particularly affected in Abcc6−/− mice fed a lipid-rich diet, though no atherosclerosis was noted in these animals (not shown). Triglyceride levels were significantly higher in all Abcc6-deficient animals as compared to Ldlr−/− or WT controls, except for the double KO on high fat diet (Table 1). Under severe atherogenic conditions (Ldlr−/− and high fat diet), experimental mice displayed a ~ tenfold increase in total cholesterol as compared to Ldlr+/+ mice but there was no significant difference between the three genotypes. However, major alterations of HDL and LDL levels were found in both Abcc6+/−;Ldlr−/− and Abcc6−/−;Ldlr−/− mice in comparison to Ldlr−/− controls (Table 1). The gradual decrease to undetectable levels of the small HDL fraction was notable (Supplemental Table 1).
Because ABCC6 deficiency did not affect atherosclerosis development in mice with an ApoE−/− background as reported30, we specifically examined the plasma levels of this apolipoprotein in Abcc6+/−;Ldlr−/− and Abcc6−/−;Ldlr−/− mice, as it may have been a contributor to the enhanced atherosclerosis phenotype we observed. However, there was no significant variation as compared to Ldlr−/− controls (Supplemental Fig. 1A).

Atherosclerosis development in Ldlr−/− mice lacking Abcc6. Atherosclerotic plaque deposition in aorta was assessed by measuring the en face surface area stained by Oil Red O staining. Littermate mice were maintained on an atherogenic diet (high fat diet) or regular chow for 16 weeks. (A) Quantification of aortic lesions is shown as percentage of the aorta surface. (B) Representative images of the Oil Red O staining. Results are shown as means ± SEM. p-values were determined by one-way ANOVA and Tukey’s post hoc test. *p < 0.05, **p < 0.01, ****p < 0.0001.
PXE patients
We followed up the murine investigation with a comprehensive analysis of the lipoproteins from PXE patients (Table 1). Total cholesterol levels were not statistically significant from healthy age- and sex-matched controls. However, the patients presented a highly significant reduction in HDL levels (− 24%, p = 0.0004) in the same proportion as Abcc6−/− vs wild type mice on a normal diet. The small and large HDL sub-fractions were also decreased as compared to healthy controls (Supplemental Table 1) but there was no change in LDL, unlike the animal models. We found no difference in plasma triglyceride levels (Table 1) or plasma ApoE concentration (Supplemental Fig. 1B) overall between PXE patients and healthy controls. However, significant differences in triglyceride levels were found between sub-groups. Triglycerides were elevated in control males (+ 82%, but still within normal range) as compared to control females (p = 0.0013, n = 10 versus n = 24). Average plasma triglyceride levels were also higher (+ 47%) in female patients vs female controls (p = 0.0066, n = 24), but, remarkably, there was no difference between male and female PXE patients (p = 0.88).
The lack of ABCC6 enhances atherosclerosis
Mice
Ldlr−/−, Abcc6+/−;Ldlr−/− and Abcc6−/−;Ldlr−/− mice were subjected to 16 weeks of high fat diet to induce atherosclerotic lesions. Quantification of aortic plaque using the en face method from the three strains of mice revealed a statistically significant increase in plaque development in mice lacking ABCC6 as determined by one-way ANOVA F (2, 20) = 9.205 p = 0.0015. Particularly noteworthy, plaque development in heterozygous Abcc6+/−;Ldlr−/− mice was as pronounced as it was in Abcc6−/−;Ldlr−/− mice (p = 0.80) despite the presence of a functional Abcc6 allele (Fig. 1). Under normal chow conditions, we did not observe any significant atherosclerosis in Ldlr−/− mice, as expected; however, Abcc6+/−;Ldlr−/− and Abcc6−/−;Ldlr−/− mice presented small but clear evidence of statistically significant lipid deposition in the aortic arch (one-way ANOVA F (2, 9) = 5.338 p = 0.0296) that mirrored calcification increases under the same conditions (Fig. 2).

Aortic calcification in Ldlr−/− mice lacking Abcc6. The level of calcification in the aorta was measured by total calcium content of the full aorta normalized to the weight of the tissue. Littermate mice were maintained on an atherogenic diet (high fat) or regular chow for 16 weeks. As shown by the blue bars, the level of vascular calcification induced by Abcc6-deficiency was not influenced by the diet. Results are shown as means ± SEM. p-values were determined by one-way ANOVA and Tukey’s post hoc test (*) or Student’s t-test (#).*, #p < 0.05, **p < 0.01, ***, ###p < 0.001.
As the highest level of Abcc6 expression is normally found in the liver12, we verified whether the lack of hepatic ABCC6 function could affect the expression of selected genes involved in reverse cholesterol transport (Abca1, Abcg1, Sr-b1, ApoA1) or cholesterol metabolism (3-hydroxy-3-methylglutaryl CoA reductase or Hmgcr and Cyp7α1). There was no significant change in Abcc6−/−;Ldlr−/− mice as compared to control Ldlr−/− mice fed normal or high fat diets except in Abcg1 expression which was significantly downregulated (p = 0.02) under high fat dietary conditions in an Abcc6-dependent manner (Supplemental Fig. 2B).
PXE patients
To determine if PXE patients are susceptible to vascular remodeling, we performed a retrospective analysis of data collected from 142 patients routinely followed at the University Hospital of Angers, France (by L.M. and G.L.). The effect of ABCC6 deficiency on the vasculature has already been extensively documented in many comparisons with healthy controls22,31,32,33,34,35. Therefore, to provide a novel disease perspective to which the effects of the PXE pathophysiology could be gauged against, we compared data from PXE patients with a cohort already at risk for significant cardiovascular anomaly i.e. 668 dysmetabolic individuals screened from the NUMEVOX cohort36.
