
Abstract
Type 2 diabetes mellitus (T2DM) influences the human health and can cause significant illnesses. The genitourinary microbiome profiles in the T2DM patients remain poorly understood. In the current study, a series of bioinformatic and statistical analyses were carried out to determine the multiple bacteria associated with the more dysbiotic genitourinary microbiomes (i.e., those with lower dysbiosis ratio) in T2DM patients, which were sequenced by Illumina-based 16S rRNA gene amplicon sequencing. All the genitourinary microbiomes from 70 patients with T2DM were clustered into three clusters of microbiome profiles, i.e., Cluster_1_T2DM, Cluster_2_T2DM and Cluster_3_T2DM, with Cluster_3_T2DM at the most dysbiotic genitourinary microbial status. The three clustered T2DM microbiomes were determined with different levels of alpha diversity indices, and driven by distinct urinalysis variables. OTU12_Clostridiales and OTU28_Oscillospira were likely to drive the T2DM microbiomes to more dysbiotic status, while OTU34_Finegoldia could play a vital role in maintaining the least dysbiotic T2DM microbiome (i.e., Cluster_1_T2DM). The functional metabolites K08300_ribonuclease E, K01223_6-phospho-beta-glucosidase and K00029_malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+) were most associated with Cluster_1_T2DM, Cluster_2_T2DM and Cluster_3_T2DM, respectively. The characteristics and multiple bacteria associated with the more dysbiotic genitourinary microbiomes in T2DM patients may help with the better diagnosis and management of genitourinary dysbiosis in T2DM patients.
Similar content being viewed by others
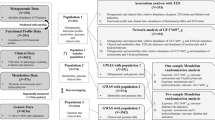


Introduction
Type 2 diabetes mellitus (T2DM) is a global health problem1. It contributes to the increasing rate of non-communicable diseases in both developed and developing counties2, and could lead to mortalities3. The associations between gut microbiome and T2DM have been well studied4,5,6,7,8,9, however, genitourinary microbiomes in T2DM patients were relatively understudied.
Recently, the genitourinary microbiomes of T2DM cohorts have been studied to achieve different objectives10,11,12. Actinobacteria, Collinsella, Desulfovibrio, Enterobacteriaceae, Flavobacteria, Flavobacteriales, Lactobacillus and Porphyromonas were capable of distinguishing the genitourinary microbiomes of T2DM patients from those of healthy subjects9,10. Lactobacillus and Prevotella were predominant in all the four cohorts of T2DM patients in a previous study12: (1) T2DM only, (2) T2DM and hypertension, (3) T2DM and hyperlipidemia, and (4) T2DM, hypertension and hyperlipidemia. Bifidobacteriaceae, Thermaceae and Shuttleworthia could contribute to the presence of interleukin-8 in the urine of T2DM patients11.
The microbiome profiles of the diseased cohorts in different disease studies have attracted increasing scientific attention13,14,15,16. Cirrhosis dysbiosis ratio was associated with the severity of liver cirrhosis17, and the dysbiosis ratio of microbiome has been used to evaluate the dysbiotic statuses of different microbiome profiles18,19, i.e., a greater dysbiosis ratio represented a less dysbiotic status. We hypothesised that there was a specific genitourinary microbiome profile of the recruited T2DM cohort was at a more dysbiotic status. In the current study, we aim to (1) determine the characteristics and dysbiotic statuses of different genitourinary microbiome profiles in the T2DM patients; (2) investigate the phylotypes associated with the more dysbiotic genitourinary microbiome in T2DM patients.
Methods and materials
Recruitment of subjects and sample collection
Seventy female patients with T2DM and 70 healthy female subjects were recruited in the present study, and the selection criteria of T2DM patients were described in our previous study12. Briefly, both patients and healthy individuals aged between 26 and 85 years old (Supplementary Fig. S1), and had similar body mass indices. Individuals with the following attributes were not included in this study: intake of antibiotics, probiotics, prebiotics or synbiotics in the past three months; menstruation during the study; with relevant genitourinary diseases or abnormal conditions.
The urine samples were collected by using a modified midstream urine collection technique involving disinfection and a four-tube collection method12. Briefly, four opened 50-ml sterile centrifuge tubes were prepared with lids upwards. The participants were pants off and squatted on a squat toilet. Antiseptic cotton balls were handled by disinfected hand to clean the far labial fold and then the near labial fold. The labia were held apart and the participant urinated into the four tubes (tubes 1–4) in order. The urine in tube 2 and 3 were aliquoted for three subsequent analyses: 15 ml for urinalysis, 1 ml for urine culture and 40 ml for Illumina sequencing.
Written informed consent was taken from all the participants prior to enrolment, and the study protocol was approved by the Institutional Review Board of the First Affiliated Hospital, School of Medicine, Zhejiang University (Zhejiang, China). The study was carried out under relevant guidelines and regulations (Declaration of Helsinki).