In both cohorts, carotid intima-media thickness (IMT) was determined using ultrasound imaging coupled with echotracking according to standardized methods while plaques were scored (i.e. presence/absence) bilaterally at the carotid, abdominal aorta and femoral bifurcations. Note that in the absence of a direct histopathology analysis, the nature of the plaque seen in PXE patients and control individuals is herein assumed to be of atheromatous nature. PXE patients showed no significant difference in intima-media thickness (IMT) as compared to the cohort at risk (617 ± 164 µm versus 650 ± 121 µm, p = 0.720). Adjustment for age and number of sites with atherosclerotic plaques did not affect the lack of statistical significance (β = − 0.0011, p = 0.312). However, PXE patients who had different (better) lipid profiles with modest dyslipidemia (average cholesterol ratio of 3.7) presented a higher number of atherosclerotic sites as compared to the NUMEVOX cohort (1 ± 3 versus 2 ± 4, p = 0.023). After adjusting for age, PXE patients still showed a significantly larger number of atherosclerotic sites than dysmetabolic controls (β = 0.39, p = 0.015), despite having less arteriosclerosis. These data (Table 2) suggest that atherosclerotic plaques at typical sites are more prevalent in PXE patients than in subjects with metabolic syndrome who are already at an increased risk for cardiovascular diseases. This suggests that the disease status of PXE patients enhances susceptibility to plaque formation.
Enhanced systemic inflammation is associated with ABCC6 dysfunction
Abcc6-deficient mice
It is now well accepted that atherosclerosis is a chronic inflammatory disease mediated by the concerted action of many circulating pro-inflammatory cytokines. Therefore, we measured selected cytokines in the plasma of Ldlr−/− and Abcc6−/−;Ldlr−/− mice fed regular chow. We found no difference or no detectable levels for IL-2, IL-4, IL-13, IL-23, TNFα, TGFβ1 or IFNγ, in both strains of mice. However, significant levels of the pro-inflammatory cytokines IL-6, IL-12 and IL17A were measured in the plasma of Abcc6−/−;Ldlr−/− mice (27.3 ± 1.9 pg/mL, 0.50 ± 0.04 pg/mL, 4.1 ± 0.6 pg/mL, n = 3, respectively) but were undetectable in Ldlr−/− controls.
PXE patients
We also profiled cytokines and additional chemokines in the plasma of PXE patients in comparison to healthy controls. There was no difference in the levels of the pro-inflammatory cytokines TNFα, IL-1β, IL-17A, CCL2, CCL4, CXCL8 and CXCL10, the anti-inflammatory cytokine IL-10 or the Th1-related cytokines IFNγ and IL-12. Remarkably, PXE patients showed elevated plasma levels of pro-inflammatory (atherogenic) cytokine IL-6 (1.11 ± 0.35 pg/mL, n = 31 versus 0.24 ± 0.08, n = 24; p = 0.03) as well as the pro-atherogenic chemokine CCL-5 (24,781 ± 3240, n = 31 versus 6374 ± 907, n = 24; p < 0.0001). Of note, because values for IL-6 obtained with the Luminex system were at the limit of detectability, plasma values were verified with a Quantikine HS ELISA Kit (R&D Systems, Minneapolis, MN) and similar values were obtained (1.91 ± 0.41 pg/mL, n = 31 versus 0.81 ± 0.09, n = 25; p = 0.023).
ABCC6 deficiency does not influence vascular calcification associated with atherosclerosis
Mice
After 16 weeks of high fat or normal chow feeding, we found elevated but equivalent levels of overall aortic calcification (2.7 fold, p < 0.05) in all three Ldlr−/−, Abcc6+/−;Ldlr−/− and Abcc6−/−;Ldlr−/− genotypes fed a high fat diet as compared to Abcc6−/− mice under the same conditions (Fig. 2). This result suggested that the increased calcification was only a consequence of atherosclerotic plaque (intimal) and not linked to the Abcc6 genotype. This conclusion is supported by the increased mineralization in Abcc6+/−;Ldlr−/− and Abcc6−/−;Ldlr−/− mice fed regular chow when compared to control Ldlr−/− and Abcc6−/− mice, as the first two mouse models presented atherosclerotic lesions whereas the two controls did not (Fig. 2).
For control purposes, we evaluated whether the absence of the Ldlr gene and a lipid-rich diet would alter the typical PXE calcification phenotype found in Abcc6−/− mice. The lack of Ldlr alone or in combination with high fat diet had no significant influence on the mineralization of the vibrissae capsule (Supplemental Fig. 3A). High fat diet can also induce cardiac calcification in mice lacking functional ABCC620. Here, there was no evidence of high fat diet-related DCC in the relatively short timeframe (16 weeks) of this study (Supplemental Fig. 3B).

Cholesterol efflux from isolated bone marrow-derived macrophages. Macrophages were pre-loaded with 1 μCi/mL of cholesterol and 50 μg/mL AcLDL in 24-well plates using media with 10% lipoprotein deficient serum for 36 h. Lipid-free ApoA1 or HDL were used as cholesterol acceptors in a 7-h incubation period. The mouse genotypes are indicated. Results are shown as means ± SEM. p-values were determined by one-way ANOVA and Tukey’s post hoc test (*). *p < 0.05.
PXE patients
As skin calcification cannot be readily quantified in PXE patients, we determined arterial calcium score as previously described34. Not surprisingly, our cohort of patients displayed significantly elevated arterial calcium score as compared to healthy controls (TEP scan units 1356 ± 430.3, n = 16 vs 137.9 ± 68.3, n = 17 respectively, p = 0.007.), however, these measures did not readily distinguish medial from intimal calcification.
The role of macrophages in dyslipidemia and atherosclerosis associated with ABCC6 deficiency in mice
Macrophages play a prominent role in the development of atherosclerotic lesions, and this has been extensively documented37,38. These immune cells normally efflux cholesterol to ApoA1, nascent and mature HDL via the ATP-binding cassette transporter A1 (ABCA1) and ABCG1, which are key steps in reverse cholesterol transport. The dysregulation of this process in vascular macrophages promotes atherogenesis39. To determine the role of macrophages in the dyslipidemia and atherosclerosis phenotypes we observed in ABCC6-deficient mice (and PXE patients), we first used bone marrow-derived macrophages isolated from our experimental mice to study cholesterol efflux. We found no difference of baseline efflux (without acceptor) or efflux towards ApoA1 between the three genotypes. However, when HDL particle acceptors were present, there was a small but significant decrease in efflux in equivalent proportions in Abcc6+/−;Ldlr−/− and Abcc6−/−;Ldlr−/− macrophages (Fig. 3).