Molecular methods
Bacterial genomic DNA of all the urine samples from all the T2DM patients and healthy subjects, as well as a DNA-free water sample (blank control), was extracted by Liu, et al.12, followed by the amplifications of barcoded 16S rDNA primers targeting the V3–V4 regions of all the extracted DNA samples. The amplicons were purified, quantified, pooled, and then sequenced on the Illumina MiSeq instrument using 2-by-300 bp chemistry12.
Processing of the sequencing data
The raw sequencing data were processed as described by Liu et al.12, including sequencing merge, chimera check, quality filtering and taxonomy assignment by using QIIME (version 1.9.0). The sample rarefaction was performed using the phyloseq package in R (version 3.6.1).
Microbial dysbiotic status of genitourinary microbiomes in T2DM and healthy cohorts
Linear discriminant analysis (LDA) effect size (LEfSe) was carried out in a program run by the Huttenhower lab to determine the OTUs associated with the genitourinary microbiomes of T2DM patients or healthy subjects.
The microbial dysbiosis ratio (MDR), i.e., the abundance ratio of “good and bad taxa”, was used to help determine the dysbiotic status of microbiomes in different disease studies17,20. In the present study, genitourinary MDR was defined as the abundance ratio of genitourinary OTUs associated with healthy cohort (n = 70) and genitourinary OTUs associated with T2DM cohort (n = 70). The genitourinary MDRs of the genitourinary microbiomes in healthy and T2DM cohorts were transformed in log10 to satisfy the assumptions of normal distribution and equal variance, before being compared by a t test.
A spearman’s test was performed to determine the correlations between the multiple variables and diversity indices (i.e., observed species, Shannon and Pielou indices) in all T2DM patients.
Clustering of the genitourinary microbiomes in T2DM patients
Partition around medoids (PAM) clustering analysis has been used in different disease studies for achieving different objectives15,21,22. In the current study, PAM analysis was carried out to cluster the genitourinary microbiomes from all the 70 T2DM patients, after the optimal number of clusters was determined by the average silhouette method23. Three clusters of T2DM microbiomes were determined, i.e., Cluster_1_T2DM, Cluster_2_T2DM and Cluster_3_T2DM.
Correlations between urinalysis variables and T2DM microbiome in each of the three clusters
The urinalysis variables with the significant effects on the genitourinary microbiomes were determined with one-way ANOVA24, and the variables with P < 0.05 were selected for the distance-based redundancy analysis (db-RDA) in Primer v7 (Primer-e Ltd., New Zealand).
Comparisons of T2DM associated OTUs between the three clustered T2DM microbiomes
To determine whether the T2DM associated OTUs had different associations with the three clusters of T2DM microbiomes, the average abundances of T2DM associated OTUs in the three clustered T2DM microbiomes were compared with Kruskal–Wallis tests. Mann–Whitney tests were carried out for the pairwise comparisons, and Bonferroni correction was used for adjusting the P values. The average abundances of the T2DM associated OTUs (that were more abundant in one or two clustered microbiomes) in the three clustered T2DM microbiomes were visualized in a heatmap in R version 3.6.1.
Differences between the three clustered T2DM microbiomes
Permutation analysis of variance (PERMANOVA) was applied in R by using adonis command25, to compare the three clustered genitourinary microbiomes from the T2DM patients. Similarity percentage (SIMPER) analysis was performed in Primer v7 to determine the similarities within each of the three clustered T2DM microbiomes, as well as the dissimilarities between the three clustered microbiomes.
The observed species (richness) of the three clustered T2DM microbiomes were transformed in square root to satisfy the assumptions of normal distribution and equal variance. A one-way analysis of variance (ANOVA) was performed to compare the transformed observed species, Shannon index (both richness and evenness) and Pielou index (evenness) of the three clustered T2DM microbiomes. T tests were used for the pairwise comparisons, with Bonferroni correction for adjusting the P values.
Genitourinary MDRs of the three clustered T2DM microbiomes were transformed in log10 before being compared with one-way ANOVA. T tests were carried out for the pairwise comparisons of genitourinary MDRs, with Bonferroni correction for adjusting the P values.
A LEfSe analysis was used to determine the representative OTUs associated with each of the three clustered T2DM microbiomes, with LDA threshold over 2.526,27. Pairwise SIMPER analyses were conducted to determine the OTUs contributing most to the dissimilarities between the most dysbiotic microbiomes (i.e., Cluster_3_T2DM) and each of the less dysbiotic T2DM microbiomes (i.e., Cluster_1_T2DM and Cluster_2_T2DM), with a cut-off of 70%28,29. The Venny program version 2.130 was used to determine whether any representative OTUs associated with Cluster_3_T2DM could also contribute most to the dissimilarities between Cluster_3_T2DM and Cluster_1_T2DM/Cluster_2_T2DM.
Kruskal–Wallis test was used to compare the multiple variables of the T2DM patients in the three clusters. Mann–Whitney test was performed for pairwise comparisons of marriage times in the three clustered patients.
Network and fragmentation analyses
Co-occurrence Network inference (CoNet) program was used to determine the correlations of the OTUs within each of the three clustered T2DM microbiomes, based on an ensemble of correlation measures as described by Faust, et al.31. The detailed manipulations followed Wagner Mackenzie, et al.21. Five coefficients, i.e., Mutual Information, Pearson, Bray Curtis, Spearman and Kullback–Leibler dissimilarities for the ensemble inference and the greatest 1000 positive and negative correlations were determined. The permutations were used to compute the preliminary individual method-specific P values, before computing the bootstraps by merging all the initial P values into a final P value by using Brown's method32.
Network fragmentation calculations and generation of a null distribution were carried out in R using the package igraph33, to determine the gatekeeper(s) in each of the three clustered T2DM microbiomes that could cause collapse of the corresponding clustered microbiomes. The details of this approach were described in Wagner Mackenzie, et al.21. A null distribution of fragmentation scores was created from 10,000 randomly constructed networks with identical node and edge distributions to the original network. Statistical significance was defined as the number of times a fragmentation score greater than that resulting from the removal of the OTU within the null distribution.
Correlations between urinalysis variables and representative OTUs in each of the three clustered T2DM microbiomes
The correlations between urinalysis variables and representative OTUs in each of the three clustered T2DM microbiomes were determined by CoNet analysis and visualized in Cytoscape software version 3.7.234. The detailed procedures of CoNet analysis were described above following Wagner Mackenzie et al.21.
Functional metabolites associated with each of the three clustered T2DM microbiomes
The functional metabolites for the three clustered T2DM microbiomes were determined with a Tax4fun package in R35. A LEfSe analysis was performed to determine the functional metabolites associated with each of the three clustered T2DM microbiomes. Those functional metabolites with LDA score over 2.5 and consistently significant across either of the three clustered T2DM microbiomes were determined being associated with the corresponding clusters of T2DM microbiomes.
Results
Microbial dysbiotic status of genitourinary microbiomes in T2DM and healthy cohorts
LEfSe analysis determined that 27 OTUs were associated with T2DM, and 71 OTUs associated with healthy cohort (Supplementary Table S1). The genitourinary MDR was greater in the genitourinary microbiomes of healthy subjects (median 34 ± SE 129) compared with T2DM patients (median 0.8 ± SE 0.9) (t test, P < 0.001), suggesting the genitourinary microbiomes in T2DM patients were at more dysbiotic status compared with those of healthy cohort at baseline.
No variable was determined as confounder to the microbiome diversity indices in T2DM cohort (− 0.37 < all correlation coefficient < 0.32) (Supplementary Table S2).
Clustering of genitourinary microbiomes in T2DM patients
Silhouette analysis determined three as the most optimal number for clustering the T2DM microbiomes (Fig. 1A). The three clustered T2DM microbiomes, i.e., Cluster_1_T2DM, Cluster_2_T2DM and Cluster_3_T2DM, contained 16, 42 and 12 individual T2DM microbiomes, respectively (Fig. 1B). The three clusters of T2DM microbiomes and healthy microbiomes were determined with different abundant families (Supplementary Fig. S2).

Clustering of the genitourinary microbiomes in T2DM patients by (A) average silhouette analysis and (B) partition around medoids clustering analysis, into three clusters of T2DM microbiomes, i.e., Cluster_1_T2DM, Cluster_2_T2DM and Cluster_3_T2DM. Note: the analyses were performed in R version 3.6.1.
Correlations between urinalysis variables and T2DM microbiomes in each of the three clusters
Different urinalysis variables were determined to influence the different microbiome profiles of T2DM patients according to the db-RDA results. Urine color, urine pH and urine protein greatly influenced the Cluster_1_T2DM (Fig. 2A). Likewise, BMI and urine crystal had great influences on Cluster_2_T2DM (Fig. 2B), while electrical conductivity and urine protein greatly influenced Cluster_3_T2DM (Fig. 2C).

The urinalysis variables determined to affect the three clustered T2DM microbiomes, i.e., (A) Cluster_1_T2DM (red), (B) Cluster_2_T2DM (yellow) and (C) Cluster_3_T2DM (blue). Note: the analysis was carried out in a PRIMER7 software.