To investigate a possible cause of this decrease in efflux, we quantified the expression of Abca1, Abcg1, ApoE, Hmgcr, Cd36 and Sr-b1 genes in both macrophages and foam cells from Ldlr−/− and Abcc6−/−;Ldlr−/− mice. The differentiation of macrophages into foam cells enhanced the expression of Abca1 and Abcg1, but there was no Abcc6-specific effect (Fig. 4A,B). These results were confirmed at the protein level as well (Fig. 4C,D,E). Similarly, the expression of Hmgcr and Sr-b1 dropped in foam cells but no Abcc6-dependent effect was observed (Fig. 4G,H,I). This result is somewhat similar to previous findings40. ApoE expression (Fig. 4F) showed no difference, which was consistent with plasma levels (Supplemental Fig. 1A), though Cd36 showed a modest (but not significant) expression decrease in Abcc6−/−;Ldlr−/− macrophages (Fig. 4H). Because the changes in gene expression were either small and/or inconsistent between macrophages and foam cells, they seemed unlikely to be physiologically relevant. The expression of Enpp1 and Nt5e was also analyzed as these genes encode proteins immediately downstream of the pathway initiated by ABCC65, 9, 10. There was no significant change for Enpp1, but the expression of Nt5e was upregulated in both macrophages and foam cells from Abcc6−/−;Ldlr−/− mice (Fig. 5A,B). Furthermore, we analyzed the expression of selected cytokine genes that play a role in atherosclerosis. We only found a significant increase in the expression of Ccl-2 (Mcp-1) in Ldlr−/−;Abcc6−/− macrophages vs Ldlr−/− controls, Fig. 5C) whereas Ccr-2 and TNF-α showed not significant change (Fig. 5D,E).

Expression of genes/proteins related to cholesterol efflux in bone marrow-derived macrophages and foam cells. Real-time RT-PCRs were performed using TaqMan probes specific for Abca1 (A), Abcg1 (B), ApoE (F), Hmgcr (G), Cd36 (H) and Sr-b1 (I) cDNAs. Units are the relative gene expression normalized to Hmbs. Most results showed expected differences between macrophages and foam cells but not between the genotypes. Results are shown as means ± SEM. p-values were determined by Student’s t-test, * p < 0.05. (C) Representative western blot images showing the levels of both ABCA1 and ABCG1 expression (red signal) in foam cells (FC) as compared to macrophages (Macro). β-actin (green) served as loading control. Data points represent individual mice from which bone marrow-derived macrophages were isolated and used for experiments. (D) Immunofluorescent detection (green signal) of ABCA1 and ABCG1 on macrophages (Macro) and foam cells (FC) derived from Abcc6−/−;Ldlr−/− and control Ldlr−/− mice. Five representative images from each condition are shown. We only observed some staining pattern variation for ABCA1 in macrophages from Abcc6−/−;Ldlr−/− mice which appeared punctated as compared to cells from control Ldlr−/− mice. (E) Negative controls for the immunofluorescent staining shows the specificity of the primary antibodies used. Nuclei were stained with DAPI.

Expression of genes related to cholesterol and extracellular purine metabolisms and cytokines/chemokines in macrophages and foam cells derived from Abcc6−/−;Ldlr−/− and control Ldlr−/− mice. Real-time RT-PCRs were performed using TaqMan probes specific for Enpp1 (A), Nt5e (B), Ccl-2 (C), Ccr-2 (D) and TNF-α (E) cDNAs. Units are the relative gene expression normalized to Hmbs. The most consistent changes and probably the most physiologically relevant are shown in panels (B,C) with Nt5 and Ccl-2 expression significantly increased in cells lacking Abcc6. Data points represent individual mice from which bone marrow-derived macrophages were isolated and used for experiments. Results are shown as means ± SEM. p-values were determined by Student’s t-test, *p < 0.05.
Remarkably, we found no significant Abcc6 expression in bone marrow-derived macrophages M1 or M2 activated or foam cells from Ldlr−/− mice (not shown).
Changes in plasma bile acids in mice lacking ABCC6
Cholesterol (as HDL) is brought to the liver via reverse transport for recycling or elimination. Because bile acids are quickly absorbed in the intestines and recycled towards the liver via the enterohepatic recirculation, we measured the levels of bile acids in the plasma of Abcc6−/−;Ldlr−/− and control Ldlr−/− mice fed either normal or high fat diets. In both dietary conditions, we found a profound decrease in the circulation of the following bile acid concentrations: cholic, α- and β-muricholic acids, chenodeoxycholic acid, deoxycholic acid, ω-muricholic acid, hyodeoxycholic acid and ursodeoxycholic acid. However, this decrease was more pronounced in mice fed high fat diet than normal chow (Fig. 6). Lithocholic acid was not detectable in our samples (not shown) but the other 8 primary, secondary and tertiary bile acids showed drastic reductions ranging from − 55 to − 99% (p < 0.0001) in Abcc6−/−;Ldlr−/− mice with cholic, β-muricholic and ω-muricholic acids being the most affected, decreasing to undetectable levels.

Plasma bile acid profiles in Ldlr−/− mice lacking Abcc6. Littermate mice were maintained on an atherogenic diet (high fat) or regular chow for 16 weeks. Panel (A) shows the primary bile acids: cholic, α- and β-muricholic acids and chenodeoxycholic acid. Panel (B) illustrates results for the secondary bile acids deoxycholic acid, ω-muricholic acid and hyodeoxycholic acid. Panel (C) represents data for the tertiary ursodeoxycholic acid. Results are shown as means ± SEM. p-values were determined by Student’s t-test. ****p < 0.0001.
In the final steps of the reverse cholesterol transport, the cholesterol half-transporters ABCG5 (ATP-binding cassette transporter G5) and ABCG8 (ATP-binding cassette transporter G8) play a critical role. Both hemi-transporters are present on the apical membrane of hepatocytes facilitating excretion of cholesterol and plant sterols into the bile and are also expressed in the intestinal epithelial cells41,42. Because elevated expression of both ABCG5/G8 in Ldlr−/− mice contributes to the attenuation of diet-induced atherosclerosis43, we examined the expression of Abcg5/8 in the liver of WT, Abcc6−/−, Ldlr−/− and Abcc6−/−;Ldlr−/− mice. We found that the deletion of the Ldlr gene induced increased Abcg5/8 expression in the liver under normal dietary conditions. There was a trend towards lower expression of Abcg5/8 in Ldlr−/− mice lacking ABCC6 but the difference was not statistically significant with the limited number of samples available for this experiment (Fig. 7A). However, with mice fed high fat diet, the hepatic expression of Abcg5 and Abcg8 decreased significantly (p = 0.020, p = 0.016 respectively, n = 4) in Abcc6−/−;Ldlr−/− mice as compared to Ldlr−/− controls (Fig. 7B), which is consistent with the reduction in plasma bile acids.

Expression of the Abcg5 and Abcg8 genes in the liver of Abcc6−/−;Ldlr−/− and control Ldlr−/− mice. Real-time RT-PCRs were performed using TaqMan probes specific for Abcg5 (A) and Abcg8 (B). Units are the relative gene expression normalized to Gapdh. Results are shown as means ± SEM. p-values were determined by Student’s t-test, *p < 0.05, ***p < 0.001.
Discussion
Several publications describing PXE patients and Abcc6-deficient mouse models have previously reported sporadic and circumstantial evidence of alteration of lipoprotein levels and arteriosclerosis16,25,44,45,46,47. However, the relationship between ABCC6, lipoproteins and calcification has never been specifically studied. This study examined this association using mouse models under normal or atherogenic conditions and compared results with human PXE patients. We found that ABCC6 deficiency caused significant changes in lipoprotein levels and enhanced atherosclerosis, which were associated with decreased cholesterol efflux from macrophages and the dysregulation of genes involved in purinergic signaling including Nt5e5,6,48,49, and in reverse cholesterol transport (ABCG5/8). The presence of pro-inflammatory conditions was likely a compounding factor. Remarkably, dyslipidemia and the development of atherosclerotic lesions occurred in a haploinsufficient manner, unlike the calcification phenotype caused by ABCC6 mutations.
Dyslipidemia and atherosclerosis occur independently
The possible effect of ABCC6 deficiency on lipoproteins was first described in PXE patients by Wand et al.47. The authors of this study reported an association between an ABCC6 variant (now considered non-pathogenic35,50,51) and elevated HDL levels and low triglycerides. However, these results are inconsistent with what others have since described22,31,33. A few years later, Gorgel et al. obtained more solid but limited lipoprotein data using mice25. The results presented here have now largely expanded upon these initial observations with a more detailed analysis of both mice and humans lacking functional ABCC6. Remarkably, we observed in our mouse model with an atherogenic background (Abcc6−/−;Ldlr−/−) the formation of atherosclerotic plaque in the aorta even under normal dietary conditions and in the absence of dyslipidemia. These results suggest that plaque development occurs in these mice independently of LDL/HDL levels, and imply that other molecular, cellular and/or physiological mechanism(s) are involved. The elevated triglycerides under these normal dietary conditions were probably one of the contributing factors52. When these mice were challenged with high fat diets, ABCC6-specific differences in lipoprotein levels and plaque development were dramatically enhanced indicating that ABCC6 influences lipoprotein levels and atherosclerosis through independent but compounding mechanisms.
ABCC6 influences plaque formation by modulating HDL
Although mice are the most common animal model used to study human diseases, the biology of rodents can be noticeably divergent53. Abcc6−/− mice, despite the known differences with the human phenotype54, have been extraordinarily useful for the study of PXE55,56 and the development of therapeutic approaches49,57,58,59,60. However, mice present singular differences in lipoproteins and atherosclerosis development61. For this reason, the overlapping characteristics between animal and PXE patient data weighed heavily in our interpretations. Even though we could only speculate as to what the link between ABCC6 molecular function and HDL metabolism could be, we distinguished in our data and the literature several arguments suggesting that ABCC6 modulates atherosclerosis by influencing HDL quantity and/or quality62, as it is the most common denominator between our mouse models and human subjects.
-
1.
We explored possible cellular mechanisms that could enhance atherosclerosis. We first examined cholesterol efflux from bone marrow-derived macrophages after lipid loading. Reduced cholesterol efflux from macrophages within the plaque is probably a significant contributing factor to the enhanced atherosclerotic plaque development we observed in the animal models and PXE patients especially in the absence of changes to LDL in humans. However, the reason behind the strictly ABCC6-dependent decrease in cholesterol efflux is unclear. Indeed, there was no significant change in Abca1 or Abcg1 expression and protein levels or in other related genes between control and experimental macrophages and foam cells. Furthermore, the notable absence of Abcc6 mRNA in macrophages likely reflected an acquired phenotype that persisted ex-vivo after bone marrow isolation and macrophage differentiation. We and others have previously described similar situations with primary skin fibroblasts, which express little to no ABCC6, from PXE patients that retain altered characteristics ex-vivo40,63,64. An alternative explanation could be that even a minimal ABCC6 presence may exert a meaningful impact on cellular functions as Hendig and co-workers suggested65,66.
-
2.