Comparisons of T2DM associated OTUs between the three clustered T2DM microbiomes
The average abundances of T2DM associated OTUs in the three clustered T2DM microbiomes were compared to determine their associations with the different clusters of T2DM microbiomes. A total of 13 OTUs associated with T2DM were determined with different average abundances between the three clustered T2DM microbiomes (Kruskal–Wallis test, all P < 0.05), nine of which were more abundant in one or two cluster(s) of T2DM microbiomes (Mann–Whitney test, all P < 0.05). OTU53_Lachnospiraceae, OTU92_Enhydrobacter and OTU142_Fusobacterium were more abundant in Cluster_1_T2DM (Fig. 3). OTU8_Coprococcus, OTU16_Lachnospiraceae and OTU108_Coprococcus were more abundant in Cluster_2_T2DM, while OTU85_Ruminococcaceae was more abundant in Cluster_3_T2DM (Fig. 3). OTU74_Dorea had greater abundances in both Cluster_1_T2DM and Cluster_3_T2DM, while OTU65_Fusobacterium had greater abundances in both Cluster_2_T2DM and Cluster_3_T2DM (Fig. 3).

Distribution of T2DM associated OTUs in the three clustered T2DM patients visualized in a heatmap. Note: the abundance of the OTUs were transformed in log2(raw read + 1) in the heatmap, and the heatmap figure was done in R version 3.6.1.
Differences between the three clustered T2DM microbiomes
PERMANOVA showed a significant difference between the three clustered T2DM microbiomes (R2 = 0.27, P < 0.001). The similarity within Cluster_2_T2DM (SIMPER average similarity = 46%) was higher than those within Cluster_1_T2DM (SIMPER average similarity = 16%) or Cluster_3_T2DM (SIMPER average similarity = 10%). The dissimilarities between the three clustered T2DM microbiomes were all over 90%.
There were significant differences in the observed species, Shannon and Pielou indices between the three clustered T2DM microbiomes (one-way ANOVA, all P < 0.001). The observed species and Shannon index were both largest in Cluster_1_T2DM compared with Cluster_2_T2DM and Cluster_3_T2DM (t test, all P < 0.02) (Table 1). The Pielou index of Cluster_1_T2DM was significantly larger than that of Cluster_2_T2DM (t test, P < 0.001), and slightly larger than Cluster_3_T2DM (t test, P = 0.053) (Table 1).
The genitourinary MDR was lower in Cluster_3_T2DM (Median 0.05 ± SE 2.38) compared with Cluster_1_T2DM (5.33 ± 2.63) and Cluster_2_T2DM (0.92 ± 4.15) (t test, both P < 0.01), while it was similar between Cluster_1_T2DM and Cluster_2_T2DM (t test, P = 0.425). This suggests that Cluster_3_T2DM was at more dysbiotic status compared to Cluster_1_T2DM and Cluster_2_T2DM.
LEfSe analysis revealed 32 representative OTUs had different associations with the three clustered T2DM microbiomes (Fig. 4). OTU12_Clostridiales, OTU28_Oscillospira, OTU348_Veillonella and OTU56_Candidatus Koribacter were the representative phylotypes more associated with Cluster_3_T2DM (Fig. 4), among which, OTU12_Clostridiales and OTU28_Oscillospira were also determined largely contributing to the dissimilarities between Cluster_3_T2DM and the two other clustered T2DM microbiomes according to the pairwise SIMPER results (Supplementary Table S3). Among the 26 OTUs more associated with Cluster_1_T2DM, OTU22_Citrobacter, OTU463_Rhizobiaceae, OTU77_Corynebacterium and OTU34_Finegoldia were the most associated representative phylotypes. In addition, OTU7_Lachnospiraceae and OTU181_Alphaproteobacteria were more associated with Cluster_2_T2DM.

The phylotypes associated with each of the three clustered T2DM microbiomes determined by Linear Discriminant Analysis (LDA) Effect Size (LEfSe). Note: the analysis was done on the Huttenhower online software (http://huttenhower.sph.harvard.edu/galaxy).
No significant difference was determined in the majority of variables (e.g., urine glucose) of the T2DM patients in the three clusters (Kruskal–Wallis test, P > 0.5), except marriage times (Supplementary Table S4). The marriage times of Cluster_3 was significantly greater than that of Cluster_2 (Mann–Whitney test, P < 0.01), and similar to that of Cluster_1 (Mann–Whitney test, P > 0.2).
Network and fragmentation analyses
The top 10 OTUs with most correlations in each of the three clustered T2DM microbiomes were largely distinct (Supplementary Table S5). Among them, OTU9_Enterobacteriaceae, OTU13_Novosphingobium and OTU778_Lachnospiraceae had most correlations in Cluster_1_T2DM, Cluster_2_T2DM and Cluster_3_T2DM, respectively.
The fragmentation analysis is used to evaluate the extent of microbiome network fragmentation. In the current study, the fragmentation level of Cluster_1_T2DM (0.611) was larger than those of Cluster_2_T2DM (0.564) and Cluster_3_T2DM (0.353). Gatekeepers were OTUs that hold together the microbiome by interacting with different parts of the network21, and they were determined by a fragmentation analysis in the present study. One OTU associated with Cluster_1_T2DM, i.e., OTU34_Finegoldia, was also determined as a gatekeeper to Cluster_1_T2DM (Fragmentation analysis, P = 0.023). By contrast, none of the OTUs associated with Cluster_2_T2DM or Cluster_3_T2DM could cause a collapse of networks of the two clustered T2DM microbiomes (Fragmentation analysis, all P > 0.05).
Correlations between urinalysis variables and T2DM associated OTUs in each of the three clusters
Only negative correlations were determined between the urinalysis variables and representative OTUs in the three clustered T2DM microbiomes. In Cluster_1_T2DM, “Genitourinary tract infections over the previous year” was negatively correlated with OTU278_Rhizobiales; Urine protein was negatively correlated with OTU337_Clostridiales, OTU356_Cytophagaceae and OTU463_Rhizobiaceae; while urine crystal was negatively correlated with OTU524_Bacillus and OTU1142_Lachnospiraceae (Fig. 5A). In Cluster_2_T2DM, asymptomatic bacteriuria was negatively correlated with OTU7_Lachnospiraceae and OTU181_Alphaproteobacteria (Fig. 5B). Urine glucose, urine mucus and sick time were negatively correlated with OTU348_Veillonella in Cluster_3_T2DM (Fig. 5C).

Correlations between the representative OTUs and urinalysis variables in (A) Cluster_1_T2DM, (B) Cluster_2_T2DM and (C) Cluster_3_T2DM. Note: the analysis was performed in Cytoscape 3.7.2. The purple and red lines represented positive and negative correlations, respectively.
Functional metabolites associated with each of the three clustered T2DM microbiomes
A total of 82 functional metabolites had different associations with the three clustered T2DM microbiomes, i.e., four for Cluster_1_T2DM, 60 for Cluster_2_T2DM and 18 for Cluster_3_T2DM (Fig. 6). Among them, K08300_ribonuclease E, K01223_6-phospho-beta-glucosidase and K00029_malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+) were most associated with Cluster_1_T2DM, Cluster_2_T2DM and Cluster_3_T2DM, respectively.

Functional metabolites associated with each of the three clustered T2DM microbiomes determined by LEfSe analysis. Note: the analysis was done on the Huttenhower online software (http://huttenhower.sph.harvard.edu/galaxy).
Discussion
Genitourinary microbiome in human beings have been well studied36,37,38. Alterations of genitourinary microbiome have been investigated in T2DM female patients and healthy female subjects, and in T2DM patients with or without other conditions (i.e., hypertension and hyperlipidemia)9,10,12. However, the genitourinary microbiome profiles in the T2DM patients remain poorly understood. The current study investigated the characteristics and multiple bacteria associated with different genitourinary microbiome profiles in T2DM patients, aiming to determine the phylotypes associated with the more dysbiotic T2DM microbiomes.
In the current study, the genitourinary T2DM microbiomes were clustered into three clusters, i.e., Cluster_1_T2DM, Cluster_2_T2DM and Cluster_3_T2DM, with great difference in the microbiome compositions. A group of nine OTUs associated with T2DM were significantly more abundant in one or two of the three clustered T2DM microbiomes, suggesting they had different associations with the three clustered T2DM microbiomes. Ruminococcaceae was enriched in the gut of mice with T2DM39. In the present study, OTU85_Ruminococcaceae was more abundant in the most dysbiotic microbiomes (i.e., Cluster_3_T2DM), suggesting Ruminococcaceae could be associated with the microbiomes in more than one organ of the T2DM cohorts. As for the T2DM associated OTUs with similar abundances in the clustered T2DM microbiomes, we acknowledge that whether they consistently contribute to all the three clustered T2DM microbiomes or have different functions needs further investigations.
Dysbiosis ratios of bacterial phylotypes are associated with multiple diseases17,20,40,41. For instance, lower cirrhosis dysbiosis ratio (i.e., abundance ratio of “good and bad taxa”) of gut microbiome was associated with more severe liver cirrhosis in the cirrhotic patients17. In the present study, the genitourinary MDR was significantly larger in the genitourinary microbiomes of healthy subjects than in T2DM microbiomes, suggesting the larger genitourinary MDR is associated with less dysbiotic status, which is consistent with the other disease studies17,20. The lowest genitourinary MDR in Cluster_3_T2DM suggests that Cluster_3_T2DM was at the most dysbiotic status, while the greatest genitourinary MDR in Cluster_1_T2DM suggested that Cluster_1_T2DM was at the least dysbiotic status. Richness and Shannon indices were both greater in the genitourinary microbiomes of healthy subjects compared with T2DM microbiomes10. In the current study, richness and Shannon indices were greatest in Cluster_1_T2DM, further suggesting Cluster_1_T2DM were at the least dysbiotic status among the three clustered T2DM microbiomes. The association of the three genitourinary microbiome profiles and T2DM severities/progression stages were not determined, as the information about T2DM severity and progression was not well recorded for the current study. We acknowledge that these need to be investigated in the future study.