A recent study of ABCC6-deficient mice in ApoE−/− background showed no significant changes in atherosclerosis (or arterial calcification) as compared to ApoE−/− controls30. The results of this study are not necessarily discordant with ours and in fact provided some support to our overall conclusion regarding HDL. ApoE−/− mice are a very useful and popular animal model. However, atherogenesis in mice is quite distinct from humans as HDL cholesterol is virtually absent in these mice67 and atherosclerosis is not a distinguishing feature of humans lacking ApoE68. In that sense, Ldlr−/− mice provide a pathophysiology somewhat closer to that of humans as plaque formation is primarily driven by higher LDL and lower HDL69 and this is what we observed in the present study (Table 1, and Fig. 1). Since Van der Veken et al. reported no difference in atherosclerosis or lipoproteins between Abcc6−/−;ApoE−/− mice and ApoE−/− controls, we first suspected that ApoE could play a role in our model. Indeed, the absence of ApoE in macrophage promotes atherosclerosis without changing plasma cholesterol and we found that atherosclerosis occurred independently of dyslipidemia70. However, we found no significant variation in the plasma of mice or in PXE patients. Another likely possibility was that ApoE−/− mice have very little or no circulating HDL. If ABCC6 modulates atherogenesis by its differential effect on HDL quantity (Table 1), and/or quality62, then ABCC6 would have less influence on plaque formation in ApoE−/− mice. In this regard, the data from Van der Veken et al. are consistent with our results and one could regret the lack of cholesterol efflux experiment in their study.
-
3.
Reverse cholesterol transport, which is primarily mediated by HDL, is a mechanism by which excess cholesterol is removed from peripheral tissues and delivered to the liver for either redistribution or excretion in the form of bile acids. About 95% of bile acids are reabsorbed during the digestive process. Thus, lower plasma bile acid level signals either a reduction of the enterohepatic recirculation (intestinal absorption and liver reabsorption) and/or decreased reverse cholesterol transport. Lower HDL plasma concentration, reduced cholesterol efflux from foam cells and the dramatic changes in plasma bile acids are all consistent with altered reverse cholesterol transport. The lack of change in Sr-b1, ApoA1 and Cyp7α expression indicated that the canonical FXR pathway was unaffected in liver and also agrees with this interpretation.
-
4.
As circumstantial evidence, a recent comparative study of polar and brown bear genomes identified ABCC6 as one of 20 genes under positive selection that allowed polar bears to adapt to a lipid-rich diet and elevated plasma LDL71.
-
5.
Furthermore, we found clear evidence for an ABCC6-dependent pro-inflammatory status both at the cellular levels in murine macrophages and foam cells with overproduction of CCL-2 as well at the systemic levels with elevated plasma levels of IL-6 both in PXE patients and our mouse models. The role of inflammation in atherosclerosis development is well-documented and thus these cytokines and chemokines are likely factors aggravating atherosclerosis associated with ABCC6 deficiency.
The pluripotent physiological function of ABCC6
The emerging model that ABCC6 is a modulator of the dynamic equilibrium of extracellular nucleotides (ATP) and adenosine5,6,9,48,49,72 is an indication that ABCC6 physiological function extends beyond the regulation of mineralization. Indeed, ABCC6 activity facilitates the cellular efflux of ATP from liver and other tissues/cells11,12, which is sequentially converted by ENPP1 and NT5E (i.e.CD73) into pyrophosphate (PPi) and adenosine, two potent inhibitors of mineralization5,7. However, ATP and adenosine have numerous other biological activities, notably towards inflammation73,74,75 and atherosclerosis76,77,78,79,80. Remarkably, the other enzymes downstream of ABCC6 (Fig. 8) that regulate ectopic calcification in PXE, GACI and CALJA4,5,7,9,13,48,49,81 have also been shown to play a role in atherosclerosis development76,77,78,79. Of note, ENTPD1 (i.e. CD39), which has a molecular function similar to that of ENPP1, influences atherosclerosis82,83, though this ectoenzyme does not regulate calcification (G. Kauffenstein, personal communication). Our current data on ABCC6 fit this pattern of dual function in this pathway.

The proteins/enzymes in this pathway regulate both calcification and atherosclerosis. ABCC6 activity facilitates the cellular efflux of ATP from liver and other tissues/cells, which is quickly converted to pyrophosphate (PPi), a potent inhibitor of mineralization. Decreased plasma PPi levels cause calcification in PXE (OMIM #264800) and GACI (OMIM #614473 and #208000). NT5E activity leads to adenosine production which has numerous biological activities towards inflammation, atherosclerosis and inhibition of TNAP synthesis. TNAP degrades PPi into inorganic phosphate (Pi), an activator of calcification which leads to vascular calcification in Calcification of Joints and Arteries patients (CALJA, OMIM #211800). In addition to ABCC6, both ENPP1 and NT5E functions have been linked to atherosclerosis development in ApoE−/− mice. ENTPD1 function overlaps with that of ENPP1 and also plays a role in atherosclerosis in mice lacking ApoE, though this ectoenzyme does not seem to regulate ectopic calcification. Symbols: ∅ : no known effect; ➚: increase; ➘: decrease.
In conclusion, the present study reveals a novel physiological role for ABCC6, influencing plasma lipoproteins and atherosclerosis in a haploinsufficient manner, with significant penetrance.
References
Ho, C. Y. & Shanahan, C. M. Medial arterial calcification: an overlooked player in peripheral arterial disease. Arterioscler. Thromb. Vasc. Biol. 36, 1475–1482. https://doi.org/10.1161/ATVBAHA.116.306717 (2016).
Ilias, A. et al. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6). J Biol Chem 277, 16860–16867 (2002).
Le Saux, O. et al. The molecular and physiological roles of ABCC6: more than meets the eye. Front. Genet. 3, 289. https://doi.org/10.3389/fgene.2012.00289 (2012).
Le Saux, O. et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 25, 223–227 (2000).
Jansen, R. S. et al. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc. Natl. Acad. Sci. U. S. A. 110, 20206–20211. https://doi.org/10.1073/pnas.1319582110 (2013).
Kauffenstein, G. et al. Alteration of extracellular nucleotide metabolism in pseudoxanthoma elasticum. J. Investig. Dermatol. 138, 1862–1870. https://doi.org/10.1016/j.jid.2018.02.023 (2018).
Ziegler, S. G. et al. Ectopic calcification in pseudoxanthoma elasticum responds to inhibition of tissue-nonspecific alkaline phosphatase. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aal1669 (2017).
Orriss, I. R., Arnett, T. R. & Russell, R. G. Pyrophosphate: a key inhibitor of mineralisation. Curr. Opin. Pharmacol. 28, 57–68. https://doi.org/10.1016/j.coph.2016.03.003 (2016).
Jansen, R. S. et al. ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arterioscler. Thromb. Vasc. Biol. 34, 1985–1989. https://doi.org/10.1161/ATVBAHA.114.304017 (2014).
Pomozi, V. et al. Pyrophosphate supplementation prevents chronic and acute calcification in ABCC6-deficient mice. Am. J. Pathol. https://doi.org/10.1016/j.ajpath.2017.02.009 (2017).
Beck, K., Hayashi, K., Dang, K., Hayashi, M. & Boyd, C. D. Analysis of ABCC6 (MRP6) in normal human tissues. Histochem. Cell Biol. 123, 517–528 (2005).
Beck, K. et al. The distribution of Abcc6 in normal mouse tissues suggests multiple functions for this ABC transporter. J. Histochem. Cytochem. 51, 887–902 (2003).
Nitschke, Y. et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am. J. Hum. Genet. 90, 25–39. https://doi.org/10.1016/j.ajhg.2011.11.020 (2012).
Hamlin, N. et al. Acquired Pseudoxanthoma elasticum-like syndrome in beta-thalassaemia patients. Br. J. Haematol. 122, 852–854 (2003).
Martin, L., Douet, V., Vanwart, C. M., Heller, M. B. & Le Saux, O. A mouse model of beta-thalassemia shows a liver-specific down-regulation of Abcc6 expression. Am. J. Pathol. 178, 774–783 (2011).
Pisciotta, L. et al. Pseudoxanthoma elasticum and familial hypercholesterolemia: a deleterious combination of cardiovascular risk factors. Atherosclerosis 210, 173–176. https://doi.org/10.1016/j.atherosclerosis.2009.11.028 (2010).
Aherrahrou, Z. et al. An alternative splice variant in Abcc6, the gene causing dystrophic calcification, leads to protein deficiency in C3H/He mice. J. Biol. Chem. 283, 7608–7615 (2008).
Brampton, C. et al. The level of hepatic ABCC6 expression determines the severity of calcification after cardiac injury. Am. J. Pathol. 184, 159–170 (2014).
Eaton, G. J., Custer, R. P., Johnson, F. N. & Stabenow, K. T. Dystrophic cardiac calcinosis in mice: genetic, hormonal, and dietary influences. Am. J. Pathol. 90, 173–186 (1978).
Everitt, J. I., Olson, L. M., Mangum, J. B. & Visek, W. J. High mortality with severe dystrophic cardiac calcinosis in C3H/OUJ mice fed high fat purified diets. Vet. Pathol. 25, 113–118 (1988).
Wang, S. S. et al. Disruption of the aortic elastic lamina and medial calcification share genetic determinants in mice. Circ. Cardiovasc. Genet. 2, 573–582. https://doi.org/10.1161/CIRCGENETICS.109.860270 (2009).
Campens, L. et al. Characterization of cardiovascular involvement in pseudoxanthoma elasticum families. Arterioscler. Thromb. Vasc. Biol. 33, 2646–2652. https://doi.org/10.1161/ATVBAHA.113.301901 (2013).
Stumpf, M. J. et al. Pseudoxanthoma elasticum - also a microvascular disease. Vasa 49, 57–62. https://doi.org/10.1024/0301-1526/a000811 (2020).
Trip, M. D. et al. Frequent mutation in the ABCC6 gene (R1141X) is associated with a strong increase in the prevalence of coronary artery disease. Circulation 106, 773–775 (2002).
Gorgels, T. G. et al. Disruption of Abcc6 in the mouse: novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum. Mol. Genet. 14, 1763–1773 (2005).
Lebwohl, M. et al. Classification of pseudoxanthoma elasticum: report of a consensus conference. J. Am. Acad. Dermatol. 30, 103–107 (1994).
McGee-Russell, S. M. Histochemical methods for calcium. J. Histochem. Cytochem. 6, 22–42. https://doi.org/10.1177/6.1.22 (1958).
Banuls, C. et al. Comparability of two different polyacrylamide gel electrophoresis methods for the classification of LDL pattern type. Clin. Chim. Acta 413, 251–257. https://doi.org/10.1016/j.cca.2011.09.047 (2012).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. https://doi.org/10.1006/meth.2001.1262 (2001).
Van der Veken, B., Roth, L., De Meyer, G. R. & Martinet, W. Development of atherosclerotic plaques in a mouse model of pseudoxanthoma elasticum. Acta Cardiol. 69, 687–692 (2014).
Germain, D. P., Boutouyrie, P., Laloux, B. & Laurent, S. Arterial remodeling and stiffness in patients with pseudoxanthoma elasticum. Arterioscler. Thromb. Vasc. Biol. 23, 836–841. https://doi.org/10.1161/01.ATV.0000067428.19031.28 (2003).
Kauffenstein, G. et al. Disseminated arterial calcification and enhanced myogenic response are associated with abcc6 deficiency in a mouse model of pseudoxanthoma elasticum. Arterioscler. Thromb. Vasc. Biol. 34, 1045–1056. https://doi.org/10.1161/ATVBAHA.113.302943 (2014).
Kornet, L. et al. In patients with pseudoxanthoma elasticum a thicker and more elastic carotid artery is associated with elastin fragmentation and proteoglycans accumulation. Ultrasound Med. Biol. 30, 1041–1048. https://doi.org/10.1016/j.ultrasmedbio.2004.06.004 (2004).
Leftheriotis, G. et al. The contribution of arterial calcification to peripheral arterial disease in pseudoxanthoma elasticum. PLoS ONE 9, e96003. https://doi.org/10.1371/journal.pone.0096003 (2014).
Leftheriotis, G. et al. The vascular phenotype in Pseudoxanthoma elasticum and related disorders: contribution of a genetic disease to the understanding of vascular calcification. Front. Genet. 4, 4. https://doi.org/10.3389/fgene.2013.00004 (2013).
Monseu, M. et al. Osteoprotegerin levels are associated with liver fat and liver markers in dysmetabolic adults. Diabetes Metab. 42, 364–367. https://doi.org/10.1016/j.diabet.2016.02.004 (2016).