A total of 32 representative phylotypes were associated with the three clustered T2DM microbiomes in this study. OTU12_Clostridiales, OTU28_Oscillospira, OTU348_Veillonella and OTU56_Candidatus Koribacter were associated with Cluster_3_T2DM. The three taxa, i.e., Clostridiales, Oscillospira and Veillonella, were also reported being associated with the other T2DM cohorts in the other studies42,43,44. Clostridiales was enriched in the gut of T2DM patients compared with healthy subjects42. Oscillospira was more abundant in the gut microbiomes of obese rodents compared with healthy cohort43. Veillonella was more abundant in the subgingival microbiomes of T2DM patients compared with nondiabetic subjects44. Among the four representative phylotypes in Cluster_3_T2DM, both OTU12_Clostridiales and OTU28_Oscillospira were also determined contributing most to the dissimilarities between Cluster_3_T2DM and the two other clustered T2DM microbiomes according to the pairwise SIMPER analyses, suggesting the two phylotypes could play vital roles in Cluster_3_T2DM. By contrast, only one of the two representative phylotypes in Cluster_2_T2DM, i.e., Alphaproteobacteria, was reported being enriched in the ocular surface of T2DM patients than healthy individuals45.
Among the 26 representative phylotypes in Cluster_1_T2DM, Citrobacter, Corynebacterium and Finegoldia were determined with different correlations with diabetes or relevant conditions46,47,48. Citrobacter was associated with asymptomatic bacteriuria in the urine of T2DM patients46. Corynebacterium was less dominant in the conjunctival microbiome of diabetes patients compared with healthy subjects47. Likewise, Finegoldia was less abundant in the gut of women with diabetes compared with healthy subjects48. In the current study, OTU34_Finegoldia acted as a gatekeeper in Cluster_1_T2DM, suggesting it may help maintain the less dysbiotic status of the T2DM patients (i.e., those patients within Cluster_1).
Fragmentation analysis has been used to investigate the fragmentation levels of microbiomes in different studies15,21. In the present study, the fragmentation level was greater in Cluster_1_T2DM compared with those of Cluster_2_T2DM or Cluster_3_T2DM, suggesting that less co-occurrence patterns and decreased biotic interactions in Cluster_1_T2DM.
The urinalysis variables were associated with the microbiomes or phylotypes in different hosts49,50,51,52. Our previous study has demonstrated that the associations between phylotypes and urinalysis variables in T2DM patients with or without hypertension or hyperlipidemia12. In the present study, the representative phylotypes had different associations with the different urinalysis variables within each of the three clustered T2DM microbiomes, suggesting that the representative phylotypes could be influenced by different urinalysis variables in each of the three clustered microbiomes. Likewise, the T2DM microbiomes in the three clusters were driven by relatively distinct urinalysis variables, suggesting the health status of the three clustered patients were largely different. Urine electrical conductivity and urine protein were determined to influence the more dysbiotic microbiomes (i.e., Cluster_3_T2DM) in this study, and the relevant mechanisms deserves further investigation. Confounding variables vary greatly in different studies53,54,55. While no confounding variable was determined in the current study, some alternative variables (e.g., hemoglobin A1c and medication regime) could be potential confounders to the microbiome diversity, which deserves further investigation. No significant difference was determined in the fasting blood glucose and glycosuria levels (i.e., urine glucose) of the three clustered T2DM cohorts, suggesting the two variables were not the driving factors for the microbiome compositions. A significant difference was determined in the marriage times among the three clustered T2DM cohorts, suggesting the marriage times was associated with the clustering of the T2DM microbiomes.
These results based on our 16S sequencing data could provide some useful information, but we acknowledge that whole genome sequencing and empirical metabolomics could be performed to verify the functional metabolites and identify the phylogenetics at a higher resolution in the future work.
In conclusion, there were great differences between the three clustered T2DM microbiomes, while Cluster_3_T2DM was at the most dysbiotic status. OTU12_Clostridiales and OTU28_Oscillospira were likely to drive the T2DM microbiomes to more dysbiotic status, while OTU34_Finegoldia could play a vital role in maintaining the stability of less dysbiotic microbiomes. The characteristics and multiple bacteria associated with the more dysbiotic genitourinary T2DM microbiomes may help with the better diagnosis and management of genitourinary dysbiosis in T2DM patients.
Data availability
The datasets for the current study are available from the corresponding author on reasonable request.
References
DeFronzo, R. A. et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 1, 15019 (2015).
Chaudhury, A. et al. Clinical review of antidiabetic drugs: Implications for type 2 diabetes mellitus management. Front. Endocrinol. 8, 6 (2017).
Zinman, B. et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 373, 2117–2128 (2015).
Forslund, K. et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528, 262 (2015).
Allin, K. H., Nielsen, T. & Pedersen, O. Mechanisms in endocrinology: Gut microbiota in patients with type 2 diabetes mellitus. Eur. J. Endocrinol. 172, 167–177 (2015).
Udayappan, S. D., Hartstra, A., Dallinga-Thie, G. & Nieuwdorp, M. Intestinal microbiota and faecal transplantation as treatment modality for insulin resistance and type 2 diabetes mellitus. Clin. Exp. Immunol. 177, 24–29 (2014).
Han, J.-L. & Lin, H.-L. Intestinal microbiota and type 2 diabetes: From mechanism insights to therapeutic perspective. World J. Gastroenterol. 20, 17737 (2014).
Larsen, N. et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 5, e9085 (2010).
Penckofer, S. et al. Characteristics of the microbiota in the urine of women with type 2 diabetes. J. Diabetes Complicat. 34, 107561 (2020).
Liu, F. et al. Dysbiosis of urinary microbiota is positively correlated with type 2 diabetes mellitus. Oncotarget 8, 3798 (2017).
Ling, Z., Liu, F., Shao, L., Cheng, Y. & Li, L. Dysbiosis of the urinary microbiota associated with urine levels of proinflammatory chemokine interleukin-8 in female type 2 diabetic patients. Front. Immunol. 8, 1032 (2017).
Liu, F. et al. Alterations of urinary microbiota in type 2 diabetes mellitus with hypertension and/or hyperlipidemia. Front. Physiol. 8, 126 (2017).
Stokholm, J. et al. Maturation of the gut microbiome and risk of asthma in childhood. Nat. Commun. 9, 141 (2018).
Seekatz, A. M., Rao, K., Santhosh, K. & Young, V. B. Dynamics of the fecal microbiome in patients with recurrent and nonrecurrent Clostridium difficile infection. Genome Med. 8, 47 (2016).
Zha, H. et al. Bacterial communities associated with tail fan necrosis in spiny lobster, Jasusedwardsii. FEMS Microbiol. Ecol. 95, fiz070 (2019).
Chen, Y. et al. Ascitic bacterial composition is associated with clinical outcomes in cirrhotic patients with culture-negative and nonneutrocytic ascites. Front. Cell. Infect. Microbiol. 8, 420 (2018).
Bajaj, J. S. et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 60, 940–947 (2014).
Zha, H. et al. Vital members in the more dysbiotic oropharyngeal microbiotas in H7N9-infected patients. Front. Med. 7, 396 (2020).
Zha, H. et al. Characteristics of three microbial colonization states in the duodenum of the cirrhotic patients. Future Microbiol. 15, 855–868 (2020).
He, C. et al. Characteristics of mucosa-associated gut microbiota during treatment in Crohn’s disease. World J. Gastroenterol. 25, 2204 (2019).
Wagner Mackenzie, B. et al. Bacterial community collapse: A meta-analysis of the sinonasal microbiota in chronic rhinosinusitis. Environ. Microbiol. 19, 381–392 (2017).
Zha, H. et al. Vital members in the gut microbiotas altered by two probiotic Bifidobacterium strains against liver damage in rats. BMC Microbiol. 20, 144 (2020).
Rousseeuw, P. J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 20, 53–65 (1987).
Meola, M., Lazzaro, A. & Zeyer, J. Diversity, resistance and resilience of the bacterial communities at two alpine glacier forefields after a reciprocal soil transplantation. Environ. Microbiol. 16, 1918–1934 (2014).
Anderson, M. J. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 26, 32–46 (2001).
Wang, X., Zhao, J. & Qin, L. VEGF-C mediated enhancement of lymphatic drainage reduces intestinal inflammation by regulating IL-9/IL-17 balance and improving gut microbiota in experimental chronic colitis. Am. J. Transl. Res. 9, 4772 (2017).
Abbas, T. et al. Anammox microbial community and activity changes in response to water and dissolved oxygen managements in a paddy-wheat soil of Southern China. Sci. Total Environ. 672, 305–313 (2019).
Brugnano, C., Granata, A., Guglielmo, L. & Zagami, G. Spring diel vertical distribution of copepod abundances and diversity in the open Central Tyrrhenian Sea (Western Mediterranean). J. Mar. Syst. 105, 207–220 (2012).
Blasi, S. et al. Soil microarthropod communities from Mediterranean forest ecosystems in Central Italy under different disturbances. Environ. Monit. Assess. 185, 1637–1655 (2013).
Oliveros, J. V. An interactive tool for comparing lists with Venn’s diagrams. https://bioinfogp.cnb.csic.es/tools/venny/index.html (2015).
Faust, K. et al. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 8, e1002606 (2012).
Brown, M. B. 400: A method for combining non-independent, one-sided tests of significance. Biometrics 31, 987–992 (1975).
Widder, S. et al. Fluvial network organization imprints on microbial co-occurrence networks. Proc. Natl. Acad. Sci. 111, 12799–12804 (2014).
Otasek, D., Morris, J. H., Bouças, J., Pico, A. R. & Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 20, 1–15 (2019).