Moore, K. J., Sheedy, F. J. & Fisher, E. A. Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 13, 709–721. https://doi.org/10.1038/nri3520 (2013).
Moore, K. J. & Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355. https://doi.org/10.1016/j.cell.2011.04.005 (2011).
Wang, X. et al. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J. Clin. Investig. 117, 2216–2224. https://doi.org/10.1172/JCI32057 (2007).
Kuzaj, P. et al. ABCC6- a new player in cellular cholesterol and lipoprotein metabolism?. Lipids Health Dis. 13, 118. https://doi.org/10.1186/1476-511X-13-118 (2014).
Yu, L. et al. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc. Natl. Acad. Sci. U. S. A. 99, 16237–16242. https://doi.org/10.1073/pnas.252582399 (2002).
Yu, X. H. et al. ABCG5/ABCG8 in cholesterol excretion and atherosclerosis. Clin. Chim. Acta 428, 82–88. https://doi.org/10.1016/j.cca.2013.11.010 (2014).
Wilund, K. R., Yu, L., Xu, F., Hobbs, H. H. & Cohen, J. C. High-level expression of ABCG5 and ABCG8 attenuates diet-induced hypercholesterolemia and atherosclerosis in Ldlr-/- mice. J. Lipid Res. 45, 1429–1436. https://doi.org/10.1194/jlr.M400167-JLR200 (2004).
Kiec-Wilk, B. et al. Acute myocardial infarction and a new ABCC6 mutation in a 16-year-old boy with pseudoxanthoma elasticum. Int. J. Cardiol. 116, 261–262. https://doi.org/10.1016/j.ijcard.2006.02.022 (2007).
Koblos, G. et al. The R1141X loss-of-function mutation of the ABCC6 gene is a strong genetic risk factor for coronary artery disease. Genet. Test. Mol. Biomarkers 14, 75–78. https://doi.org/10.1089/gtmb.2009.0094 (2010).
Peloso, G. M. et al. Common genetic variation in multiple metabolic pathways influences susceptibility to low HDL-cholesterol and coronary heart disease. J. Lipid Res. 51, 3524–3532. https://doi.org/10.1194/jlr.P008268 (2010).
Wang, J., Near, S., Young, K., Connelly, P. W. & Hegele, R. A. ABCC6 gene polymorphism associated with variation in plasma lipoproteins. J. Hum. Genet. 46, 699–705 (2001).
Miglionico, R. et al. Dysregulation of gene expression in ABCC6 knockdown HepG2 cells. Cell. Mol. Biol. Lett. 19, 517–526. https://doi.org/10.2478/s11658-014-0208-2 (2014).
Pomozi, V. et al. Pyrophosphate supplementation prevents chronic and acute calcification in ABCC6-deficient mice. Am. J. Pathol. 187, 1258–1272. https://doi.org/10.1016/j.ajpath.2017.02.009 (2017).
Hu, X. et al. Analysis of the frequent R1141X mutation in the ABCC6 gene in pseudoxanthoma elasticum. Investig. Ophthalmol. Vis. Sci. 44, 1824–1829 (2003).
Morcher, M., Hausser, I., Brandt, T. & Grond-Ginsbach, C. Heterozygous carriers of Pseudoxanthoma elasticum were not found among patients with cervical artery dissections. J. Neurol. 250, 983–986. https://doi.org/10.1007/s00415-003-1139-4 (2003).
Talayero, B. G. & Sacks, F. M. The role of triglycerides in atherosclerosis. Curr. Cardiol. Rep. 13, 544–552. https://doi.org/10.1007/s11886-011-0220-3 (2011).
Perlman, R. L. Mouse models of human disease: an evolutionary perspective. Evol. Med. Public Health https://doi.org/10.1093/emph/eow014 (2016).
Klement, J. F. et al. Targeted ablation of the abcc6 gene results in ectopic mineralization of connective tissues. Mol. Cell. Biol. 25, 8299–8310 (2005).
Brampton, C. et al. Vitamin K does not prevent soft tissue mineralization in a mouse model of pseudoxanthoma elasticum. Cell Cycle 10, 1810–1820 (2011).
Li, Q. et al. Mouse models for pseudoxanthoma elasticum: genetic and dietary modulation of the ectopic mineralization phenotypes. PLoS ONE 9, e89268. https://doi.org/10.1371/journal.pone.0089268 (2014).
Dedinszki, D. et al. Oral administration of pyrophosphate inhibits connective tissue calcification. EMBO Mol. Med. 9, 1463–1470. https://doi.org/10.15252/emmm.201707532 (2017).
Pomozi, V. et al. Analysis of pseudoxanthoma elasticum-causing missense mutants of ABCC6 in vivo; pharmacological correction of the mislocalized proteins. J. Investig. Dermatol. 134, 946–953. https://doi.org/10.1038/jid.2013.482 (2014).
Pomozi, V. et al. Functional rescue of ABCC6 deficiency by 4-phenylbutyrate therapy reduces dystrophic calcification in Abcc6(-/-) mice. J. Investig. Dermatol. 137, 595–602. https://doi.org/10.1016/j.jid.2016.10.035 (2017).
Pomozi, V. et al. Dietary pyrophosphate modulates calcification in a mouse model of pseudoxanthoma elasticum: implication for treatment of patients. J. Investig. Dermatol. 139, 1082–1088. https://doi.org/10.1016/j.jid.2018.10.040 (2019).
Getz, G. S. & Reardon, C. A. Animal models of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 1104–1115. https://doi.org/10.1161/ATVBAHA.111.237693 (2012).
Pirillo, A., Catapano, A. L. & Norata, G. D. Biological consequences of dysfunctional HDL. Curr. Med. Chem. 26, 1644–1664. https://doi.org/10.2174/0929867325666180530110543 (2019).
Le Saux, O. et al. Serum factors from pseudoxanthoma elasticum patients alter elastic fiber formation in vitro. J. Investig. Dermatol. 126, 1497–1505 (2006).
Quaglino, D. et al. Abnormal phenotype of in vitro dermal fibroblasts from patients with Pseudoxanthoma elasticum (PXE). Biochim. Biophys. Acta 1501, 51–62 (2000).