Aßhauer, K. P., Wemheuer, B., Daniel, R. & Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 31, 2882–2884 (2015).
Siddiqui, H., Nederbragt, A. J., Lagesen, K., Jeansson, S. L. & Jakobsen, K. S. Assessing diversity of the female urine microbiota by high throughput sequencing of 16S rDNA amplicons. BMC Microbiol. 11, 244 (2011).
Siddiqui, H., Lagesen, K., Nederbragt, A. J., Jeansson, S. L. & Jakobsen, K. S. Alterations of microbiota in urine from women with interstitial cystitis. BMC Microbiol. 12, 205 (2012).
Xie, J. et al. Profiling the urinary microbiome in men with calcium-based kidney stones. BMC Microbiol. 20, 1–10 (2020).
Wang, H.-Y. et al. Polysaccharide from tuberous roots of Ophiopogon japonicus regulates gut microbiota and its metabolites during alleviation of high-fat diet-induced type-2 diabetes in mice. J. Funct. Foods 63, 103593 (2019).
De Minicis, S. et al. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology 59, 1738–1749 (2014).
Lu, Y. et al. Mucosal adherent bacterial dysbiosis in patients with colorectal adenomas. Sci. Rep. 6, 26337 (2016).
Remely, M. et al. Abundance and diversity of microbiota in type 2 diabetes and obesity. J. Diabetes Metab. 4, 253 (2013).
Jiao, N. et al. High-fat diet increases clostridium clusters XIVa in obese rodents: A meta-analysis. Gastroenterology 152, S1012 (2017).
Casarin, R. et al. Subgingival biodiversity in subjects with uncontrolled type-2 diabetes and chronic periodontitis. J. Periodont. Res. 48, 30–36 (2013).
Li, S. et al. How ocular surface microbiota debuts in type 2 diabetes mellitus. Front. Cell. Infect. Microbiol. 9, 202 (2019).
Bharti, A. et al. Asymptomatic bacteriuria among the patients of type 2 diabetes mellitus. J. Fam. Med. Prim. Care 8, 539–543 (2019).
Bilen, H. et al. Conjunctival flora in patients with type 1 or type 2 diabetes mellitus. Adv. Ther. 24, 1028–1035 (2007).
Moon, J.-Y. et al. Gut microbiota and plasma metabolites associated with diabetes in women with, or at high risk for HIV infection. EBioMedicine 37, 392–400 (2018).
De Theije, C. G. et al. Altered gut microbiota and activity in a murine model of autism spectrum disorders. Brain Behav. Immun. 37, 197–206 (2014).
Nakayama, J. et al. Impact of westernized diet on gut microbiota in children on Leyte island. Front. Microbiol. 8, 197 (2017).
Kreisinger, J. et al. Fecal microbiota associated with phytohaemagglutinin-induced immune response in nestlings of a passerine bird. Ecol. Evol. 8, 9793–9802 (2018).
Wu, W. et al. Protective effect of Akkermansia muciniphila against immune-mediated liver injury in a mouse model. Front. Microbiol. 8, 1804 (2017).
Aho, V. T. et al. Gut microbiota in Parkinson’s disease: Temporal stability and relations to disease progression. EBioMedicine 44, 691–707 (2019).
Hermes, G., Zoetendal, E. & Smidt, H. Molecular ecological tools to decipher the role of our microbial mass in obesity. Benef. Microbes 6, 61–81 (2015).
Mbakwa, C. A. et al. Gut microbiota and body weight in school-aged children: The KOALA birth cohort study. Obesity 26, 1767–1776 (2018).
Acknowledgements
Thanks to Yonghong Xiao, Qing Yang, Baohong Wang for their help in the laboratory. This work was supported by the National Natural Science Foundation of China (82003441), the National Key Research and Development Program of China (2018YFC2000500), the National Natural Science Foundation of China (81790631).
Author information
Authors and Affiliations
Contributions
H.Z. and L.L. designed the study. H.Z. completed all the analyses, the draft and revision of the manuscript. F.L. provided the raw data. F.L. and Z.L. helped with the revision of the manuscript. K.C. and J.Y. reviewed and corrected the bioinformatic and statistical analyses. All authors have reviewed and approved the submitted version of manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zha, H., Liu, F., Ling, Z. et al. Multiple bacteria associated with the more dysbiotic genitourinary microbiomes in patients with type 2 diabetes mellitus. Sci Rep 11, 1824 (2021). https://doi.org/10.1038/s41598-021-81507-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-81507-x
This article is cited by
-
Characteristics of the intestinal bacterial microbiota profiles in Bifidobacterium pseudocatenulatum LI09 pre-treated rats with D-galactosamine-induced liver injury
World Journal of Microbiology and Biotechnology (2023)
-
Time-course alterations of gut microbiota and short-chain fatty acids after short-term lincomycin exposure in young swine
Applied Microbiology and Biotechnology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