Hendig, D. et al. Gene expression profiling of ABC transporters in dermal fibroblasts of pseudoxanthoma elasticum patients identifies new candidates involved in PXE pathogenesis. Lab. Investig. 88, 1303–1315 (2008).
Kuzaj, P. et al. Large-scaled metabolic profiling of human dermal fibroblasts derived from pseudoxanthoma elasticum patients and healthy controls. PLoS ONE 9, e108336. https://doi.org/10.1371/journal.pone.0108336 (2014).
Moghadasian, M. H. et al. Pathophysiology of apolipoprotein E deficiency in mice: relevance to apo E-related disorders in humans. FASEB J. 15, 2623–2630. https://doi.org/10.1096/fj.01-0463com (2001).
Schaefer, E. J. et al. Familial apolipoprotein E deficiency. J. Clin. Investig. 78, 1206–1219. https://doi.org/10.1172/JCI112704 (1986).
Ishibashi, S. et al. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Investig. 92, 883–893. https://doi.org/10.1172/JCI116663 (1993).
Fazio, S. et al. Increased atherosclerosis in mice reconstituted with apolipoprotein E null macrophages. Proc. Natl. Acad. Sci. U. S. A. 94, 4647–4652. https://doi.org/10.1073/pnas.94.9.4647 (1997).
Liu, S. et al. Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell 157, 785–794. https://doi.org/10.1016/j.cell.2014.03.054 (2014).
Ostuni, A. et al. Inhibition of ABCC6 transporter modifies cytoskeleton and reduces motility of HepG2 cells via purinergic pathway. Cells https://doi.org/10.3390/cells9061410 (2020).
Dosch, M., Gerber, J., Jebbawi, F. & Beldi, G. Mechanisms of ATP Release by inflammatory cells. Int. J. Mol. Sci. https://doi.org/10.3390/ijms19041222 (2018).
Hasko, G. & Cronstein, B. Regulation of inflammation by adenosine. Front. Immunol. 4, 85. https://doi.org/10.3389/fimmu.2013.00085 (2013).
Silva-Vilches, C., Ring, S. & Mahnke, K. ATP and its metabolite adenosine as regulators of dendritic cell activity. Front. Immunol. 9, 2581. https://doi.org/10.3389/fimmu.2018.02581 (2018).
Buchheiser, A. et al. Inactivation of CD73 promotes atherogenesis in apolipoprotein E-deficient mice. Cardiovasc. Res. 92, 338–347. https://doi.org/10.1093/cvr/cvr218 (2011).
Jalkanen, J., Hollmen, M., Jalkanen, S. & Hakovirta, H. Regulation of CD73 in the development of lower limb atherosclerosis. Purinergic Signal 13, 127–134. https://doi.org/10.1007/s11302-016-9545-0 (2017).
Nitschke, Y., Weissen-Plenz, G., Terkeltaub, R. & Rutsch, F. Npp1 promotes atherosclerosis in ApoE knockout mice. J. Cell. Mol. Med. 15, 2273–2283. https://doi.org/10.1111/j.1582-4934.2011.01327.x (2011).
Sutton, N. R. et al. CD73 Promotes age-dependent accretion of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 40, 61–71. https://doi.org/10.1161/ATVBAHA.119.313002 (2020).
Takenaka, M. C., Robson, S. & Quintana, F. J. Regulation of the T Cell response by CD39. Trends Immunol, 37, 427–439. https://doi.org/10.1016/j.it.2016.04.009 (2016).
Markello, T. C. et al. Vascular pathology of medial arterial calcifications in NT5E deficiency: implications for the role of adenosine in pseudoxanthoma elasticum. Mol. Genet. Metab. 103, 44–50. https://doi.org/10.1016/j.ymgme.2011.01.018 (2011).
De Giorgi, M. et al. Complete deletion of Cd39 is atheroprotective in apolipoprotein E-deficient mice. J. Lipid Res. 58, 1292–1305. https://doi.org/10.1194/jlr.M072132 (2017).
Kanthi, Y. et al. Flow-dependent expression of ectonucleotide tri(di)phosphohydrolase-1 and suppression of atherosclerosis. J. Clin. Investig. 125, 3027–3036. https://doi.org/10.1172/JCI79514 (2015).
Acknowledgements
We thank Dr. Stéphanie Billon-Crossouard of the Plateforme de Spectrometrie de Masse Faculté de Médecine, Université de Nantes, France for the bile acids measurements. Financial support to OLS came from National Institutes of Health HL108249, P20GM113134, G12 MD007601, The American Heart Association grant 11GRNT5840005, the Ingeborg v.F. McKee (15ADVC-74403), the Victoria S. And Bradley L. Geist Funds (18CON-90814) of the Hawaii Community Foundation and PXE International Inc. The Hungarian grant OTKA 128003 provided financial support to VP. The funding agencies were not involved in the design or execution of this study.
Author information
Authors and Affiliations
Contributions
O.L.S. designed and funded the project, and also wrote the manuscript with significant contribution from C.B., V.P., J.Z., G.L. and L.M. L.-H.C., A.A., S.M. and W.A.B. contributed data related to mouse and bone marrow-derived macrophage experiments as shown in Fig. 1–3 and the supplemental figures. C.B., V.P. and J.Z. contributed to Fig. 1–5 as well as the Tables. G.L. provided data for Fig. 6. G.L. and L.M. contributed human samples and data (Table 1, 2, Supplemental Table 1) as well as related interpretation and manuscript preparation. D.H., S.B. and S.K. performed data compilation, analysis and interpretation with text and table editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brampton, C., Pomozi, V., Chen, LH. et al. ABCC6 deficiency promotes dyslipidemia and atherosclerosis. Sci Rep 11, 3881 (2021). https://doi.org/10.1038/s41598-021-82966-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-82966-y
This article is cited by
-
Various vascular malformations are prevalent in Finnish pseudoxanthoma elasticum (PXE) patients: a national registry study
Orphanet Journal of Rare Diseases (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
